
Large-Scale Rice Mutant Establishment and High-Throughput Mutant Manipulation Help Advance Rice Functional Genomics

 , ,
, ,  and
and 
Abstract
1. Introduction
2. Approaches for Establishment of Loss-of-Function Mutants
2.1. Chemical and Physical Mutagenesis
2.2. T-DNA Insertional Mutagenesis
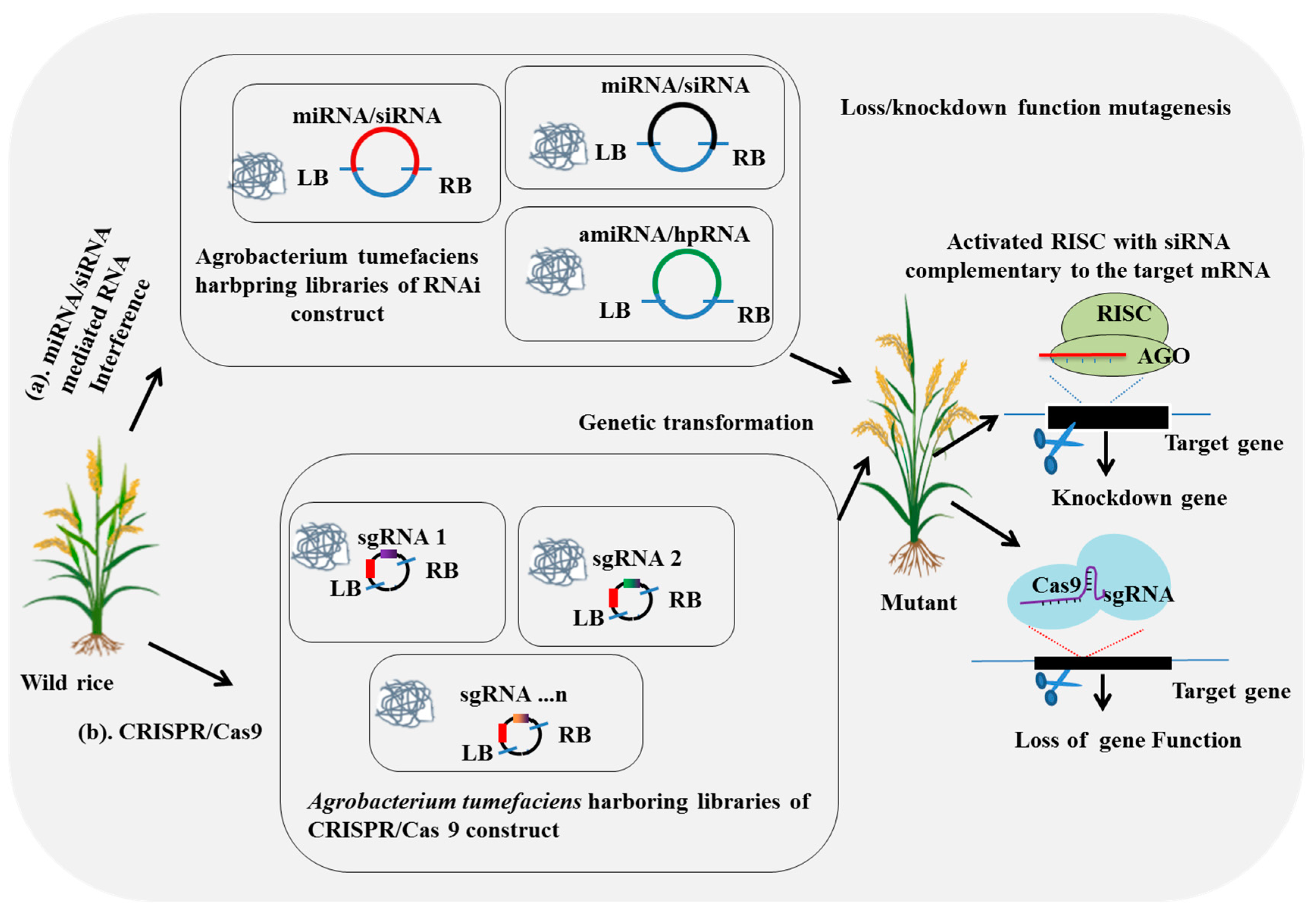
2.3. Large-Scale CRISPR/Cas9-Mediated Mutagenesis
2.4. Large-Scale RNA-Interference-Mediated Mutagenesis
3. Approaches for Establishment of Gain-of-Function Mutants
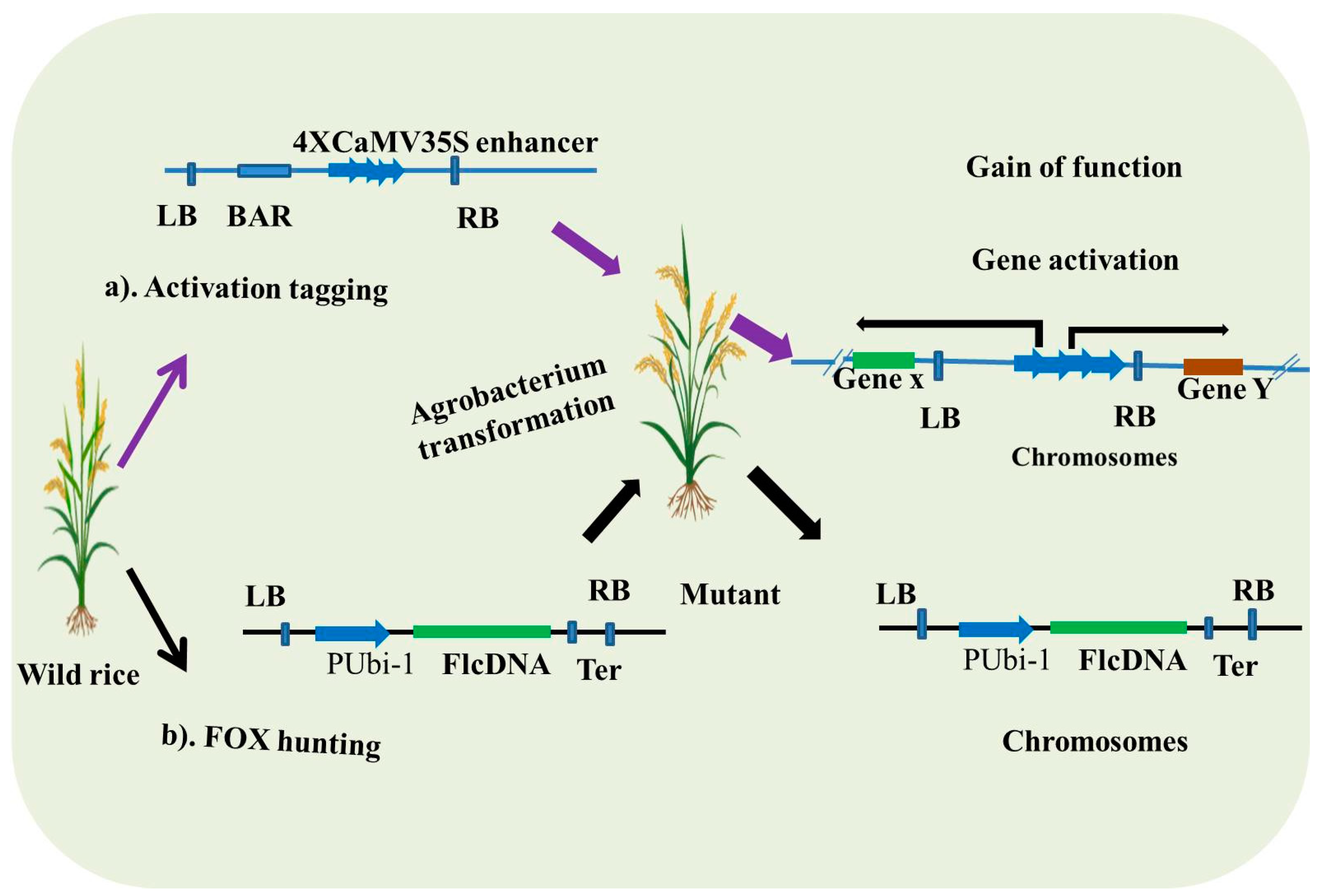
3.1. Activation Tagging
3.2. FOX (Full-Length cDNA Overexpression) Hunting
4. Genetic Resources Required for a Gain of Function
4.1. Full-Length cDNA Library
4.2. Wild Relatives of Rice
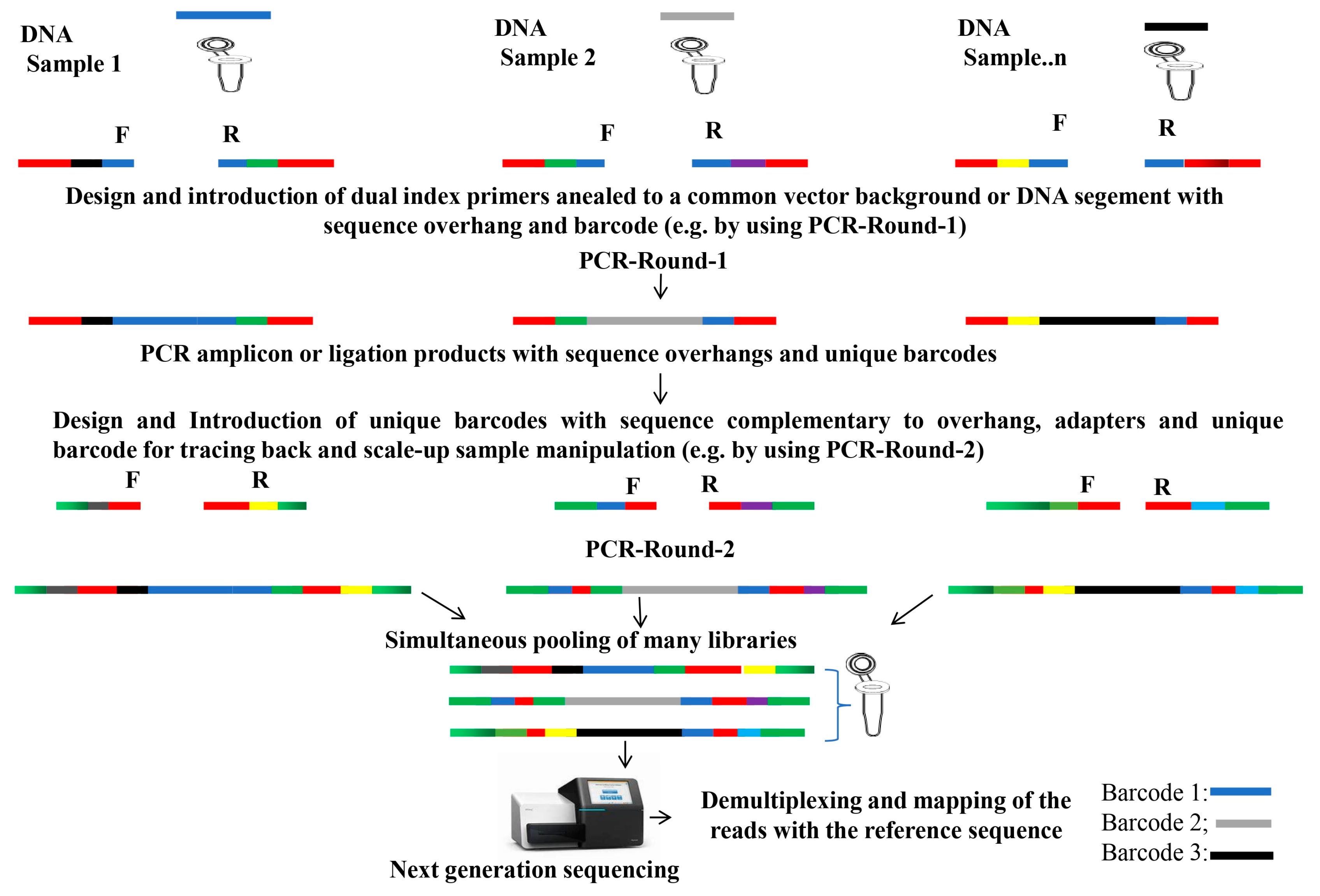
5. DNA Barcoding for Large-Scale Mutagenesis to Accelerate Functional Genomics
5.1. Fast and High-Throughput Genotyping
5.2. High-Througput Phenotyping and OMICS Integration to Accelerate Functional Genomics
6. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Solis, C.A.; Yong, M.T.; Vinarao, R.; Jena, K.; Holford, P.; Shabala, L.; Zhou, M.; Shabala, S.; Chen, Z.-H. Back to the wild: On a quest for donors toward salinity tolerant rice. Front. Plant Sci. 2020, 11, 323. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Soto, A.; Echeverría-Beirute, F.; Abdelnour-Esquivel, A.; Valdez-Melara, M.; Boch, J.; Gatica-Arias, A. Rice breeding in the new era: Comparison of useful agronomic traits. Curr. Plant Biol. 2021, 27, 100211. [Google Scholar] [CrossRef]
- Mizuno, H.; Ito, K.; Wu, J.; Tanaka, T.; Kanamori, H.; Katayose, Y.; Sasaki, T.; Matsumoto, T. Identification and mapping of expressed genes, simple sequence repeats and transposable elements in centromeric regions of rice chromosomes. DNA Res. 2006, 13, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Tian, D.; Zhang, Z.; Hu, S.; Yu, J. Rice genomics: Over the past two decades and into the future. Genom. Proteom. Bioinform. 2018, 16, 397–404. [Google Scholar] [CrossRef]
- Wang, C.; Han, B. Twenty years of rice genomics research: From sequencing and functional genomics to quantitative genomics. Mol. Plant 2022, 15, 593–619. [Google Scholar] [CrossRef]
- IRGS. The map-based sequence of the rice genome. Nature 2005, 436, 793–800. [Google Scholar] [CrossRef]
- Abe, K.; Ichikawa, H. Gene overexpression resources in cereals for functional genomics and discovery of useful genes. Front. Plant Sci. 2016, 7, 1359. [Google Scholar] [CrossRef]
- Lo, S.F.; Fan, M.J.; Hsing, Y.I.; Chen, L.J.; Chen, S.; Wen, I.C.; Liu, Y.L.; Chen, K.T.; Jiang, M.J.; Lin, M.K. Genetic resources offer efficient tools for rice functional genomics research. Plant Cell Environ. 2016, 39, 998–1013. [Google Scholar] [CrossRef]
- MVD. Joint FAO/IAEA Mutant Variety Database. 2022. Available online: https://mvd.iaea.org/ (accessed on 15 September 2024).
- Luz, V.K.d.; Silveira, S.F.d.S.; Fonseca, G.M.d.; Groli, E.L.; Figueiredo, R.G.; Baretta, D.; Kopp, M.M.; Magalhães, A.M.d.; Maia, L.C.d.; Oliveira, A.C.d. Identification of variability for agronomically important traits in rice mutant families. Bragantia 2016, 75, 41–50. [Google Scholar] [CrossRef]
- Manikandan, V.; Vanniarajan, C. Induced macromutational spectrum and frequency of viable mutants in M2 generation of rice (Oryza sativa L.). Int. J. Curr. Microbiol. Appl. Sci. 2017, 6, 1825–1834. [Google Scholar] [CrossRef]
- Oladosu, Y.; Rafii, M.Y.; Abdullah, N.; Hussin, G.; Ramli, A.; Rahim, H.A.; Miah, G.; Usman, M. Principle and application of plant mutagenesis in crop improvement: A review. Biotechnol. Biotechnol. Equip. 2016, 30, 1–16. [Google Scholar] [CrossRef]
- Viana, V.E.; Pegoraro, C.; Busanello, C.; Costa de Oliveira, A. Mutagenesis in rice: The basis for breeding a new super plant. Front. Plant Sci. 2019, 10, 1326. [Google Scholar] [CrossRef]
- Parry, M.A.; Madgwick, P.J.; Bayon, C.; Tearall, K.; Hernandez-Lopez, A.; Baudo, M.; Rakszegi, M.; Hamada, W.; Al-Yassin, A.; Ouabbou, H. Mutation discovery for crop improvement. J. Exp. Bot. 2009, 60, 2817–2825. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Cheng, S.; Wu, D.; Xia, Y.; Shu, Q. Mutagenic Improvement of Shattering Characteristic of the Restorer Line of a Hybrid Rice “Xieyou 9308”. Rice Sci. 2004, 11, 222. [Google Scholar]
- Shen, S.; Wu, D.; Gao, M.; Xia, Y.; Shu, Q. An elite restorer mutant R3027 induced by the integral application of {gamma}-rays irradiation and somaclonal variation techniques. Acta Agric. Nucleatae Sin. 2003, 17, 165–170. [Google Scholar]
- Talebi, A.B.; Shahrokhifar, B. Ethyl methane sulphonate (EMS) induced mutagenesis in Malaysian rice (cv. MR219) for lethal dose determination. Am. J. Plant Sci. 2012, 3, 1661–1665. [Google Scholar] [CrossRef]
- Nawaz, Z.; Shu, Q. Molecular nature of chemically and physically induced mutants in plants: A review. Plant Genet. Resour. 2014, 12 (Suppl. S1), S74–S78. [Google Scholar] [CrossRef]
- Li, G.; Jain, R.; Chern, M.; Pham, N.T.; Martin, J.A.; Wei, T.; Schackwitz, W.S.; Lipzen, A.M.; Duong, P.Q.; Jones, K.C. The sequences of 1504 mutants in the model rice variety Kitaake facilitate rapid functional genomic studies. Plant Cell 2017, 29, 1218–1231. [Google Scholar] [CrossRef]
- Li, Y.; Xiao, J.; Chen, L.; Huang, X.; Cheng, Z.; Han, B.; Zhang, Q.; Wu, C. Rice functional genomics research: Past decade and future. Mol. Plant 2018, 11, 359–380. [Google Scholar] [CrossRef]
- Wu, J.-L.; Wu, C.; Lei, C.; Baraoidan, M.; Bordeos, A.; Madamba, M.R.S.; Ramos-Pamplona, M.; Mauleon, R.; Portugal, A.; Ulat, V.J. Chemical-and irradiation-induced mutants of indica rice IR64 for forward and reverse genetics. Plant Mol. Biol. 2005, 59, 85–97. [Google Scholar] [CrossRef]
