
Insights into the Red Seaweed Asparagopsis taxiformis Using an Integrative Multi-Omics Analysis

 ,
, 
 , , , , , ,
, , , , , ,  and
and
Abstract
1. Introduction
2. Results and Discussion
2.1. Phylogeny of A. taxiformis and Description of the Sample Used for Genomic Analysis
2.2. Genomic Features of Asparagopsis taxiformis (L6)
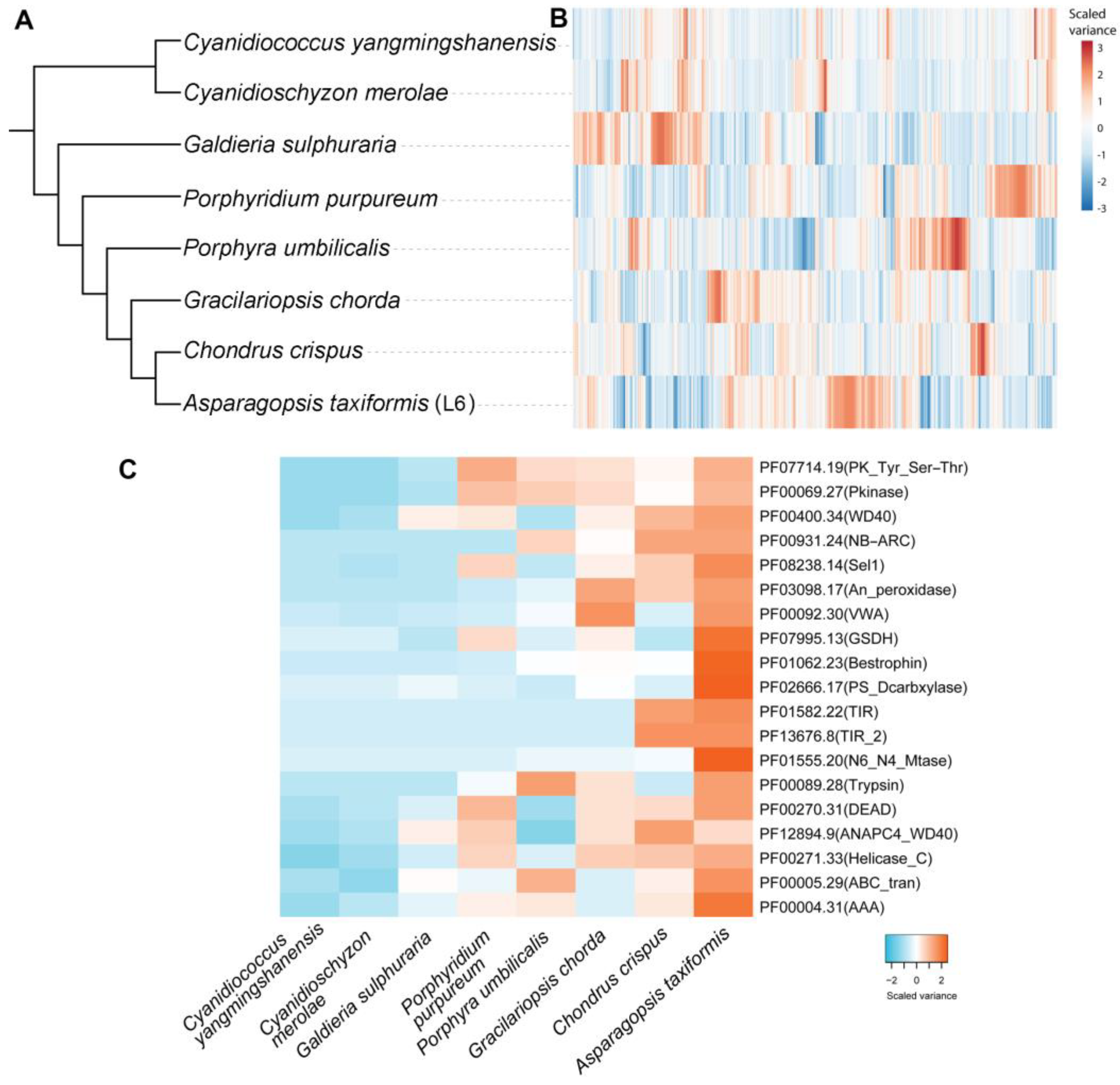
2.3. Comparative and Functional Features of the A. taxiformis (L6) Genome
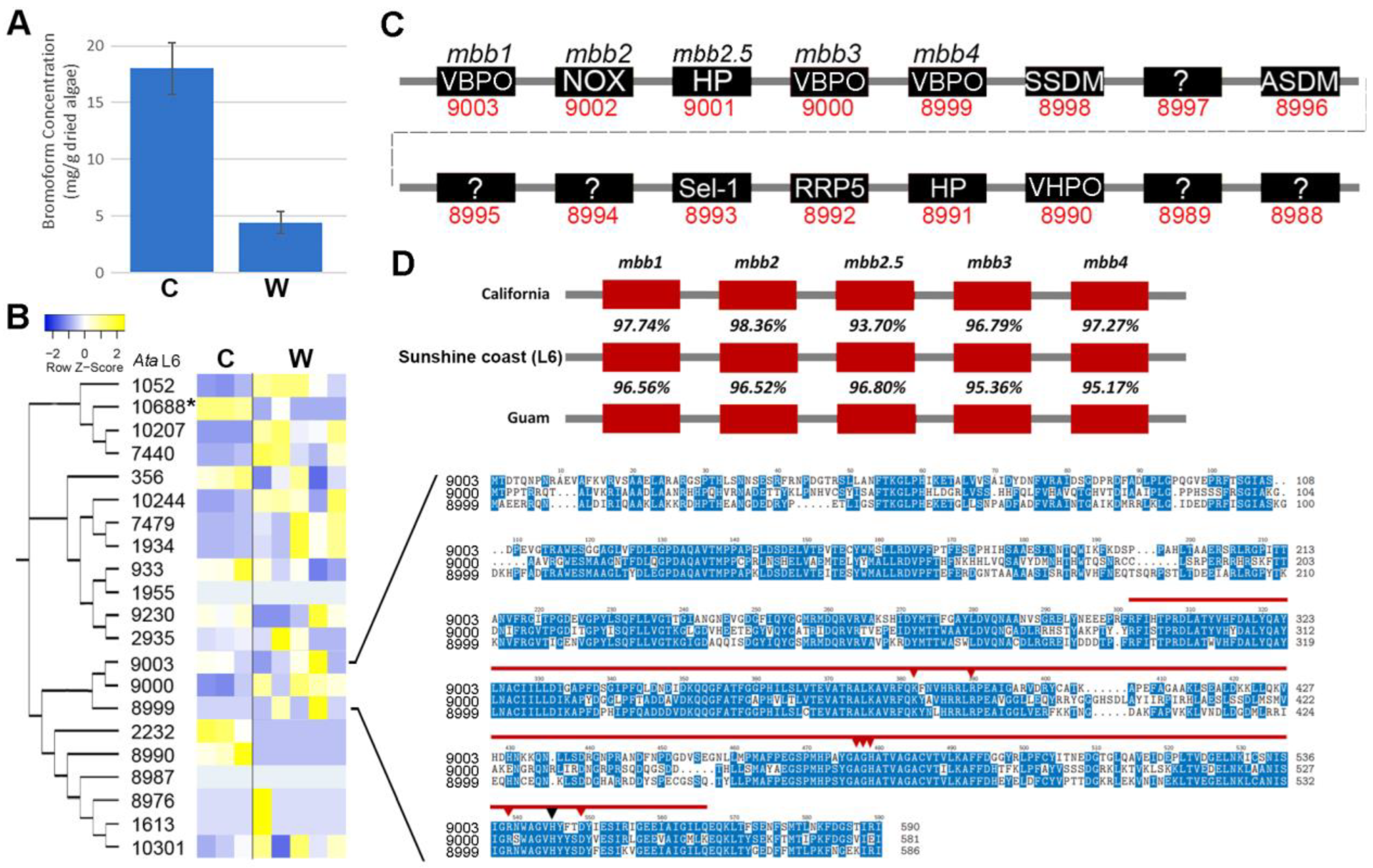
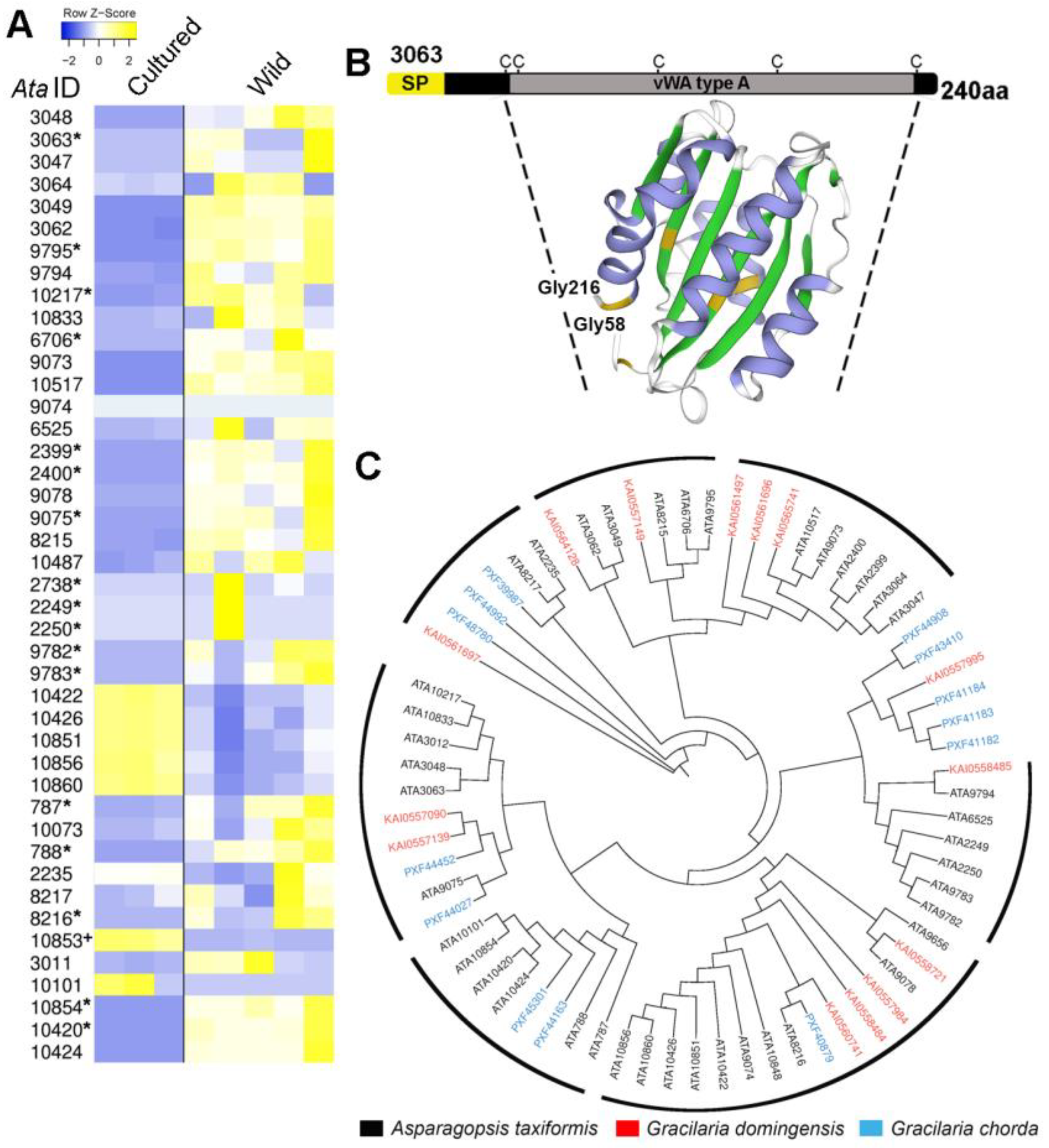
2.4. A. taxiformis (L6) Cultured and Wild Sporophyte: An Integrative Omics Analysis
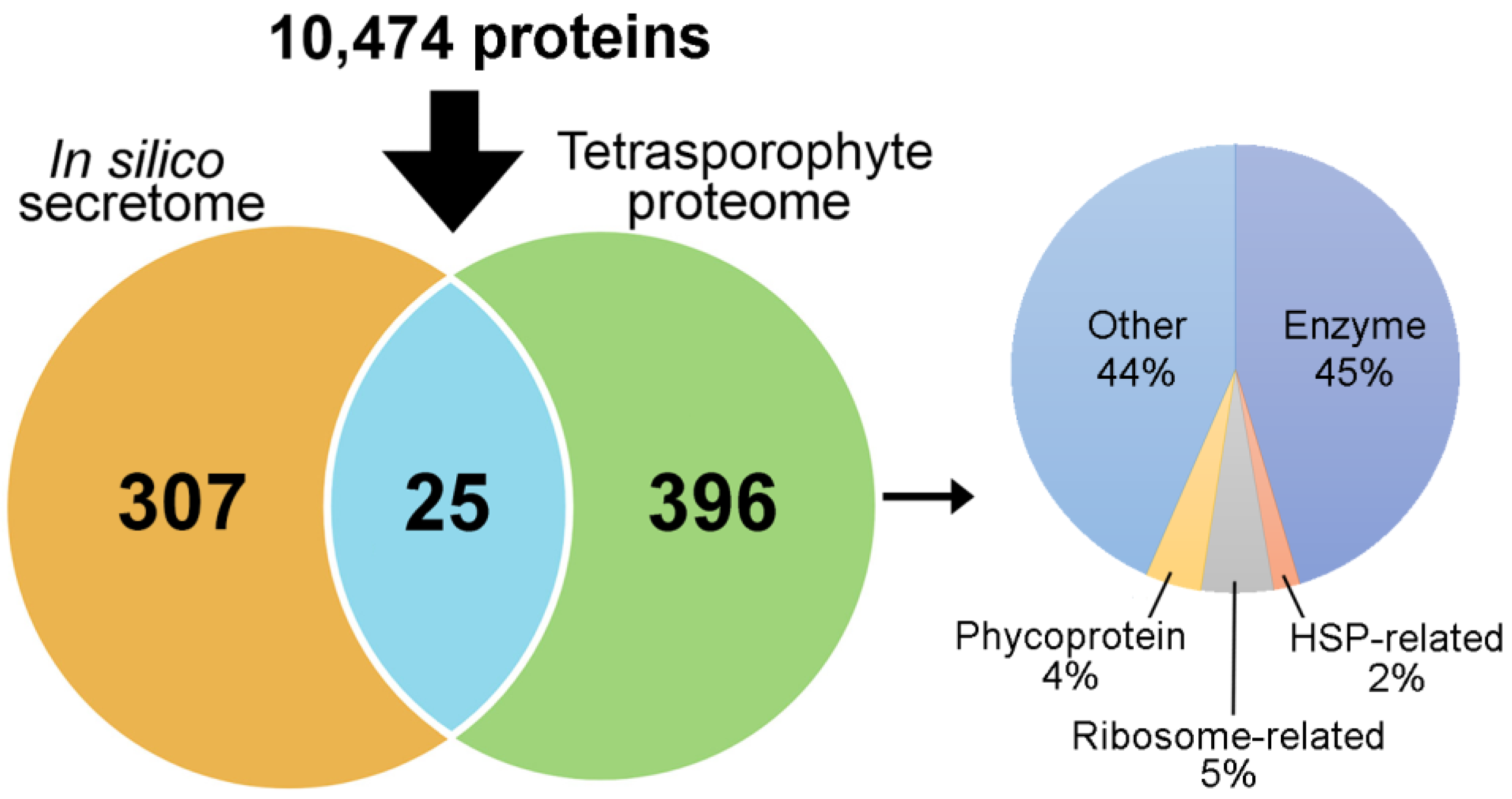
2.5. A. taxiformis Tetrasporophyte Proteomics
3. Materials and Methods
3.1. Asparagopsis taxiformis Tissue Collection
3.2. Library Construction and Sequencing
3.3. Genome Assembly and Functional Evaluation
3.4. Repeat Masking and Transposable Elements
3.5. Gene Prediction and Functional Annotation
3.6. Genome Decontamination
3.7. Comparative Genome Analysis
3.8. Targeted Gene Annotation
3.9. Cultured and Wild Sporophyte Samples for Comparative Bromoform Analysis and RNA-Seq Gene Quantification
3.10. Asparagopsis taxiformis Sporophyte Proteomics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| GO | Gene ontology |
| HSP | Heat shock protein |
| L6 | Lineage 6 |
| NOX | NADPH-oxidase |
| NR | Non-redundant |
| PAP2 | Type 2 phosphatidic acid phosphatases |
| rRNA | Ribosomal RNA |
| SSW | Sterile seawater |
| TF | Transcription factor |
| TTF | Trihelix transcription factor |
| VBPO | Vanadium-dependent bromoperoxidase |
| VHPO | Vanadium haloperoxidases |
| VWA | von Willebrand factor type A |
References
- Brawley, S.H.; Blouin, N.A.; Ficko-Blean, E.; Wheeler, G.L.; Lohr, M.; Goodson, H.V.; Jenkins, J.W.; Blaby-Haas, C.E.; Helliwell, K.E.; Chan, C.X.; et al. Insights into the red algae and eukaryotic evolution from the genome of Porphyra umbilicalis (Bangiophyceae, Rhodophyta). Proc. Natl. Acad. Sci. USA 2017, 114, E6361–E6370. [Google Scholar] [CrossRef]
- Barrows, G.; Sexton, S.; Zilberman, D. Agricultural biotechnology: The promise and prospects of genetically modified crops. J. Econ. Perspect. 2014, 28, 99–120. [Google Scholar]
- Climate Change 2014—Synthesis Report; IPCC: Geneva, Switzerland, 2015; p. 151.
- National Greenhouse Gas Inventory; Center for Global Environmental Research, Earth System Division, National Institute for Environmental Studies: Tsukuba, Japan, 2025.
- Machado, L.; Magnusson, M.; Paul, N.A.; Kinley, R.; de Nys, R.; Tomkins, N. Dose-response effects of Asparagopsis taxiformis and Oedogonium sp. on in vitro fermentation and methane production. J. Appl. Phycol. 2016, 28, 1443–1452. [Google Scholar]
