
Transcriptome Profiling Identifies Key Regulators of Tuber Skin Color in Potato


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Phenotypic and Genotypic Characterization
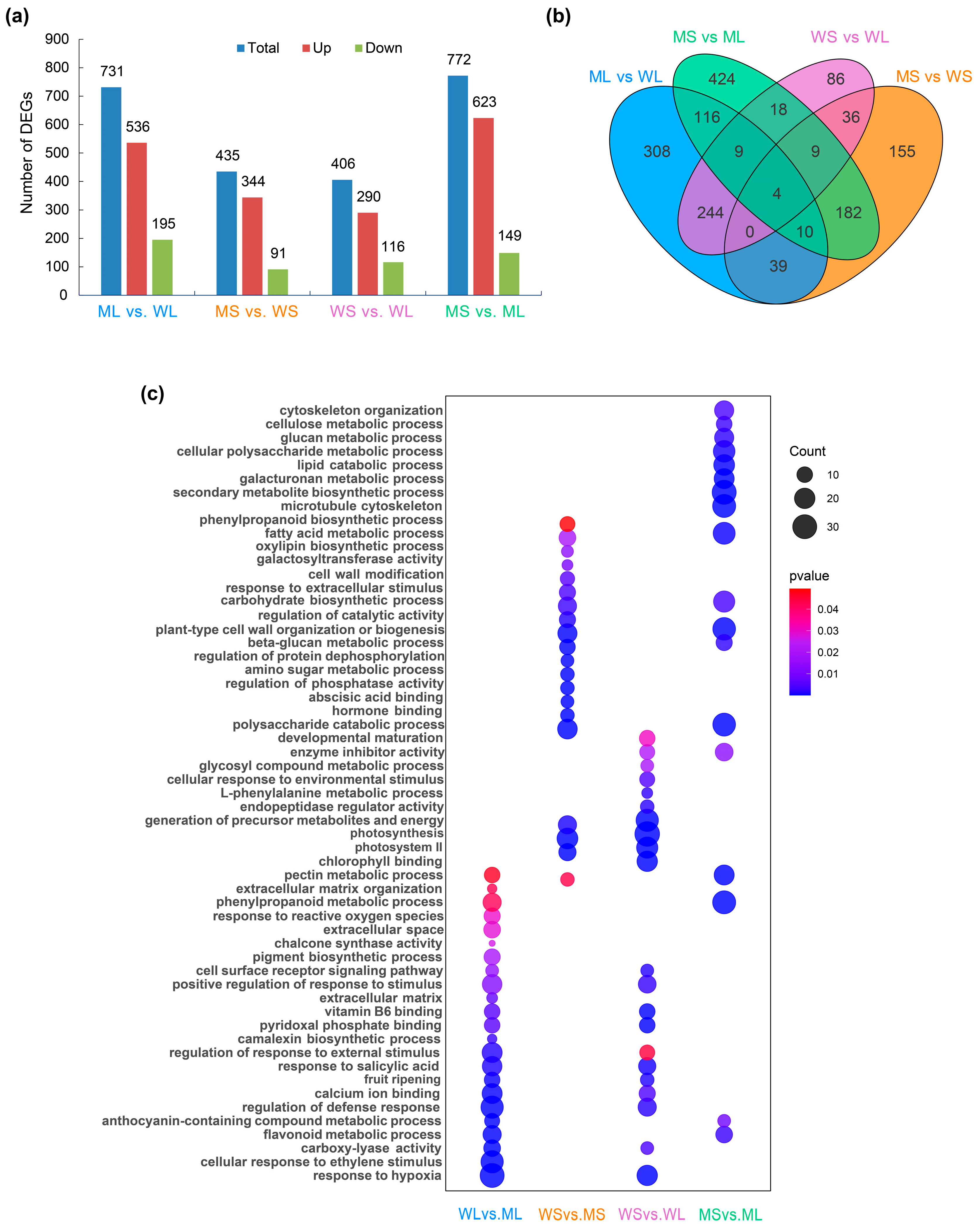
2.2. Transcriptome Profiling and Functional Enrichment Analysis of Differentially Expressed Genes (DEGs)
2.3. Identification of Tuber Skin Color-Associated Modules via Weighted Gene Co-Expression Network Analysis (WGCNA)
2.4. Expression Clustering Reveals an Anthocyanin-Associated Gene Module
2.5. Gene Expression Changes in the Anthocyanin Biosynthetic Pathway
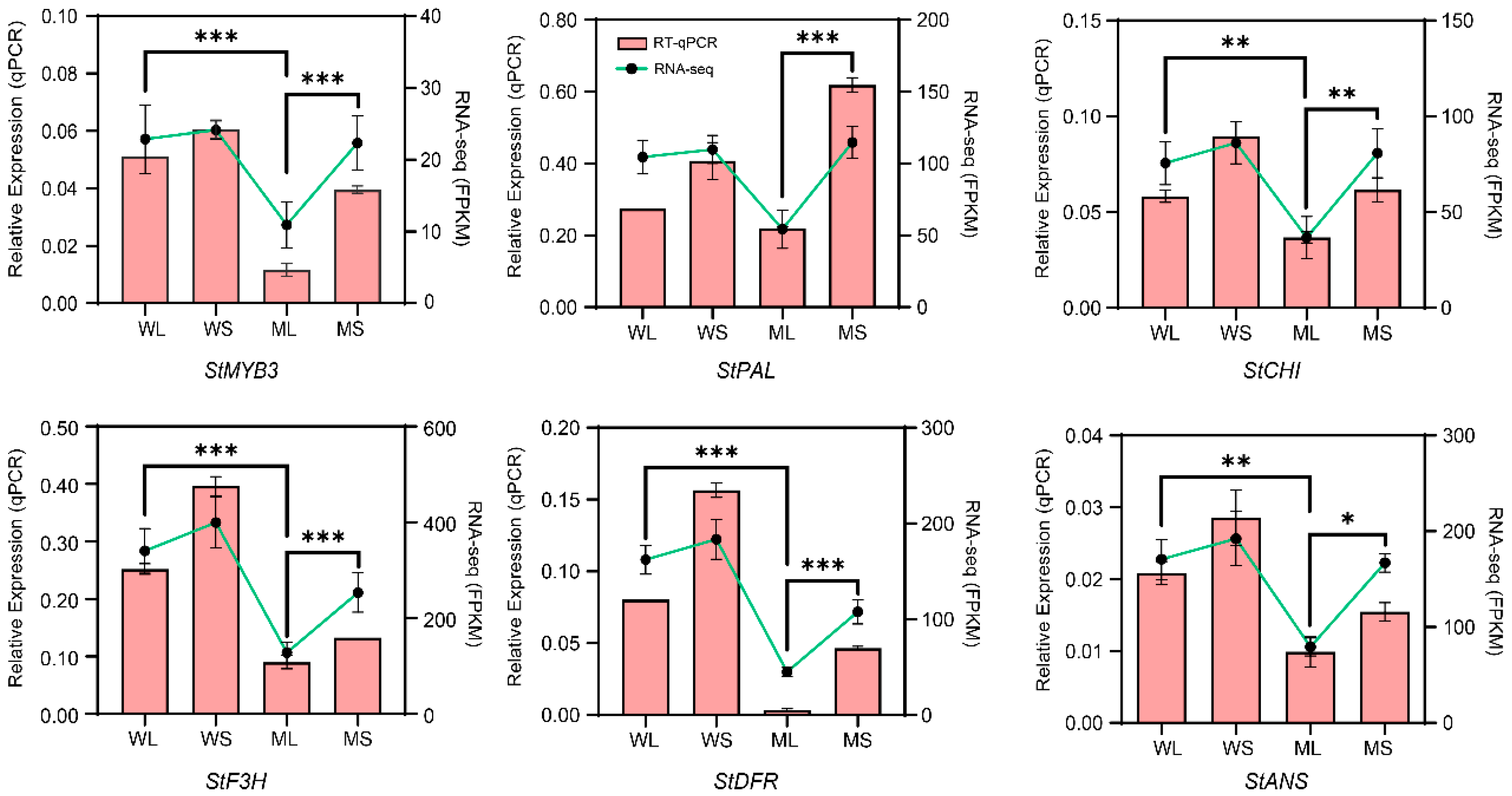
2.6. Quantitative Real-Time PCR Validation of Gene Expression
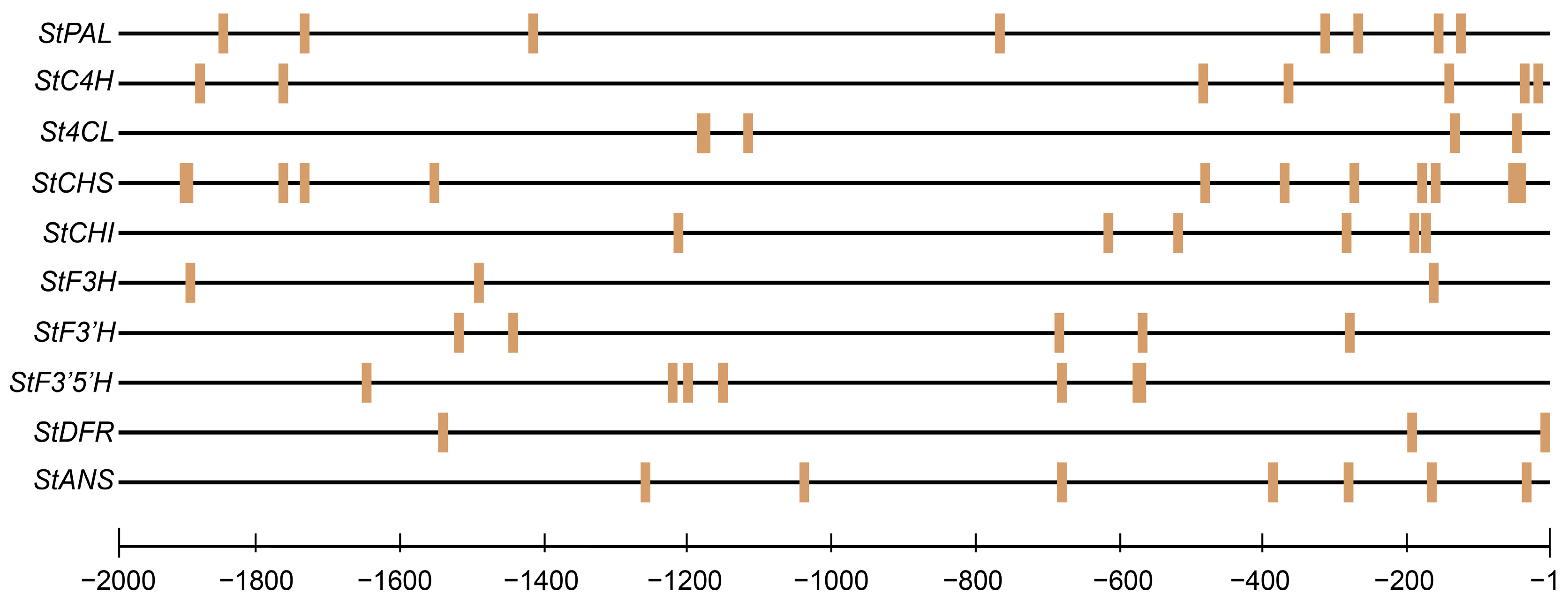
2.7. Prediction of StMYB3 Binding Sites in Anthocyanin Biosynthetic Gene Promoters
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Growth Conditions
4.2. DNA Extraction and SSR Analysis
4.3. Determination of Anthocyanin Type and Content
4.4. Differentially Expressed Gene (DEG) Expression and Gene Ontology (GO) Enrichment Analyses
4.5. Gene Co-Expression Analysis and Network Visualization
4.6. K-Means Clustering and Correlation Network Analysis of DEGs
4.7. Quantitative Real-Time PCR (RT-qPCR) Analysis
4.8. Promoter Sequence Extraction and Cis-Element Prediction
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Mohammadi, N.; Farrell, M.; O’Sullivan, L.; Langan, A.; Franchin, M.; Azevedo, L.; Granato, D. Effectiveness of anthocyanin-containing foods and nutraceuticals in mitigating oxidative stress, inflammation, and cardiovascular health-related biomarkers: A systematic review of animal and human interventions. Food Funct. 2024, 15, 3274–3299. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, X.; Gao, Y.; Xian, X.; Zhang, D.; Zhao, W.; Wang, X.; Wang, Y. Orchestrating anthocyanin biosynthesis in fruit of fruit trees: Transcriptional, post-transcriptional, and post-translational regulation. Int. J. Biol. Macromol. 2025, 307, 141835. [Google Scholar] [CrossRef]