- Krishnan, A.; Guiderdoni, E.; An, G.; Hsing, Y.I.; Han, C.D.; Lee, M.C.; Yu, S.M.; Upadhyaya, N.; Ramachandran, S.; Zhang, Q.; et al. Mutant resources in rice for functional genomics of the grasses. Plant Physiol. 2009, 149, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Ram, H.; Soni, P.; Salvi, P.; Gandass, N.; Sharma, A.; Kaur, A.; Sharma, T.R. Insertional Mutagenesis Approaches and Their Use in Rice for Functional Genomics. Plants 2019, 8, 310. [Google Scholar] [CrossRef] [PubMed]
- Kuromori, T.; Takahashi, S.; Kondou, Y.; Shinozaki, K.; Matsui, M. Phenome analysis in plant species using loss-of-function and gain-of-function mutants. Plant Cell Physiol. 2009, 50, 1215–1231. [Google Scholar] [CrossRef]
- Wang, N.; Long, T.; Yao, W.; Xiong, L.; Zhang, Q.; Wu, C. Mutant resources for the functional analysis of the rice genome. Mol. Plant 2013, 6, 596–604. [Google Scholar] [CrossRef] [PubMed]
- Baharum, S.N.; Azizan, K.A. Metabolomics in systems biology. In Omics Applications for Systems Biology; Springer: Berlin/Heidelberg, Germany, 2018; pp. 51–68. [Google Scholar]
- Jalil, S.; Ali, Q.; Khan, A.U.; Nazir, M.M.; Ali, S.; Zulfiqar, F.; Javed, M.A.; Jin, X. Molecular and biochemical characterization of rice developed through conventional integration of nDart1-0 transposon gene. Sci. Rep. 2023, 13, 8139. [Google Scholar] [CrossRef]
- Jeon, J.S.; Lee, S.; Jung, K.H.; Jun, S.H.; Jeong, D.H.; Lee, J.; Kim, C.; Jang, S.; Yang, K.; Nam, J.; et al. T-DNA insertional mutagenesis for functional genomics in rice. Plant J. 2000, 22, 561–570. [Google Scholar] [CrossRef]
- Pucker, B.; Kleinbölting, N.; Weisshaar, B. Large scale genomic rearrangements in selected Arabidopsis thaliana T-DNA lines are caused by T-DNA insertion mutagenesis. BMC Genom. 2021, 22, 599. [Google Scholar] [CrossRef]
- Figueroa-Bossi, N.; Balbontín, R.; Bossi, L. Mapping transposon insertion sites by inverse polymerase chain reaction and Sanger sequencing. Cold Spring Harb. Protoc. 2024, 2024, pdb-prot108197. [Google Scholar]
- Sha, Y.; Li, S.; Pei, Z.; Luo, L.; Tian, Y.; He, C. Generation and flanking sequence analysis of a rice T-DNA tagged population. Theor. Appl. Genet. 2004, 108, 306–314. [Google Scholar] [CrossRef]
- Thole, V.; Worland, B.; Wright, J.; Bevan, M.W.; Vain, P. Distribution and characterization of more than 1000 T-DNA tags in the genome of Brachypodium distachyon community standard line Bd21. Plant Biotechnol.J. 2010, 8, 734–747. [Google Scholar] [CrossRef]
- Gentile, A.; D’Alessandro, L.; Medico, E. Gene trapping: A multi-purpose tool for functional genomics. Biotechnol. Genet. Eng 2003, 20, 77–100. [Google Scholar] [CrossRef] [PubMed]
- Serrania, J.; Johner, T.; Rupp, O.; Goesmann, A.; Becker, A. Massive parallel insertion site sequencing of an arrayed Sinorhizobium meliloti signature-tagged mini-Tn 5 transposon mutant library. J. Biotechnol. 2017, 257, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Wan, S.; Wu, J.; Zhang, Z.; Sun, X.; Lv, Y.; Gao, C.; Ning, Y.; Ma, J.; Guo, Y.; Zhang, Q.; et al. Activation tagging, an efficient tool for functional analysis of the rice genome. Plant Mol. Biol. 2009, 69, 69–80. [Google Scholar] [CrossRef]
- Jeong, D.H.; An, S.; Park, S.; Kang, H.G.; Park, G.G.; Kim, S.R.; Sim, J.; Kim, Y.O.; Kim, M.K.; Kim, S.R. Generation of a flanking sequence-tag database for activation-tagging lines in japonica rice. Plant J. 2006, 45, 123–132. [Google Scholar] [CrossRef]
- Johnson, A.A.; Hibberd, J.M.; Gay, C.; Essah, P.A.; Haseloff, J.; Tester, M.; Guiderdoni, E. Spatial control of transgene expression in rice (Oryza sativa L.) using the GAL4 enhancer trapping system. Plant J. 2005, 41, 779–789. [Google Scholar] [CrossRef] [PubMed]
- Sallaud, C.; Gay, C.; Larmande, P.; Bès, M.; Piffanelli, P.; Piégu, B.; Droc, G.; Regad, F.; Bourgeois, E.; Meynard, D.; et al. High throughput T-DNA insertion mutagenesis in rice: A first step towards in silico reverse genetics. Plant J. 2004, 39, 450–464. [Google Scholar] [CrossRef]
- Zhang, J.; Li, C.; Wu, C.; Xiong, L.; Chen, G.; Zhang, Q.; Wang, S. RMD: A rice mutant database for functional analysis of the rice genome. Nucleic Acids Res. 2006, 34, D745–D748. [Google Scholar] [CrossRef]
- Chern, C.-G.; Fan, M.-J.; Yu, S.-M.; Hour, A.-L.; Lu, P.-C.; Lin, Y.-C.; Wei, F.-J.; Huang, S.-C.; Chen, S.; Lai, M.-H. A rice phenomics study—Phenotype scoring and seed propagation of a T-DNA insertion-induced rice mutant population. Plant Mol.Biol. 2007, 65, 427–438. [Google Scholar] [CrossRef]
- Wang, L.; Zheng, J.; Luo, Y.; Xu, T.; Zhang, Q.; Zhang, L.; Xu, M.; Wan, J.; Wang, M.B.; Zhang, C.; et al. Construction of a genomewide RNAi mutant library in rice. Plant Biotechnol. J. 2013, 11, 997–1005. [Google Scholar] [CrossRef]
- Upadhyaya, N.M.; Zhu, Q.H.; Bhat, R.S. Transposon insertional mutagenesis in rice. Methods Mol. Biol. 2011, 678, 147–177. [Google Scholar]
- Takano, M.; Kanegae, H.; Shinomura, T.; Miyao, A.; Hirochika, H.; Furuya, M. Isolation and characterization of rice phytochrome A mutants. Plant Cell 2001, 13, 521–534. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.H.; Ramm, K.; Shivakkumar, R.; Dennis, E.S.; Upadhyaya, N.M. The ANTHER INDEHISCENCE1 gene encoding a single MYB domain protein is involved in anther development in rice. Plant Physiol. 2004, 135, 1514–1525. [Google Scholar] [CrossRef]
- Ramamoorthy, R.; Jiang, S.Y.; Ramachandran, S. Oryza sativa cytochrome P450 family member OsCYP96B4 reduces plant height in a transcript dosage dependent manner. PLoS ONE 2011, 6, e28069. [Google Scholar] [CrossRef] [PubMed]
- Margis-Pinheiro, M.; Zhou, X.-R.; Zhu, Q.-H.; Dennis, E.S.; Upadhyaya, N.M. Isolation and characterization of a Ds-tagged rice (Oryza sativa L.) GA-responsive dwarf mutant defective in an early step of the gibberellin biosynthesis pathway. Plant Cell Rep. 2005, 23, 819–833. [Google Scholar] [CrossRef]