- Machado, L.; Magnusson, M.; Paul, N.A.; de Nys, R.; Tomkins, N. Effects of marine and freshwater macroalgae on in vitro total gas and methane production. PLoS ONE 2014, 9, e85289. [Google Scholar]
- Fenical, W. Natural products chemistry in the marine environment. Science 1982, 215, 923–928. [Google Scholar] [CrossRef]
- Paul, N.A.; de Nys, R.; Steinberg, P.D. Chemical defence against bacteria in the red alga Asparagopsis armata: Linking structure with function. Mol. Ecol. Prog. Ser. 2006, 306, 87–101. [Google Scholar]
- Tomkins, N.; Colegate, S.; Hunter, R.A. A bromochloromethane formulation reduces enteric methanogenesis in cattle fed grain-based diets. Anim. Prod. Sci. 2009, 49, 1053–1058. [Google Scholar]
- Davison, T.; Hill, J.; Savage, S. Reducing Emissions from Livestock Research Program; Meat & Livestock Australia Ltd.: Sydney, Australia, 2012. [Google Scholar]
- Zhou, M.; Hünerberg, M.; Chen, Y.; Reuter, T.; McAllister, T.A.; Evans, F.; Critchley, A.T.; Guan, L.L. Air-dried brown seaweed, Ascophyllum nodosum, alters the rumen microbiome in a manner that changes rumen fermentation profiles and lowers the prevalence of foodborne pathogens. mSphere 2018, 3, e00017-18. [Google Scholar]
- Manley, S.L. Phytogenesis of halomethanes: A product ofselection or a metabolic accident? Biogeochemistry 2002, 60, 163–180. [Google Scholar]
- Mata, L.; Lawton, R.J.; Magnusson, M.; Andreakis, N.; de Nys, R.; Paul, N.A. Within-species and temperature-related variation in the growth and natural products of the red alga Asparagopsis taxiformis. J. Appl. Phycol. 2017, 29, 1437–1447. [Google Scholar]
- Egan, S.; Harder, T.; Burke, C.; Steinberg, P.; Kjelleberg, S.; Thomas, T. The seaweed holobiont: Understanding seaweed-bacteria interactions. FEMS Microbiol. Rev. 2013, 37, 462–476. [Google Scholar]
- Wahl, M.; Goecke, F.; Labes, A.; Dobretsov, S.; Weinberger, F.J. The second skin: Ecological role of epibiotic biofilms on marine organisms. Front. Microbiol. 2012, 3, 292. [Google Scholar]
- Vergés, A.; Paul, N.A.; Steinberg, P.D. Sex and life-history stage alter herbivore responses to a chemically defended red alga. Ecology 2008, 89, 1334–1343. [Google Scholar]
- Thapa, H.R.; Lin, Z.; Yi, D.; Smith, J.E.; Schmidt, E.W.; Agarwal, V. Genetic and Biochemical Reconstitution of Bromoform Biosynthesis in Asparagopsis Lends Insights into Seaweed Reactive Oxygen Species Enzymology. ACS Chem. Biol. 2020, 15, 1662–1670. [Google Scholar] [CrossRef]
- Hollants, J.; Leliaert, F.; De Clerck, O.; Willems, A. What we can learn from sushi: A review on seaweed–bacterial associations. FEMS Microbiol. Ecol. 2013, 83, 1–16. [Google Scholar]