- Parra-Galindo, M.A.; Soto-Sedano, J.C.; Mosquera-Vásquez, T.; Roda, F. Pathway-based analysis of anthocyanin diversity in diploid potato. PLoS ONE 2021, 16, e0250861. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Liu, L.; Qiang, X.; Meng, Y.; Li, Z.; Huang, F. Integrated Metabolomic and Transcriptomic Profiles Provide Insights into the Mechanisms of Anthocyanin and Carotenoid Biosynthesis in Petals of Medicago sativa ssp. sativa and Medicago sativa ssp. falcata. Plants 2024, 13, 700. [Google Scholar] [CrossRef] [PubMed]
- Ke, Y.; Ashraf, U.; Wang, D.; Hassan, W.; Zou, Y.; Qi, Y.; Zhou, Y.; Abbas, F. Function of Anthocyanin and Chlorophyll Metabolic Pathways in the Floral Sepals Color Formation in Different Hydrangea Cultivars. Plants 2025, 14, 742. [Google Scholar] [CrossRef]
- Liu, W.; Feng, Y.; Yu, S.; Fan, Z.; Li, X.; Li, J.; Yin, H. The Flavonoid Biosynthesis Network in Plants. Int. J. Mol. Sci. 2021, 22, 12824. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, Y.; Liu, Z.; Wang, L.; Bi, Z.; Sun, C.; Yao, P.; Zhang, J.; Bai, J.; Zeng, Y. Integrated transcriptomic and metabolomic analysis revealed altitude-related regulatory mechanisms on flavonoid accumulation in potato tubers. Food Res. Int. 2023, 170, 112997. [Google Scholar] [CrossRef]
- Mo, F.; Li, L.; Zhang, C.; Yang, C.; Chen, G.; Niu, Y.; Si, J.; Liu, T.; Sun, X.; Wang, S.; et al. Genome-Wide Analysis and Expression Profiling of the Phenylalanine Ammonia-Lyase Gene Family in Solanum tuberosum. Int. J. Mol. Sci. 2022, 23, 6833. [Google Scholar] [CrossRef] [PubMed]