- Jiang, S.-Y.; Cai, M.; Ramachandran, S. ORYZA SATIVA MYOSIN XI B controls pollen development by photoperiod-sensitive protein localizations. Dev.Bio. 2007, 304, 579–592. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; You, C.; Li, C.; Long, T.; Chen, G.; Byrne, M.E.; Zhang, Q. RID1, encoding a Cys2/His2-type zinc finger transcription factor, acts as a master switch from vegetative to floral development in rice. Proc. Natl. Acad. Sci. USA 2008, 105, 12915–12920. [Google Scholar] [CrossRef]
- Sun, Q.; Zhou, D.X. Rice jmjC domain-containing gene JMJ706 encodes H3K9 demethylase required for floral organ development. Proc. Natl. Acad. Sci. USA 2008, 105, 13679–13684. [Google Scholar] [CrossRef]
- Wei, S.; Hu, W.; Deng, X.; Zhang, Y.; Liu, X.; Zhao, X.; Luo, Q.; Jin, Z.; Li, Y.; Zhou, S.; et al. A rice calcium-dependent protein kinase OsCPK9 positively regulates drought stress tolerance and spikelet fertility. BMC Plant Biol. 2014, 14, 133. [Google Scholar] [CrossRef]
- Ning, J.; Zhang, B.; Wang, N.; Zhou, Y.; Xiong, L. Increased leaf angle1, a Raf-like MAPKKK that interacts with a nuclear protein family, regulates mechanical tissue formation in the Lamina joint of rice. Plant Cell 2011, 23, 4334–4347. [Google Scholar] [CrossRef]
- Du, H.; Wang, N.; Cui, F.; Li, X.; Xiao, J.; Xiong, L. Characterization of the beta-carotene hydroxylase gene DSM2 conferring drought and oxidative stress resistance by increasing xanthophylls and abscisic acid synthesis in rice. Plant Physiol. 2010, 154, 1304–1318. [Google Scholar] [CrossRef]
- Hasegawa, K.; Kamada, S.; Takehara, S.; Takeuchi, H.; Nakamura, A.; Satoh, S.; Iwai, H. Rice Putative Methyltransferase Gene OsPMT16 Is Required for Pistil Development Involving Pectin Modification. Front. Plant Sci. 2020, 11, 475. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.; Lee, Y.S.; Lee, D.Y.; Cho, M.H.; Jeon, J.S.; An, G. OsMPK6 plays a critical role in cell differentiation during early embryogenesis in Oryza sativa. J. Exp. Bot. 2016, 67, 2425–2437. [Google Scholar] [CrossRef]
- Yi, J.; Moon, S.; Lee, Y.S.; Zhu, L.; Liang, W.; Zhang, D.; Jung, K.H.; An, G. Defective Tapetum Cell Death 1 (DTC1) Regulates ROS Levels by Binding to Metallothionein during Tapetum Degeneration. Plant Physiol. 2016, 170, 1611–1623. [Google Scholar] [CrossRef]
- Kang, H.G.; Park, S.; Matsuoka, M.; An, G. White-core endosperm floury endosperm-4 in rice is generated by knockout mutations in the C4-type pyruvate orthophosphate dikinase gene (OsPPDKB). Plant J. 2005, 42, 901–911. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Long, T.; Wu, C. Effort and contribution of T-DNA Insertion mutant library for rice functional genomics research in China: Review and perspective. J. Integr. Plant Biol. 2012, 54, 953–966. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Li, X.; Yuan, W.; Chen, G.; Kilian, A.; Li, J.; Xu, C.; Li, X.; Zhou, D.X.; Wang, S. Development of enhancer trap lines for functional analysis of the rice genome. Plant J. 2003, 35, 418–427. [Google Scholar] [CrossRef]
- Wei, F.J.; Tsai, Y.C.; Hsu, Y.M.; Chen, Y.A.; Huang, C.T.; Wu, H.P.; Huang, L.T.; Lai, M.H.; Kuang, L.Y.; Lo, S.F.; et al. Lack of Genotype and Phenotype Correlation in a Rice T-DNA Tagged Line Is Likely Caused by Introgression in the Seed Source. PLoS ONE 2016, 11, e0155768. [Google Scholar] [CrossRef]
- Jiang, Y.; Cai, Z.; Xie, W.; Long, T.; Yu, H.; Zhang, Q. Rice functional genomics research: Progress and implications for crop genetic improvement. Biotechnol. Adv. 2012, 30, 1059–1070. [Google Scholar] [CrossRef]
- Zhang, J.; Guo, D.; Chang, Y.; You, C.; Li, X.; Dai, X.; Weng, Q.; Zhang, J.; Chen, G.; Li, X. Non-random distribution of T-DNA insertions at various levels of the genome hierarchy as revealed by analyzing 13 804 T-DNA flanking sequences from an enhancer-trap mutant library. Plant J. 2007, 49, 947–959. [Google Scholar] [CrossRef]
- Gong, W.; Zhou, Y.; Wang, R.; Wei, X.; Zhang, L.; Dai, Y.; Zhu, Z. Analysis of T-DNA integration events in transgenic rice. J. Plant Physiol. 2021, 266, 153527. [Google Scholar] [CrossRef]
- Hong, W.J.; Kim, Y.J.; Kim, E.J.; Kumar Nalini Chandran, A.; Moon, S.; Gho, Y.S.; Yoou, M.H.; Kim, S.T.; Jung, K.H. CAFRI-Rice: CRISPR applicable functional redundancy inspector to accelerate functional genomics in rice. Plant J. 2020, 104, 532–545. [Google Scholar] [CrossRef] [PubMed]
- Gaillochet, C.; Develtere, W.; Jacobs, T.B. CRISPR screens in plants: Approaches, guidelines, and future prospects. Plant Cell 2021, 33, 794–813. [Google Scholar] [CrossRef]
- Park, S.R.; Son, S. CRISPR/Cas9-based mutant library screening: The discovery of novel genes regulating immune responses in cotton and rice. Front. Plant Sci. 2024, 15, 1501092. [Google Scholar] [CrossRef]
- Arora, L.; Narula, A. Gene Editing and Crop Improvement Using CRISPR-Cas9 System. Front. Plant Sci. 2017, 8, 1932. [Google Scholar] [CrossRef]
- Lowder, L.G.; Zhou, J.; Zhang, Y.; Malzahn, A.; Zhong, Z.; Hsieh, T.F.; Voytas, D.F.; Zhang, Y.; Qi, Y. Robust Transcriptional Activation in Plants Using Multiplexed CRISPR-Act2.0 and mTALE-Act Systems. Mol. Plant 2018, 11, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Char, S.N.; Li, R.; Yang, B. CRISPR/Cas9 for mutagenesis in rice. In Transgenic Plants: Methods and Protocols; Springer: Berlin/Heidelberg, Germany, 2019; pp. 279–293. [Google Scholar]
- Chen, F.; Chen, L.; Yan, Z.; Xu, J.; Feng, L.; He, N.; Guo, M.; Zhao, J.; Chen, Z.; Chen, H.; et al. Recent advances of CRISPR-based genome editing for enhancing staple crops. Front. Plant Sci. 2024, 15, 1478398. [Google Scholar] [CrossRef]
- Sun, L.; Alariqi, M.; Wang, Y.; Wang, Q.; Xu, Z.; Zafar, M.N.; Yang, G.; Jia, R.; Hussain, A.; Chen, Y.; et al. Construction of Host Plant Insect-Resistance Mutant Library by High-Throughput CRISPR/Cas9 System and Identification of A Broad-Spectrum Insect Resistance Gene. Adv. Sci. 2024, 11, e2306157. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Liang, S.; Wang, G.; Hu, T.; Fu, C.; Wang, Q.; Xu, Z.; Fan, Y.; Che, L.; Min, L.; et al. CRISPR-Cas9-mediated construction of a cotton CDPK mutant library for identification of insect-resistance genes. Plant Commun. 2024, 5, 101047. [Google Scholar] [CrossRef]