- Kinley, R.; Fredeen, A.H. In vitro evaluation of feeding North Atlantic stormtoss seaweeds on ruminal digestion. J. Appl. Phycol. 2015, 27, 2387–2393. [Google Scholar]
- Greff, S.; Aires, T.; Serrao, E.A.; Engelen, A.H.; Thomas, O.P.; Perez, T. The interaction between the proliferating macroalga Asparagopsis taxiformis and the coral Astroides calycularis induces changes in microbiome and metabolomic fingerprints. Sci. Rep. 2017, 7, 42625. [Google Scholar] [CrossRef]
- Gregersen, S.; Pertseva, M.; Marcatili, P.; LøvstadHoldt, S.; Jacobsen, C.; García-Morenod, P.J.; BechHansen, E.; ToftOvergaard, M. Proteomic characterization of pilot scale hot-water extracts from the industrial carrageenan red seaweed Eucheuma denticulatum. Algal Res. 2022, 62, 102619. [Google Scholar]
- Wang, D.; You, W.; Chen, N.; Cao, M.; Tang, X.; Guan, X.; Qu, W.; Chen, R.; Mao, Y.; Poetsch, A. Comparative Quantitative Proteomics Reveals the Desiccation Stress Responses of the Intertidal Seaweed NEOPORPHYRA haitanensis. J. Phycol. 2020, 56, 1664–1675. [Google Scholar]
- Andreakis, N.; Costello, P.; Zanolla, M.; Saunders, G.W.; Mata, L. Endemic or introduced? Phylogeography of Asparagopsis (Florideophyceae) in Australia reveals multiple introductions and a new mitochondrial lineage. J. Phycol. 2016, 52, 141–147. [Google Scholar] [CrossRef]
- Collén, J.; Porcel, B.; Carré, W.; Ball, S.G.; Chaparro, C.; Tonon, T.; Barbeyron, T.; Michel, G.; Noel, B.; Valentin, K.; et al. Genome structure and metabolic features in the red seaweed Chondrus crispus shed light on evolution of the Archaeplastida. Proc. Natl. Acad. Sci. USA 2013, 110, 5247–5252. [Google Scholar]
- Johnson, D.G.; Walker, C.L. Cyclins and cell cycle checkpoints. Annu. Rev. Pharmacol. Toxicol. 1999, 39, 295–312. [Google Scholar]
- Preuss, M.; Nelson, W.A.; D’Archino, R. Cryptic diversity and phylogeographic patterns in the Asparagopsis armata species complex (Bonnemaisoniales, Rhodophyta) from New Zealand. Phycologia 2022, 61, 89–96. [Google Scholar] [CrossRef]
- Verbruggen, H.; Maggs, C.A.; Saunders, G.W.; Le Gall, L.; Yoon, H.S.; De Clerck, O. Data mining approach identifies research priorities and data requirements for resolving the red algal tree of life. BMC Evol. Biol. 2010, 10, 16. [Google Scholar] [CrossRef]
- Xu, C.; Min, J. Structure and function of WD40 domain proteins. Protein Cell. 2011, 2, 202–214. [Google Scholar]
- Ambawat, S.; Sharma, P.; Yadav, N.R.; Yadav, R.C. MYB transcription factor genes as regulators for plant responses: An overview. Physiol. Mol. Biol. Plants 2013, 19, 307–321. [Google Scholar] [CrossRef]