- de Vries, S.; Fürst-Jansen, J.M.R.; Irisarri, I.; Dhabalia Ashok, A.; Ischebeck, T.; Feussner, K.; Abreu, I.N.; Petersen, M.; Feussner, I.; de Vries, J. The evolution of the phenylpropanoid pathway entailed pronounced radiations and divergences of enzyme families. Plant J. 2021, 107, 975–1002. [Google Scholar] [CrossRef]
- Nie, T.; Sun, X.; Wang, S.; Wang, D.; Ren, Y.; Chen, Q. Genome-Wide Identification and Expression Analysis of the 4-Coumarate: CoA Ligase Gene Family in Solanum tuberosum. Int. J. Mol. Sci. 2023, 24, 1642. [Google Scholar] [CrossRef]
- Tang, X.; Ren, C.; Hu, J.; Chen, J.; Wang, J.; Wang, R.; Wu, Q.; Liao, W.; Pei, J. Cloning, expression and activity analysises of chalcone synthase genes in Carthamus tinctorius. Chin. Herb. Med. 2023, 15, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Park, S.I.; Park, H.L.; Bhoo, S.H.; Lee, S.W.; Cho, M.H. Biochemical and Molecular Characterization of the Rice Chalcone Isomerase Family. Plants 2021, 10, 2064. [Google Scholar] [CrossRef]
- Xu, P.; Li, X.; Fan, J.; Tian, S.; Cao, M.; Lin, A.; Gao, Q.; Xiao, K.; Wang, C.; Kuang, H.; et al. An arginine-to-histidine mutation in flavanone-3-hydroxylase results in pink strawberry fruits. Plant Physiol. 2023, 193, 1849–1865. [Google Scholar] [CrossRef]
- McClean, P.E.; Lee, R.; Howe, K.; Osborne, C.; Grimwood, J.; Levy, S.; Haugrud, A.P.; Plott, C.; Robinson, M.; Skiba, R.M.; et al. The Common Bean V Gene Encodes Flavonoid 3′5′ Hydroxylase: A Major Mutational Target for Flavonoid Diversity in Angiosperms. Front. Plant Sci. 2022, 13, 869582. [Google Scholar] [CrossRef]
- Chen, L.Z.; Tian, X.C.; Feng, Y.Q.; Qiao, H.L.; Wu, A.Y.; Li, X.; Hou, Y.J.; Ma, Z.H. The Genome-Wide Identification of the Dihydroflavonol 4-Reductase (DFR) Gene Family and Its Expression Analysis in Different Fruit Coloring Stages of Strawberry. Int. J. Mol. Sci. 2024, 25, 9911. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhao, X.; Zhang, J.; Yang, B.; Yu, Y.; Liu, T.; Nie, B.; Song, B. Functional analysis of an anthocyanin synthase gene StANS in potato. Sci. Hortic. 2020, 272, 109569. [Google Scholar] [CrossRef]