- Lu, Y.; Ye, X.; Guo, R.; Huang, J.; Wang, W.; Tang, J.; Tan, L.; Zhu, J.K.; Chu, C.; Qian, Y. Genome-wide Targeted Mutagenesis in Rice Using the CRISPR/Cas9 System. Mol. Plant 2017, 10, 1242–1245. [Google Scholar] [CrossRef]
- Till, B.J.; Cooper, J.; Tai, T.H.; Colowit, P.; Greene, E.A.; Henikoff, S.; Comai, L. Discovery of chemically induced mutations in rice by TILLING. BMC Plant Biol. 2007, 7, 19. [Google Scholar] [CrossRef]
- Kolesnik, T.; Szeverenyi, I.; Bachmann, D.; Kumar, C.S.; Jiang, S.; Ramamoorthy, R.; Cai, M.; Ma, Z.G.; Sundaresan, V.; Ramachandran, S. Establishing an efficient Ac/Ds tagging system in rice: Large-scale analysis of Ds flanking sequences. Plant J. 2004, 37, 301–314. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Ke, R.; Du, M.; Yi, Y.; Chen, Y.; Wang, X.; Yao, L.; Liu, H.; Hou, X.; Xiong, L.; et al. A FLASH pipeline for arrayed CRISPR library construction and the gene function discovery of rice receptor-like kinases. Mol. Plant 2022, 15, 243–257. [Google Scholar] [CrossRef]
- Son, S.; Song, G.; Nam, S.; Lee, G.; Im, J.; Lee, K.S.; Park, Y.J.; Suh, E.J.; Park, S.R. CRISPR/Cas9-mediated mutagenesis of rice NAC transcription factor genes results in altered innate immunity. Plant Physiol. 2024, 195, 1138–1142. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Luo, Y.Z.; Zhang, L.; Jiao, X.M.; Wang, M.B.; Fan, Y.L. Rolling circle amplification-mediated hairpin RNA (RMHR) library construction in plants. Nucleic Acids Res. 2008, 36, e149. [Google Scholar] [CrossRef] [PubMed]
- Weigel, D.; Ahn, J.H.; Blázquez, M.A.; Borevitz, J.O.; Christensen, S.K.; Fankhauser, C.; Ferrándiz, C.; Kardailsky, I.; Malancharuvil, E.J.; Neff, M.M. Activation tagging in Arabidopsis. Plant Physiol. 2000, 122, 1003–1014. [Google Scholar] [CrossRef]
- Ichikawa, T.; Nakazawa, M.; Kawashima, M.; Iizumi, H.; Kuroda, H.; Kondou, Y.; Tsuhara, Y.; Suzuki, K.; Ishikawa, A.; Seki, M. The FOX hunting system: An alternative gain-of-function gene hunting technique. Plant J. 2006, 48, 974–985. [Google Scholar] [CrossRef]
- Nakamura, H.; Hakata, M.; Amano, K.; Miyao, A.; Toki, N.; Kajikawa, M.; Pang, J.; Higashi, N.; Ando, S.; Toki, S. A genome-wide gain-of-function analysis of rice genes using the FOX-hunting system. Plant Mol. Biol. 2007, 65, 357–371. [Google Scholar] [CrossRef]
- Meng, X.; Yu, H.; Zhang, Y.; Zhuang, F.; Song, X.; Gao, S.; Gao, C.; Li, J. Construction of a Genome-Wide Mutant Library in Rice Using CRISPR/Cas9. Mol. Plant 2017, 10, 1238–1241. [Google Scholar] [CrossRef]
- Zhao, D.; Chen, S.; Han, Y.; Liu, G.; Liu, J.; Yang, Q.; Zhang, T.; Shen, J.; Fan, X.; Zhang, C.; et al. A CRISPR/Cas9-mediated mutant library of seed-preferred genes in rice. Plant Biotechnol. J. 2024, 22, 3012–3014. [Google Scholar] [CrossRef]
- Younis, A.; Siddique, M.I.; Kim, C.-K.; Lim, K.-B. RNA interference (RNAi) induced gene silencing: A promising approach of hi-tech plant breeding. Int. J.Biol. Sci. 2014, 10, 1150. [Google Scholar] [CrossRef]
- Guo, Q.; Liu, Q.; Smith, N.A.; Liang, G.; Wang, M.B. RNA Silencing in Plants: Mechanisms, Technologies and Applications in Horticultural Crops. Curr. Genom. 2016, 17, 476–489. [Google Scholar] [CrossRef]
- Malakondaiah, S.; Julius, A.; Ponnambalam, D.; Gunthoti, S.S.; Ashok, J.; Krishana, P.S.; Rebecca, J. Gene silencing by RNA interference: A review. Genome Instab. Dis. 2024, 5, 225–241. [Google Scholar] [CrossRef]
- Hirochika, H.; Guiderdoni, E.; An, G.; Hsing, Y.-I.; Eun, M.Y.; Han, C.-D.; Upadhyaya, N.; Ramachandran, S.; Zhang, Q.; Pereira, A. Rice mutant resources for gene discovery. Plant Mol. Biol. 2004, 54, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Mei, Q.; Fu, Y.W.; Li, T.M.; Xuan, Y.H. Ac/Ds-Induced Receptor-like Kinase Genes Deletion Provides Broad-Spectrum Resistance to Bacterial Blight in Rice. Int. J. Mol. Sci. 2022, 23, 4561. [Google Scholar] [CrossRef]
- Huang, C.K.; Sie, Y.S.; Chen, Y.F.; Huang, T.S.; Lu, C.A. Two highly similar DEAD box proteins, OsRH2 and OsRH34, homologous to eukaryotic initiation factor 4AIII, play roles of the exon junction complex in regulating growth and development in rice. BMC Plant Biol. 2016, 16, 84. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Woo, H.R.; Guo, H. Genetic redundancy of senescence-associated transcription factors in Arabidopsis. J. Exp. Bot. 2018, 69, 811–823. [Google Scholar] [CrossRef]
- Ling, J.; Li, R.; Nwafor, C.C.; Cheng, J.; Li, M.; Xu, Q.; Wu, J.; Gan, L.; Yang, Q.; Liu, C. Development of iFOX-hunting as a functional genomic tool and demonstration of its use to identify early senescence-related genes in the polyploid Brassica napus. Plant Biotechnol. J. 2018, 16, 591–602. [Google Scholar] [CrossRef]
- Cheng, H.; Gao, J.; Cai, H.; Zhu, J.; Huang, H. Gain-of-function in Arabidopsis (GAINA) for identifying functional genes in Hevea brasiliensis. SpringerPlus 2016, 5, 1853. [Google Scholar] [CrossRef]
- Pathak, B.; Maurya, C.; Faria, M.C.; Alizada, Z.; Nandy, S.; Zhao, S.; Jamsheer, K.M.; Srivastava, V. Targeting TOR and SnRK1 Genes in Rice with CRISPR/Cas9. Plants 2022, 11, 1453. [Google Scholar] [CrossRef]
- Zhao, S.; Luo, J.; Zeng, X.; Li, K.; Yuan, R.; Zhu, L.; Li, X.; Wu, G.; Yan, X. Rolling Circle Amplification (RCA)-Mediated Genome-Wide ihpRNAi Mutant Library Construction in Brassica napus. Int. J. Mol. Sci. 2020, 21, 7243. [Google Scholar] [CrossRef]