- Cheng, X.; Xiong, R.; Yan, H.; Gao, Y.; Liu, H.; Wu, M.; Xiang, Y. The trihelix family of transcription factors: Functional and evolutionary analysis in Moso bamboo (Phyllostachys edulis). BMC Plant Biol. 2019, 19, 154. [Google Scholar] [CrossRef]
- Rensing, S.A. Gene duplication as a driver of plant morphogenetic evolution. Curr. Opin. Plant Biol. 2014, 17, 43–48. [Google Scholar] [CrossRef]
- Pallanca, J.E.; Smirnoff, N. Ascorbic acid metabolism in pea seedlings. A comparison of D-glucosone, L-sorbosone, and L-galactono-1,4-lactone as ascorbate precursors. Plant Physiol. 1999, 120, 453–462. [Google Scholar] [CrossRef]
- Trotter, P.J.; Pedretti, J.; Yates, R.; Voelker, D.R. Phosphatidylserine decarboxylase 2 of Saccharomyces cerevisiae. Cloning and mapping of the gene, heterologous expression, and creation of the null allele. J. Biol. Chem. 1995, 270, 6071–6080. [Google Scholar] [CrossRef]
- Thépot, V.; Campbell, A.H.; Paul, N.A.; Rimmer, M.A. Seaweed dietary supplements enhance the innate immune response of the mottled rabbitfish, Siganus fuscescens. Fish. Shellfish. Immunol. 2021, 113, 176–184. [Google Scholar]
- Park, C.J.; Seo, Y.S. Heat Shock Proteins: A Review of the Molecular Chaperones for Plant Immunity. Plant Pathol. J. 2015, 31, 323–333. [Google Scholar]
- Uji, T.; Gondaira, Y.; Fukuda, S.; Mizuta, H.; Saga, N. Characterization and expression profiles of small heat shock proteins in the marine red alga Pyropia yezoensis. Cell Stress and Chaperones 2019, 24, 223–233. [Google Scholar]
- Höfler, G.T.; But, A.; Hollmann, F. Haloperoxidases as catalysts in organic synthesis. Org. Biomol. Chem. 2019, 17, 9267–9274. [Google Scholar]
- Isupov, M.N.; Dalby, A.R.; Brindley, A.A.; Izumi, Y.; Tanabe, T.; Murshudov, G.N.; Littlechild, J.A. Crystal structure of dodecameric vanadium-dependent bromoperoxidase from the red algae Corallina officinalis. J. Mol. Biol. 2000, 299, 1035–1049. [Google Scholar] [CrossRef]
- Wang, X.; Chung, K.P.; Lin, W.; Jiang, L. Protein secretion in plants: Conventional and unconventional pathways and new techniques. J. Exp. Bot. 2017, 69, 21–37. [Google Scholar]
- Suhre, M.H.; Scheibel, T.; Steegborn, C.; Gertz, M. Crystallization and preliminary X-ray diffraction analysis of proximal thread matrix protein 1 (PTMP1) from Mytilus galloprovincialis. Acta Crystallogr. F Struct. Biol. Commun. 2014, 70, 769–772. [Google Scholar] [CrossRef]
- Waite, J.H.; Lichtenegger, H.C.; Stucky, G.D.; Hansma, P. Exploring molecular and mechanical gradients in structural bioscaffolds. Biochemistry 2004, 43, 7653–7662. [Google Scholar] [CrossRef]