- Chen, S.; Wang, S. GLABRA2, A Common Regulator for Epidermal Cell Fate Determination and Anthocyanin Biosynthesis in Arabidopsis. Int. J. Mol. Sci. 2019, 20, 4997. [Google Scholar] [CrossRef]
- Chen, L.; Hu, B.; Qin, Y.; Hu, G.; Zhao, J. Advance of the negative regulation of anthocyanin biosynthesis by MYB transcription factors. Plant Physiol. Biochem. 2019, 136, 178–187. [Google Scholar] [CrossRef]
- Mondal, S.K.; Roy, S. Genome-wide sequential, evolutionary, organizational and expression analyses of phenylpropanoid biosynthesis associated MYB domain transcription factors in Arabidopsis. J. Biomol. Struct. Dyn. 2018, 36, 1577–1601. [Google Scholar] [CrossRef]
- Du, H.; Zhai, Z.; Pu, J.; Liang, J.; Wang, R.; Zhang, Z.; Wang, P.; Zhu, Y.; Huang, L.; Li, D.; et al. Two tandem R2R3 MYB transcription factor genes cooperatively regulate anthocyanin accumulation in potato tuber flesh. Plant Biotechnol. J. 2025, 23, 1521–1534. [Google Scholar] [CrossRef]
- Sampaio, S.L.; Lonchamp, J.; Dias, M.I.; Liddle, C.; Petropoulos, S.A.; Glamočlija, J.; Alexopoulos, A.; Santos-Buelga, C.; Ferreira, I.; Barros, L. Anthocyanin-rich extracts from purple and red potatoes as natural colourants: Bioactive properties, application in a soft drink formulation and sensory analysis. Food Chem. 2021, 342, 128526. [Google Scholar] [CrossRef] [PubMed]
- van Eck, H.J.; Jacobs, J.M.; van Dijk, J.; Stiekema, W.J.; Jacobsen, E. Identification and mapping of three flower colour loci of potato (S. tuberosum L.) by RFLP analysis. Theor. Appl. Genet. 1993, 86, 295–300. [Google Scholar] [CrossRef]