- Hauser, F.; Ceciliato, P.H.O.; Lin, Y.C.; Guo, D.; Gregerson, J.D.; Abbasi, N.; Youhanna, D.; Park, J.; Dubeaux, G.; Shani, E.; et al. A seed resource for screening functionally redundant genes and isolation of new mutants impaired in CO2 and ABA responses. J.Exp.Bot. 2019, 70, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Hauser, F.; Chen, W.; Deinlein, U.; Chang, K.; Ossowski, S.; Fitz, J.; Hannon, G.J.; Schroeder, J.I. A genomic-scale artificial microRNA library as a tool to investigate the functionally redundant gene space in Arabidopsis. Plant Cell 2013, 25, 2848–2863. [Google Scholar] [CrossRef]
- Jover-Gil, S.; Paz-Ares, J.; Micol, J.L.; Ponce, M.R. Multi-gene silencing in Arabidopsis: A collection of artificial microRNAs targeting groups of paralogs encoding transcription factors. Plant J. 2014, 80, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Moin, M.; Bakshi, A.; Saha, A.; Dutta, M.; Kirti, P. Gain-of-function mutagenesis approaches in rice for functional genomics and improvement of crop productivity. Brief. Funct. Genom 2017, 16, 238–247. [Google Scholar] [CrossRef]
- Deng, M.; Wang, Y.; Kuzma, M.; Chalifoux, M.; Tremblay, L.; Yang, S.; Ying, J.; Sample, A.; Wang, H.M.; Griffiths, R. Activation tagging identifies Arabidopsis transcription factor AtMYB68 for heat and drought tolerance at yield determining reproductive stages. Plant J. 2020, 104, 1535–1550. [Google Scholar] [CrossRef]
- Kota, V.R.; Gundra, S.R.; Vudem, D.R.; Pulugurtha, B.K.; Khareedu, V.R. Development of a large population of activation-tagged mutants in an elite indica rice variety. Plant Breed. 2020, 139, 328–343. [Google Scholar] [CrossRef]
- Plemmons, A.N.; Askins, A.R.; Mendoza, S.M.; Hancock, C.N. A Transposon-Based Activation Tag System for Functional Genomics in Cereals: Detection of Mping-Based Activation Tag Mobilization in Wheat. In Accelerated Breeding of Cereal Crops; Springer: Berlin/Heidelberg, Germany, 2022; pp. 195–207. [Google Scholar]
- Mahmoud, M.; Zhou, Z.; Kaur, R.; Bekele, W.; Tinker, N.A.; Singh, J. Toward the development of Ac/Ds transposon-mediated gene tagging system for functional genomics in oat (Avena sativa L.). Funct. Integr. Genom 2022, 22, 669–681. [Google Scholar] [CrossRef]
- Mathews, H.; Clendennen, S.K.; Caldwell, C.G.; Liu, X.L.; Connors, K.; Matheis, N.; Schuster, D.K.; Menasco, D.; Wagoner, W.; Lightner, J. Activation tagging in tomato identifies a transcriptional regulator of anthocyanin biosynthesis, modification, and transport. Plant Cell 2003, 15, 1689–1703. [Google Scholar] [CrossRef]
- Ayliffe, M.A.; Pryor, A.J. Activation tagging and insertional mutagenesis in barley. In Plant Reverse Genetics Methods and Protocols; Springer: Berlin/Heidelberg, Germany, 2011; pp. 107–128. [Google Scholar]
- An, S.; Park, S.; Jeong, D.-H.; Lee, D.-Y.; Kang, H.-G.; Yu, J.-H.; Hur, J.; Kim, S.-R.; Kim, Y.-H.; Lee, M. Generation and analysis of end sequence database for T-DNA tagging lines in rice. Plant Physiol. 2003, 133, 2040–2047. [Google Scholar] [CrossRef]
- Chen, S.; Jin, W.; Wang, M.; Zhang, F.; Zhou, J.; Jia, Q.; Wu, Y.; Liu, F.; Wu, P. Distribution and characterization of over 1000 T-DNA tags in rice genome. Plant J. 2003, 36, 105–113. [Google Scholar] [CrossRef]
- Hsing, Y.-I.; Chern, C.-G.; Fan, M.-J.; Lu, P.-C.; Chen, K.-T.; Lo, S.-F.; Sun, P.-K.; Ho, S.-L.; Lee, K.-W.; Wang, Y.-C. A rice gene activation/knockout mutant resource for high throughput functional genomics. Plant Mol. Biol. 2007, 63, 351–364. [Google Scholar] [CrossRef] [PubMed]
- Mori, M.; Tomita, C.; Sugimoto, K.; Hasegawa, M.; Hayashi, N.; Dubouzet, J.G.; Ochiai, H.; Sekimoto, H.; Hirochika, H.; Kikuchi, S. Isolation and molecular characterization of a Spotted leaf 18 mutant by modified activation-tagging in rice. Plant Mol. Biol. 2007, 63, 847–860. [Google Scholar] [CrossRef]
- Qu, S.; Desai, A.; Wing, R.; Sundaresan, V. A versatile transposon-based activation tag vector system for functional genomics in cereals and other monocot plants. Plant Physiol. 2008, 146, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Dutta, M.; Moin, M.; Saha, A.; Dutta, D.; Bakshi, A.; Kirti, P. Gain-of-function mutagenesis through activation tagging identifies XPB2 and SEN1 helicase genes as potential targets for drought stress tolerance in rice. Theor. Appl. Genet. 2021, 134, 2253–2272. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Wang, X.; Liu, J.; Yu, K.; Gao, Y.; Liu, H.; Wang, C.; Wang, W.; Wang, G.; Liu, M.; et al. Application of T-DNA activation tagging to identify glutamate receptor-like genes that enhance drought tolerance in plants. Plant Cell Rep. 2014, 33, 617–631. [Google Scholar] [CrossRef]
- Gandikota, M.; Krishnakanth Yadav, T.; Maram, R.R.; Kalluru, S.; Sena, M.B.; Siddiq, E.; Kalinati Narasimhan, Y.; Vemireddy, L.R.; Ghanta, A. Development of activation-tagged gain-of-functional mutants in indica rice line (BPT 5204) for sheath blight resistance. Mo. Biol. Rep. 2024, 51, 381. [Google Scholar] [CrossRef]
- Kondou, Y.; Higuchi, M.; Takahashi, S.; Sakurai, T.; Ichikawa, T.; Kuroda, H.; Yoshizumi, T.; Tsumoto, Y.; Horii, Y.; Kawashima, M. Systematic approaches to using the FOX hunting system to identify useful rice genes. Plant J. 2009, 57, 883–894. [Google Scholar] [CrossRef]
- Albinsky, D.; Kusano, M.; Higuchi, M.; Hayashi, N.; Kobayashi, M.; Fukushima, A.; Mori, M.; Ichikawa, T.; Matsui, K.; Kuroda, H. Metabolomic screening applied to rice FOX Arabidopsis lines leads to the identification of a gene-changing nitrogen metabolism. Mol. Plant 2010, 3, 125–142. [Google Scholar] [CrossRef]
- Higashi, Y.; Ohama, N.; Ishikawa, T.; Katori, T.; Shimura, A.; Kusakabe, K.; Yamaguchi-Shinozaki, K.; Ishida, J.; Tanaka, M.; Seki, M. HsfA1d, a protein identified via FOX hunting using Thellungiella salsuginea cDNAs improves heat tolerance by regulating heat-stress-responsive gene expression. Mol. Plant 2013, 6, 411–422. [Google Scholar] [CrossRef]
- Taji, T.; Sakurai, T.; Mochida, K.; Ishiwata, A.; Kurotani, A.; Totoki, Y.; Toyoda, A.; Sakaki, Y.; Seki, M.; Ono, H. Large-scale collection and annotation of full-length enriched cDNAs from a model halophyte, Thellungiella halophila. BMC Plant Biol. 2008, 8, 115. [Google Scholar] [CrossRef]