- Whittaker, C.A.; Hynes, R.O. Distribution and evolution of von Willebrand/integrin A domains: Widely dispersed domains with roles in cell adhesion and elsewhere. Mol. Biol. Cell. 2002, 13, 3369–3387. [Google Scholar]
- Kerrison, P.D.; Stanley, M.S.; De Smet, D.; Buyle, G.; Hughes, A.D. Holding (not so) fast: Surface chemistry constrains kelp bioadhesion. Eur. J. Phycol. 2019, 54, 291–299. [Google Scholar]
- Charoensiddhi, S.; Abraham, R.E.; Su, P.; Zhang, W. Seaweed and seaweed-derived metabolites as prebiotics. Adv. Food Nutr. Res. 2020, 91, 97–156. [Google Scholar]
- Sonani, R.R.; Rastogi, R.P.; Patel, R.; Madamwar, D. Recent advances in production, purification and applications of phycobiliproteins. World J. Biol. Chem. 2016, 7, 100–109. [Google Scholar]
- Marcais, G.; Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Simao, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Flynn, J.M.; Hubley, R.; Goubert, C.; Rosen, J.; Clark, A.G.; Feschotte, C.; Smit, A.F. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 2020, 117, 9451–9457. [Google Scholar] [CrossRef]
- Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. 2004, 4, 4–10. [Google Scholar] [CrossRef]
- Stanke, M.; Keller, O.; Gunduz, I.; Hayes, A.; Waack, S.; Morgenstern, B. AUGUSTUS: Ab initio prediction of alternative transcripts. Nucleic Acids Res. 2006, 34, W435–W439. [Google Scholar] [CrossRef]
- Conesa, A.; Gotz, S. Blast2GO: A comprehensive suite for functional analysis in plant genomics. Int. J. Plant Genom. 2008, 2008, 619832. [Google Scholar] [CrossRef]
- Coordinators, N.R. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2016, 44, D7–D19. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef]
- Mistry, J.; Finn, R.D.; Eddy, S.R.; Bateman, A.; Punta, M. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 2013, 41, e121. [Google Scholar] [CrossRef]
- Astashyn, A.; Tvedte, E.S.; Sweeney, D.; Sapojnikov, V.; Bouk, N.; Joukov, V.; Mozes, E.; Strope, P.K.; Sylla, P.M.; Wagner, L.; et al. Rapid and sensitive detection of genome contamination at scale with FCS-GX. Genome Biol. 2024, 25, 60. [Google Scholar] [CrossRef]
- Buchfink, B.; Reuter, K.; Drost, H.-G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 2021, 18, 366–368. [Google Scholar] [CrossRef]
- Junker, V.; Contrino, S.; Fleischmann, W.; Hermjakob, H.; Lang, F.; Magrane, M.; Martin, M.J.; Mitaritonna, N.; O’Donovan, C.; Apweiler, R. The role SWISS-PROT and TrEMBL play in the genome research environment. J. Biotechnol. 2000, 78, 221–234. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. STAG: Species Tree Inference from All Genes. bioRxiv 2018. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree: Computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 2009, 26, 1641–1650. [Google Scholar] [CrossRef]