- Jung, C.S.; Griffiths, H.M.; De Jong, D.M.; Cheng, S.; Bodis, M.; Kim, T.S.; De Jong, W.S. The potato developer (D) locus encodes an R2R3 MYB transcription factor that regulates expression of multiple anthocyanin structural genes in tuber skin. Theor. Appl. Genet. 2009, 120, 45–57. [Google Scholar] [CrossRef]
- Jung, C.S.; Griffiths, H.M.; De Jong, D.M.; Cheng, S.; Bodis, M.; De Jong, W.S. The potato P locus codes for flavonoid 3′,5′-hydroxylase. Theor. Appl. Genet. 2005, 110, 269–275. [Google Scholar] [CrossRef] [PubMed]
- De Jong, W.S.; Eannetta, N.T.; De Jong, D.M.; Bodis, M. Candidate gene analysis of anthocyanin pigmentation loci in the Solanaceae. Theor. Appl. Genet. 2004, 108, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Hina, A.; Razzaq, M.K.; Abbasi, A.; Shehzad, M.B.; Arshad, M.; Sanaullah, T.; Arshad, K.; Raza, G.; Ali, H.M.; Hayat, F.; et al. Genomic blueprints of soybean (Glycine max) pathogen resistance: Revealing the key genes for sustainable agriculture. Funct. Plant Biol. 2024, 51, FP23295. [Google Scholar] [CrossRef]
- Plessis, A.; Hafemeister, C.; Wilkins, O.; Gonzaga, Z.J.; Meyer, R.S.; Pires, I.; Müller, C.; Septiningsih, E.M.; Bonneau, R.; Purugganan, M. Multiple abiotic stimuli are integrated in the regulation of rice gene expression under field conditions. eLife 2015, 4, e08411. [Google Scholar] [CrossRef]
- Piofczyk, T.; Jeena, G.; Pecinka, A. Arabidopsis thaliana natural variation reveals connections between UV radiation stress and plant pathogen-like defense responses. Plant Physiol. Biochem. 2015, 93, 34–43. [Google Scholar] [CrossRef]
- Cong, Q.Q.; Wang, Y.; Liu, J.; Lan, Y.F.; Guo, Z.K.; Yang, J.G.; Li, X.D.; Tian, Y.P. Evaluation of Potato virus X mild mutants for cross protection against severe infection in China. Virol. J. 2019, 16, 36. [Google Scholar] [CrossRef]
- Liu, F.; Yang, Y.; Gao, J.; Ma, C.; Bi, Y. A comparative transcriptome analysis of a wild purple potato and its red mutant provides insight into the mechanism of anthocyanin transformation. PLoS ONE 2018, 13, e0191406. [Google Scholar] [CrossRef]