- Hakata, M.; Nakamura, H.; Iida-Okada, K.; Miyao, A.; Kajikawa, M.; Imai-Toki, N.; Pang, J.; Amano, K.; Horikawa, A.; Tsuchida-Mayama, T. Production and characterization of a large population of cDNA-overexpressing transgenic rice plants using Gateway-based full-length cDNA expression libraries. Breed. Sci. 2010, 60, 575–585. [Google Scholar] [CrossRef]
- Ogawa, Y.; Dansako, T.; Yano, K.; Sakurai, N.; Suzuki, H.; Aoki, K.; Noji, M.; Saito, K.; Shibata, D. Efficient and high-throughput vector construction and Agrobacterium-mediated transformation of Arabidopsis thaliana suspension-cultured cells for functional genomics. Plant Cell Physiol. 2008, 49, 242–250. [Google Scholar] [CrossRef]
- Papdi, C.; Abrahám, E.; Joseph, M.P.; Popescu, C.; Koncz, C.; Szabados, L. Functional identification of Arabidopsis stress regulatory genes using the controlled cDNA overexpression system. Plant Physiol. 2008, 147, 528–542. [Google Scholar] [CrossRef] [PubMed]
- Rauschendorfer, J.; Yordanov, Y.; Dobrev, P.; Vankova, R.; Sykes, R.; Külheim, C.; Busov, V. Overexpression of a developing xylem cDNA library in transgenic poplar generates high mutation rate specific to wood formation. Plant Biotechnol. J. 2020, 18, 1434–1443. [Google Scholar] [CrossRef]
- Kong, L.; Li, Z.; Song, Q.; Li, X.; Luo, K. Construction of a full-length cdna over-expressing library to identify valuable genes from Populus tomentosa. Int. J. Mol. Sci. 2021, 22, 3448. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Cai, W.; Du, Z.; Zhang, W.; Xu, Q.; Sun, W.; Chen, M. Establishing a System for Functional Characterization of Full-Length cDNAs of Camellia sinensis. Int. J. Mol. Sci. 2019, 20, 5929. [Google Scholar] [CrossRef]
- Wu, J.; Zhang, Z.; Zhang, Q.; Liu, Y.; Zhu, B.; Cao, J.; Li, Z.; Han, L.; Jia, J.; Zhao, G. Generation of wheat transcription factor FOX rice lines and systematic screening for salt and osmotic stress tolerance. PLoS ONE 2015, 10, e0132314. [Google Scholar] [CrossRef]
- Fujita, M.; Mizukado, S.; Fujita, Y.; Ichikawa, T.; Nakazawa, M.; Seki, M.; Matsui, M.; Yamaguchi-Shinozaki, K.; Shinozaki, K. Identification of stress-tolerance-related transcription-factor genes via mini-scale Full-length cDNA Over-eXpressor (FOX) gene hunting system. Biochem. Biophys. Res. Commun. 2007, 364, 250–257. [Google Scholar] [CrossRef]
- Kikuchi, S.; Satoh, K.; Nagata, T.; Kawagashira, N.; Doi, K.; Kishimoto, N.; Yazaki, J.; Ishikawa, M.; Yamada, H.; Ooka, H. Collection, mapping, and annotation of over 28,000 cDNA clones from japonica rice. Science 2003, 301, 376–379. [Google Scholar] [CrossRef]
- Liu, X.; Lu, T.; Yu, S.; Li, Y.; Huang, Y.; Huang, T.; Zhang, L.; Zhu, J.; Zhao, Q.; Fan, D. A collection of 10,096 indica rice full-length cDNAs reveals highly expressed sequence divergence between Oryza sativa indica and japonica subspecies. Plant Mol. Biol. 2007, 65, 403–415. [Google Scholar] [CrossRef]
- Xie, K.; Zhang, J.; Xiang, Y.; Feng, Q.; Han, B.; Chu, Z.; Wang, S.; Zhang, Q.; Xiong, L. Isolation and annotation of 10828 putative full length cDNAs from indica rice. Sci. China Ser. C Life Sci. 2005, 48, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Yu, S.; Fan, D.; Mu, J.; Shangguan, Y.; Wang, Z.; Minobe, Y.; Lin, Z.; Han, B. Collection and comparative analysis of 1888 full-length cDNAs from wild rice Oryza rufipogon Griff. W1943. DNA Res. 2008, 15, 285–295. [Google Scholar] [CrossRef]
- Jain, M.; Khurana, P.; Tyagi, A.K.; Khurana, J.P. Genome-wide analysis of intronless genes in rice and Arabidopsis. Funct. Integr. Genom. 2008, 8, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Aviña-Padilla, K.; Ramírez-Rafael, J.A.; Herrera-Oropeza, G.E.; Muley, V.Y.; Valdivia, D.I.; Díaz-Valenzuela, E.; García-García, A.; Varela-Echavarría, A.; Hernández-Rosales, M. Evolutionary perspective and expression analysis of intronless genes highlight the conservation of their regulatory role. Front. Genet. 2021, 12, 654256. [Google Scholar] [CrossRef]
- Shabalina, S.A.; Ogurtsov, A.Y.; Spiridonov, A.N.; Novichkov, P.S.; Spiridonov, N.A.; Koonin, E.V. Distinct patterns of expression and evolution of intronless and intron-containing mammalian genes. Mol. Biol. Evol. 2010, 27, 1745–1749. [Google Scholar] [CrossRef]
- Ariga, H.; Tanaka, T.; Ono, H.; Sakata, Y.; Hayashi, T.; Taji, T. CSP41b, a protein identified via FOX hunting using Eutrema salsugineum cDNAs, improves heat and salinity stress tolerance in transgenic Arabidopsis thaliana. Biochem. Biophys. Res. Commun. 2015, 464, 318–323. [Google Scholar] [CrossRef]
- Atwell, B.J.; Wang, H.; Scafaro, A.P. Could abiotic stress tolerance in wild relatives of rice be used to improve Oryza sativa? Plant Sci. 2014, 215–216, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Bohra, A.; Kilian, B.; Sivasankar, S.; Caccamo, M.; Mba, C.; McCouch, S.R.; Varshney, R.K. Reap the crop wild relatives for breeding future crops. Trends Biotechnol. 2022, 40, 412–431. [Google Scholar] [CrossRef]
- Wisniewski, M.; Artlip, T.; Liu, J.; Ma, J.; Burchard, E.; Norelli, J.; Dardick, C. Fox hunting in wild apples: Searching for novel genes in Malus sieversii. Int. J. Mol. Sci. 2020, 21, 9516. [Google Scholar] [CrossRef]
- Kozich, J.J.; Westcott, S.L.; Baxter, N.T.; Highlander, S.K.; Schloss, P.D. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. App. Environ. Microbiol. 2013, 79, 5112–5120. [Google Scholar] [CrossRef]
- Bohmann, K.; Elbrecht, V.; Carøe, C.; Bista, I.; Leese, F.; Bunce, M.; Yu, D.W.; Seymour, M.; Dumbrell, A.J.; Creer, S. Strategies for sample labelling and library preparation in DNA metabarcoding studies. Mol. Ecol. Resour. 2022, 22, 1231–1246. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Li, Q.; Deng, J.; Liu, Q.; Huang, X. The devil is in the details: Problems in DNA barcoding practices indicated by systematic evaluation of insect barcodes. Front. Ecol. Evol. 2023, 11, 1149839. [Google Scholar] [CrossRef]