- Jin, J.; Tian, F.; Yang, D.C.; Meng, Y.Q.; Kong, L.; Luo, J.; Gao, G. PlantTFDB 4.0: Toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 2017, 45, D1040–D1045. [Google Scholar] [CrossRef]
- Almagro Armenteros, J.J.; Tsirigos, K.D.; Sonderby, C.K.; Petersen, T.N.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019, 37, 420–423. [Google Scholar] [CrossRef]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef]
- Marchler-Bauer, A.; Bo, Y.; Han, L.; He, J.; Lanczycki, C.J.; Lu, S.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; et al. CDD/SPARCLE: Functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 2017, 45, D200–D203. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar]
- Romanazzi, D.; Sanchez-Garcia, C.; Svenson, J.; Mata, L.; Pes, K.; Hayman, C.M.; Wheeler, T.T.; Magnusson, M. Rapid Analytical Method for the Quantification of Bromoform in the Red Seaweeds Asparagopsis armata and Asparagopsis taxiformis Using Gas Chromatography–Mass Spectrometry. ACS Agric. Sci. Technol. 2021, 1, 436–442. [Google Scholar] [CrossRef]
- Götz, S.; García-Gómez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talón, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef]
- Metsalu, T.; Vilo, J. ClustVis: A web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 2015, 43, W566–W570. [Google Scholar] [CrossRef]
- Hall, M.R.; Kocot, K.M.; Baughman, K.W.; Fernandez-Valverde, S.L.; Gauthier, M.E.; Hatleberg, W.L.; Krishnan, A.; McDougall, C.; Motti, C.A.; Shoguchi, E.J.N. The crown-of-thorns starfish genome as a guide for biocontrol of this coral reef pest. Nature 2017, 544, 231–234. [Google Scholar]
- Kupper, F.C.; Leblanc, C.; Meyer-Klaucke, W.; Potin, P.; Feiters, M.C. Different speciation for bromine in brown and red algae, revealed by in vivo X-ray absorption spectroscopic studies. J. Phycol. 2014, 50, 652–664. [Google Scholar] [CrossRef]
- Thépot, V.; Campbell, A.H.; Rimmer, M.A.; Jelocnik, M.; Johnston, C.; Evans, B.; Paul, N.A. Dietary inclusion of the red seaweed Asparagopsis taxiformis boosts production, stimulates immune response and modulates gut microbiota in Atlantic salmon, Salmo salar. Aquaculture 2022, 546, 737286. [Google Scholar] [CrossRef]
- Félix, R.; Dias, P.; Félix, C.; Cerqueira, T.; Andrade, P.B.; Valentão, P.; Lemos, M.F. The biotechnological potential of Asparagopsis armata: What is known of its chemical composition, bioactivities and current market? Algal Res. 2021, 60, 102534. [Google Scholar]
- Pinteus, S.; Lemos, M.F.; Alves, C.; Silva, J.; Pedrosa, R. The marine invasive seaweeds Asparagopsis armata and Sargassum muticum as targets for greener antifouling solutions. Sci. Total Environ. 2021, 750, 141372. [Google Scholar]