- Zhang, H.; Yu, L.; Zhao, Y.; Zhang, Z.; Zhang, Z.; Pei, M.; Song, B. Tissue-Specific Expression of StMYB3 Gene Involves Regulating Potato Tuber Skin and Vascular Bundle Anthocyanin Biosynthesis. Potato Res. 2023, 66, 159–178. [Google Scholar] [CrossRef]
- Kim, D.; Jeon, S.J.; Yanders, S.; Park, S.C.; Kim, H.S.; Kim, S. MYB3 plays an important role in lignin and anthocyanin biosynthesis under salt stress condition in Arabidopsis. Plant Cell Rep. 2022, 41, 1549–1560. [Google Scholar] [CrossRef]
- Gambhir, P.; Raghuvanshi, U.; Kumar, R.; Sharma, A.K. Transcriptional regulation of tomato fruit ripening. Physiol. Mol. Biol. Plants 2024, 30, 289–303. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Yue, M.; Liu, Y.; Zhang, N.; Lin, Y.; Zhang, Y.; Wang, Y.; Li, M.; Luo, Y.; Zhang, Y.; et al. A novel R2R3-MYB transcription factor FaMYB5 positively regulates anthocyanin and proanthocyanidin biosynthesis in cultivated strawberries (Fragaria × ananassa). Plant Biotechnol. J. 2023, 21, 1140–1158. [Google Scholar] [CrossRef]
- Xhulaj, D.; Gixhari, B. In vitro micropropagation of potato (Solanum tuberosum L). cultivars. J. Agric. For. 2018, 64, 105–112. [Google Scholar] [CrossRef]
- Allen, G.C.; Flores-Vergara, M.A.; Krasynanski, S.; Kumar, S.; Thompson, W.F. A modified protocol for rapid DNA isolation from plant tissues using cetyltrimethylammonium bromide. Nat. Protoc. 2006, 1, 2320–2325. [Google Scholar] [CrossRef]
- Jian, W.; Lu, H.; Ruo-yu, W.; Miao-miao, H.; Qing-chang, L. Genetic diversity and population structure of 288 potato(Solanum tuberosum L.) germplasms revealed by SSR and AFLP markers. J. Integr. Agric. 2017, 16, 2434–2443. [Google Scholar]