- Pochon, X.; Bott, N.J.; Smith, K.F.; Wood, S.A. Evaluating detection limits of next-generation sequencing for the surveillance and monitoring of international marine pests. PLoS ONE 2013, 8, e73935. [Google Scholar] [CrossRef]
- Gous, A.; Swanevelder, D.Z.H.; Eardley, C.D.; Willows-Munro, S. Plant-pollinator interactions over time: Pollen metabarcoding from bees in a historic collection. Evol. Appl. 2019, 12, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Lucas, A.; Bodger, O.; Brosi, B.J.; Ford, C.R.; Forman, D.W.; Greig, C.; Hegarty, M.; Jones, L.; Neyland, P.J.; de Vere, N. Floral resource partitioning by individuals within generalised hoverfly pollination networks revealed by DNA metabarcoding. Sci. Rep. 2018, 8, 5133. [Google Scholar] [CrossRef]
- Pjevac, P.; Hausmann, B.; Schwarz, J.; Kohl, G.; Herbold, C.; Loy, A.; Berry, D. An Economical and Flexible Dual Barcoding, Two-Step PCR Approach for Highly Multiplexed Amplicon Sequencing. Front. Microbiol. 2021, 12, 669776. [Google Scholar] [CrossRef]
- Lyons, E.; Sheridan, P.; Tremmel, G.; Miyano, S.; Sugano, S. Large-scale DNA barcode library generation for biomolecule identification in high-throughput screens. Sci. Rep. 2017, 7, 13899. [Google Scholar] [CrossRef]
- Carøe, C.; Bohmann, K. Tagsteady: A metabarcoding library preparation protocol to avoid false assignment of sequences to samples. Mol. Ecol. Resour. 2020, 20, 1620–1631. [Google Scholar] [CrossRef]
- Liu, Q.; Wang, C.; Jiao, X.; Zhang, H.; Song, L.; Li, Y.; Gao, C.; Wang, K. Hi-TOM: A platform for high-throughput tracking of mutations induced by CRISPR/Cas systems. Sci. China Life Sci. 2019, 62, 1–7. [Google Scholar] [CrossRef]
- Zhu, S.; Cao, Z.; Liu, Z.; He, Y.; Wang, Y.; Yuan, P.; Li, W.; Tian, F.; Bao, Y.; Wei, W. Guide RNAs with embedded barcodes boost CRISPR-pooled screens. Genome Biol. 2019, 20, 20. [Google Scholar] [CrossRef]
- Liu, H.-J.; Jian, L.; Xu, J.; Zhang, Q.; Zhang, M.; Jin, M.; Peng, Y.; Yan, J.; Han, B.; Liu, J. High-throughput CRISPR/Cas9 mutagenesis streamlines trait gene identification in maize. Plant Cell 2020, 32, 1397–1413. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.-R.; Hale, D.C.; Ciolek, P.J.; Runge, K.W. Generation and analysis of a barcode-tagged insertion mutant library in the fission yeast Schizosaccharomyces pombe. BMC Genom. 2012, 13, 161. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Miller, D.; Liu, X.; Tosi, L.; Chkaiban, L.; Mei, H.; Hung, P.-H.; Parekkadan, B.; Sherlock, G.; Levy, S.F. Arrayed in vivo barcoding for multiplexed sequence verification of plasmid DNA and demultiplexing of pooled libraries. Nucleic Acids Res. 2024, 52, e47. [Google Scholar] [CrossRef]
- Tripathi, S.; Voogdt, C.G.P.; Bassler, S.O.; Anderson, M.; Huang, P.H.; Sakenova, N.; Capraz, T.; Jain, S.; Koumoutsi, A.; Bravo, A.M.; et al. Randomly barcoded transposon mutant libraries for gut commensals I: Strategies for efficient library construction. Cell Rep. 2024, 43, 113517. [Google Scholar] [CrossRef]
- Piffanelli, P.; Droc, G.; Mieulet, D.; Lanau, N.; Bès, M.; Bourgeois, E.; Rouvière, C.; Gavory, F.; Cruaud, C.; Ghesquière, A.; et al. Large-scale characterization of Tos17 insertion sites in a rice T-DNA mutant library. Plant Mol. Biol. 2007, 65, 587–601. [Google Scholar] [CrossRef] [PubMed]
- Uzbas, F.; Opperer, F.; Sönmezer, C.; Shaposhnikov, D.; Sass, S.; Krendl, C.; Angerer, P.; Theis, F.J.; Mueller, N.S.; Drukker, M. BART-Seq: Cost-effective massively parallelized targeted sequencing for genomics, transcriptomics, and single-cell analysis. Genome Biol. 2019, 20, 155. [Google Scholar] [CrossRef]
- Chen, W.; Wang, P.; Wang, L.; Zhang, D.; Han, M.; Han, M.; Song, L. Low-complexity and highly robust barcodes for error-rich single molecular sequencing. 3 Biotech 2021, 11, 78. [Google Scholar] [CrossRef]
- Srivathsan, A.; Meier, R. Scalable, Cost-Effective, and Decentralized DNA Barcoding with Oxford Nanopore Sequencing. Methods Mol. Biol. 2024, 2744, 223–238. [Google Scholar]
- Duan, L.; Huang, C.; Chen, G.; Xiong, L.; Liu, Q.; Yang, W. Determination of rice panicle numbers during heading by multi-angle imaging. Crop J. 2015, 3, 211–219. [Google Scholar] [CrossRef]
- Fahlgren, N.; Gehan, M.A.; Baxter, I. Lights, camera, action: High-throughput plant phenotyping is ready for a close-up. Curr. Opin. Plant Biol. 2015, 24, 93–99. [Google Scholar] [CrossRef]
- Pratap, A.; Gupta, S.; Nair, R.M.; Gupta, S.; Schafleitner, R.; Basu, P.; Singh, C.M.; Prajapati, U.; Gupta, A.K.; Nayyar, H. Using plant phenomics to exploit the gains of genomics. Agronomy 2019, 9, 126. [Google Scholar] [CrossRef]
- Song, P.; Wang, J.; Guo, X.; Yang, W.; Zhao, C. High-throughput phenotyping: Breaking through the bottleneck in future crop breeding. Crop J. 2021, 9, 633–645. [Google Scholar] [CrossRef]
- Jangra, S.; Chaudhary, V.; Yadav, R.C.; Yadav, N.R. High-throughput phenotyping: A platform to accelerate crop improvement. Phenomics 2021, 1, 31–53. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Guo, Z.; Huang, C.; Duan, L.; Chen, G.; Jiang, N.; Fang, W.; Feng, H.; Xie, W.; Lian, X. Combining high-throughput phenotyping and genome-wide association studies to reveal natural genetic variation in rice. Nat. Commun. 2014, 5, 5087. [Google Scholar] [CrossRef]
- Qin, Z.; Zhang, Z.; Hua, X.; Yang, W.; Liang, X.; Zhai, R.; Huang, C. Cereal grain 3D point cloud analysis method for shape extraction and filled/unfilled grain identification based on structured light imaging. Sci. Rep. 2022, 12, 3145. [Google Scholar] [CrossRef]
- Sun, M.; Huang, S.; Lu, Z.; Wang, M.; Zhang, S.; Yang, K.; Tang, B.; Yang, W.; Huang, C. A novel method for intelligent analysis of rice yield traits based on LED transmission imaging and cloud computing. Measurement 2023, 217, 113017. [Google Scholar] [CrossRef]