- Streftaris, N.; Zenetos, A. Alien Marine Species in the Mediterranean—The 100 ‘Worst Invasives’ and their Impact. Mediterr. Mar. Sci. 2006, 7, 87–118. [Google Scholar] [CrossRef]
- Navarro-Barranco, C.; Florido, M.; Ros, M.; González-Romero, P.; Guerra-García, J.M. Impoverished mobile epifaunal assemblages associated with the invasive macroalga Asparagopsis taxiformis in the Mediterranean Sea. Mar. Environ. Res. 2018, 141, 44–52. [Google Scholar] [CrossRef]
- Guerra-García, J.M.; Ros, M.; Izquierdo, D.; Soler-Hurtado, M.M. The invasive Asparagopsis armata versus the native Corallina elongata: Differences in associated peracarid assemblages. J. Exp. Mar. Biol. Ecol. 2012, 416–417, 121–128. [Google Scholar] [CrossRef]
- Moigne, J.Y. Use of Algae Extracts as Antibacterial and/or Antifungal Agent and Composition Containing Same. WIPO WO1998010656A1, 19 March 1998. [Google Scholar]
- Neethu, P.V.; Suthindhiran, K.; Jayasri, M.A. Antioxidant and Antiproliferative Activity of Asparagopsis taxiformis. Pharmacogn. Res. 2017, 9, 238–246. [Google Scholar]
- Nunes, N.; Valente, S.; Ferraz, S.; Barreto, M.C.; Pinheiro de Carvalho, M.A.A. Nutraceutical potential of Asparagopsis taxiformis (Delile) Trevisan extracts and assessment of a downstream purification strategy. Heliyon 2018, 4, e00957. [Google Scholar] [CrossRef]
- Pegler, J.L.; Nguyen, D.Q.; Oultram, J.M.J.; Grof, C.P.L.; Eamens, A.L. Molecular Manipulation of the miR396 and miR399 Expression Modules Alters the Response of Arabidopsis thaliana to Phosphate Stress. Plants 2021, 10, 2570. [Google Scholar] [CrossRef]
- Litholdo, C.G., Jr.; Eamens, A.L.; Waterhouse, P.M. The phenotypic and molecular assessment of the non-conserved Arabidopsis MICRORNA163/S-ADENOSYL-METHYLTRANSFERASE regulatory module during biotic stress. Mol. Genet. Genom. 2018, 293, 503–523. [Google Scholar] [CrossRef]
- Chow, H.T.; Ng, D.W. Regulation of miR163 and its targets in defense against Pseudomonas syringae in Arabidopsis thaliana. Sci. Rep. 2017, 7, 46433. [Google Scholar] [CrossRef]
- Eamens, A.L.; McHale, M.; Waterhouse, P.M. The use of artificial microRNA technology to control gene expression in Arabidopsis thaliana. Methods Mol. Biol. 2014, 1062, 211–224. [Google Scholar] [CrossRef]
- McHale, M.; Eamens, A.L.; Finnegan, E.J.; Waterhouse, P.M. A 22-nt artificial microRNA mediates widespread RNA silencing in Arabidopsis. Plant J. 2013, 76, 519–529. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Assembly Statistics | |
|---|---|
| Total base pairs (bp) | 142,472,235 |
| Number of contigs | 3308 |