- Hu, J.; Mei, M.; Jin, F.; Xu, J.; Duan, S.; Bian, C.; Li, G.; Wang, X.; Jin, L. Phenotypic variability and genetic diversity analysis of cultivated potatoes in China. Front. Plant Sci. 2022, 13, 954162. [Google Scholar] [CrossRef]
- Wang, H.; Sun, S.; Zhou, Z.; Qiu, Z.; Cui, X. Rapid analysis of anthocyanin and its structural modifications in fresh tomato fruit. Food Chem. 2020, 333, 127439. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. Clusterprofiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Kumar, L.; Matthias, E.F. Mfuzz: A software package for soft clustering of microarray data. Bioinformation 2007, 2, 5–7. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Zhang, N.; Si, H.; Calderón-Urrea, A. Selection and validation of reference genes for RT-qPCR analysis in potato under abiotic stress. Plant Methods 2017, 13, 85. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Yang, Y.; Guo, X.; Jin, L.; Xiong, X.; Yang, X.; Li, G. Exogenous SA Initiated Defense Response and Multi-signaling Pathway in Tetraploid Potato SD20. Hortic. Plant J. 2020, 6, 99–110. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, B.; Wang, S.; Hu, J.; Jin, L.; Xu, J. Transcriptome Profiling Identifies Key Regulators of Tuber Skin Color in Potato. Plants 2025, 14, 1544. https://doi.org/10.3390/plants14101544
Li B, Wang S, Hu J, Jin L, Xu J. Transcriptome Profiling Identifies Key Regulators of Tuber Skin Color in Potato. Plants. 2025; 14(10):1544. https://doi.org/10.3390/plants14101544
Chicago/Turabian StyleLi, Boshu, Shuo Wang, Jun Hu, Liping Jin, and Jianfei Xu. 2025. "Transcriptome Profiling Identifies Key Regulators of Tuber Skin Color in Potato" Plants 14, no. 10: 1544. https://doi.org/10.3390/plants14101544
APA StyleLi, B., Wang, S., Hu, J., Jin, L., & Xu, J. (2025). Transcriptome Profiling Identifies Key Regulators of Tuber Skin Color in Potato. Plants, 14(10), 1544. https://doi.org/10.3390/plants14101544