- Guo, Z.; Yang, W.; Chang, Y.; Ma, X.; Tu, H.; Xiong, F.; Jiang, N.; Feng, H.; Huang, C.; Yang, P. Genome-wide association studies of image traits reveal genetic architecture of drought resistance in rice. Mol. Plant 2018, 11, 789–805. [Google Scholar] [CrossRef] [PubMed]
- Ricachenevsky, F.K.; Punshon, T.; Lee, S.; Oliveira, B.H.N.; Trenz, T.S.; Maraschin, F.d.S.; Hindt, M.N.; Danku, J.; Salt, D.E.; Fett, J. Elemental profiling of rice FOX lines leads to characterization of a new Zn plasma membrane transporter, OsZIP7. Front. Plant Sci. 2018, 9, 865. [Google Scholar] [CrossRef]
- Wairich, A.; Vitali, A.; Adamski, J.M.; Lopes, K.L.; Duarte, G.L.; Ponte, L.R.; Costa, H.K.; Menguer, P.K.; Santos, R.P.d.; Fett, J.P.J. Enhanced expression of OsNAC5 leads to up-regulation of OsNAC6 and changes rice (Oryza sativa L.) ionome. Genet. Mol. Biol. 2022, 46 (Suppl. S1), e20220190. [Google Scholar] [CrossRef]
- Cao, Z.Z.; Lin, X.Y.; Yang, Y.J.; Guan, M.Y.; Xu, P.; Chen, M.X. Gene identification and transcriptome analysis of low cadmium accumulation rice mutant (lcd1) in response to cadmium stress using MutMap and RNA-seq. BMC Plant Biol. 2019, 19, 250. [Google Scholar] [CrossRef]
- Ishikawa, S.; Ae, N.; Yano, M. Chromosomal regions with quantitative trait loci controlling cadmium concentration in brown rice (Oryza sativa). New Phytol. 2005, 168, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Xu, Y.; Liu, H.; Mao, Z.; Zhang, C.; Ma, Y.; Zhang, Q.; Meng, Z.; Chong, K. The interaction between OsMADS57 and OsTB1 modulates rice tillering via DWARF14. Nat. Commun. 2013, 4, 1566. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutagenesis Tools | Mutation Type | Advantages | Challenges | Precision | Scalability | References |
|---|---|---|---|---|---|---|
| Chemical (e.g., EMS) | Random | Simple non-transgenic with high mutagenesis efficiency | Requires extensive screening | Low | Low | [73] |
| T-DNA Insertion | Random | Genome-wide coverage; barcode-compatible. Thousands of mutants analyzed in parallel | Random insertion disrupts non-target genes.Individual mutant screening via PCR or phenotyping | Low | High | [24,28] |
| Transposons (e.g., Ac/Ds) | Random | Mobility enables regional mutagenesis.Reusable systems | Requires transposase control; lower efficiency Individual mutant screening via PCR and phenotyping | Moderate | Moderate | [74] |
| CRISPR/Cas9 | Targeted knockouts/editing | High precision; multiplex editing of redundant genes | Complex design for large libraries | High | High | [64,75,76] |
| RNAi | Targeted knockdown | Rapid; scalable with barcoded vectors (e.g., pooled RNAi screens) for redundant genes | Requires efficient design for target silencing | High | Moderate | [41,64,77] |
| Activation tagging | Gain-of-function | Identifies dominant alleles | random activation may cause pleiotropy Low mutant frequency | Low | Moderate | [35,78] |
| FOX hunting | cDNA overexpression | Precise overexpression. Possible to use barcoding to trace back the cDNA clones; links phenotype to known genes | High cost of cDNA library construction. Complex phenotyping | High | High | [79,80] |
| Approaches for Integration of DNA Barcoding | References | |
|---|---|---|
| FOX hunting | Pooled cDNA clone libraries or mutants with unique barcodes can be synthesized using amplification primers or sequencing primers | [148] |
| Activation tagging | Integrates unique barcodes into the T-DNA construct | [24] |
| T-DNA | Embeds unique barcodes into the mutagenic T-DNA construct | [24] |
| Transposon (Ac/Ds) | Ds transposons tagged with barcodes for tracking reinsertion sites | [24,149] |
| Retrotransposon (Tos17) | Unique barcode embedded with long terminal repeat (LTR) for tracking insertion sites | [150] |
| RNAi | Multiplexed shRNA or amiRNA libraries with unique barcodes | [41,77] |
| CRISPR/Cas9 | Multiplexed sgRNA libraries with unique barcodes | [75,76,82] |
| Important features of DNA barcoding | ||
| Tracking and screening | Scalable: sequencing identifies barcodes linked to samples | [141,151] |
| Scalability | Thousands of mutants can be analyzed simultaneously | [24,41,70,75,76,77,82] |
| Precision | High: pooled libraries enable genome-wide screening (e.g., CRISPR-Cas9 with barcoded sgRNAs; and shRNA or amiRNA libraries with unique barcodes) | [41,75,76,77,144] |
| Applications | Large-scale functional genomics, pooled screens | [24,41,70,75,76,77,82] |
| Challenges | Requires careful design and integration of DNA barcode and next-generation sequencing data analysis | [136,152,153] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wolella, E.K.; Cheng, Z.; Li, M.; Xia, D.; Zhang, J.; Duan, L.; Liu, L.; Li, Z.; Zhang, J. Large-Scale Rice Mutant Establishment and High-Throughput Mutant Manipulation Help Advance Rice Functional Genomics. Plants 2025, 14, 1492. https://doi.org/10.3390/plants14101492
Wolella EK, Cheng Z, Li M, Xia D, Zhang J, Duan L, Liu L, Li Z, Zhang J. Large-Scale Rice Mutant Establishment and High-Throughput Mutant Manipulation Help Advance Rice Functional Genomics. Plants. 2025; 14(10):1492. https://doi.org/10.3390/plants14101492
Chicago/Turabian StyleWolella, Eyob Kassaye, Zhen Cheng, Mengyuan Li, Dandan Xia, Jianwei Zhang, Liu Duan, Li Liu, Zhiyong Li, and Jian Zhang. 2025. "Large-Scale Rice Mutant Establishment and High-Throughput Mutant Manipulation Help Advance Rice Functional Genomics" Plants 14, no. 10: 1492. https://doi.org/10.3390/plants14101492
APA StyleWolella, E. K., Cheng, Z., Li, M., Xia, D., Zhang, J., Duan, L., Liu, L., Li, Z., & Zhang, J. (2025). Large-Scale Rice Mutant Establishment and High-Throughput Mutant Manipulation Help Advance Rice Functional Genomics. Plants, 14(10), 1492. https://doi.org/10.3390/plants14101492