| Average contig length (bp) | 43,069 |
| Largest contig length (bp) | 523,412 |
| N50 (bp) | 55,006 |
| Gaps | 0 |
| BUSCO overview | |
| Overall coverage (C/Total) | 81.60% |
| Complete BUSCOs (C) | 208 |
| Complete and single-copy BUSCOs (S) | 183 |
| Complete and duplicated BUSCOs (D) | 25 |
| Fragmented BUSCOs (F) | 11 |
| Missing BUSCOs (M) | 36 |
| Total BUSCO groups searched | 255 |
| Category | Number | Examples | Cultured | Wild |
|---|---|---|---|---|
| Halogen metabolism | 49 | PAP2/vanadium haloperoxidases, non-animal heme peroxidases | 1 | 5 |
| Potential pathogen receptors | 391 | WD40-repeat containing proteins, LRR and TRR domain proteins, Sel1 repeat-containing proteins | 8 | 6 |
| Potential defence effectors | 8 | SGT1 ortholog, apoptosis-inducing factor, exportin | 0 | 0 |
| Stress genes ROS scavenging | 27 | Ascorbate peroxidase, Cu/Zn superoxide dismutase, glutathione reductase, peroxiredoxin | 4 | 1 |
| Stress genes heat shock proteins | 59 | HSP20, HSP40, HSP90, HSP100, HSP70 binding protein, heat shock transcription factor | 4 | 7 |
| Stress-related genes | 59 | T-complex alpha/beta/gamma, tubulin binding complex B/C/D, peptidyl-prolyl cis-trans isomerase | 4 | 5 |
| Cytochrome P450 (CYP450) | 13 | CYP51G, CYP97G, CYP80B | 3 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, M.; Lang, T.; Patwary, Z.; Eamens, A.L.; Wang, T.; Webb, J.; Zuccarello, G.C.; Wegner-Thépot, A.; O’Grady, C.; Heyne, D.; et al. Insights into the Red Seaweed Asparagopsis taxiformis Using an Integrative Multi-Omics Analysis. Plants 2025, 14, 1523. https://doi.org/10.3390/plants14101523
Zhao M, Lang T, Patwary Z, Eamens AL, Wang T, Webb J, Zuccarello GC, Wegner-Thépot A, O’Grady C, Heyne D, et al. Insights into the Red Seaweed Asparagopsis taxiformis Using an Integrative Multi-Omics Analysis. Plants. 2025; 14(10):1523. https://doi.org/10.3390/plants14101523
Chicago/Turabian StyleZhao, Min, Tomas Lang, Zubaida Patwary, Andrew L. Eamens, Tianfang Wang, Jessica Webb, Giuseppe C. Zuccarello, Ana Wegner-Thépot, Charlotte O’Grady, David Heyne, and et al. 2025. "Insights into the Red Seaweed Asparagopsis taxiformis Using an Integrative Multi-Omics Analysis" Plants 14, no. 10: 1523. https://doi.org/10.3390/plants14101523
APA StyleZhao, M., Lang, T., Patwary, Z., Eamens, A. L., Wang, T., Webb, J., Zuccarello, G. C., Wegner-Thépot, A., O’Grady, C., Heyne, D., McKinnie, L., Pascelli, C., Satoh, N., Shoguchi, E., Campbell, A. H., Paul, N. A., & Cummins, S. F. (2025). Insights into the Red Seaweed Asparagopsis taxiformis Using an Integrative Multi-Omics Analysis. Plants, 14(10), 1523. https://doi.org/10.3390/plants14101523