
Unmasking the Aliphatic Repertoire: New Polyunsaturated Metabolites in Bupleurum falcatum sensu lato Provide Chemotaxonomic Insights


Abstract
1. Introduction
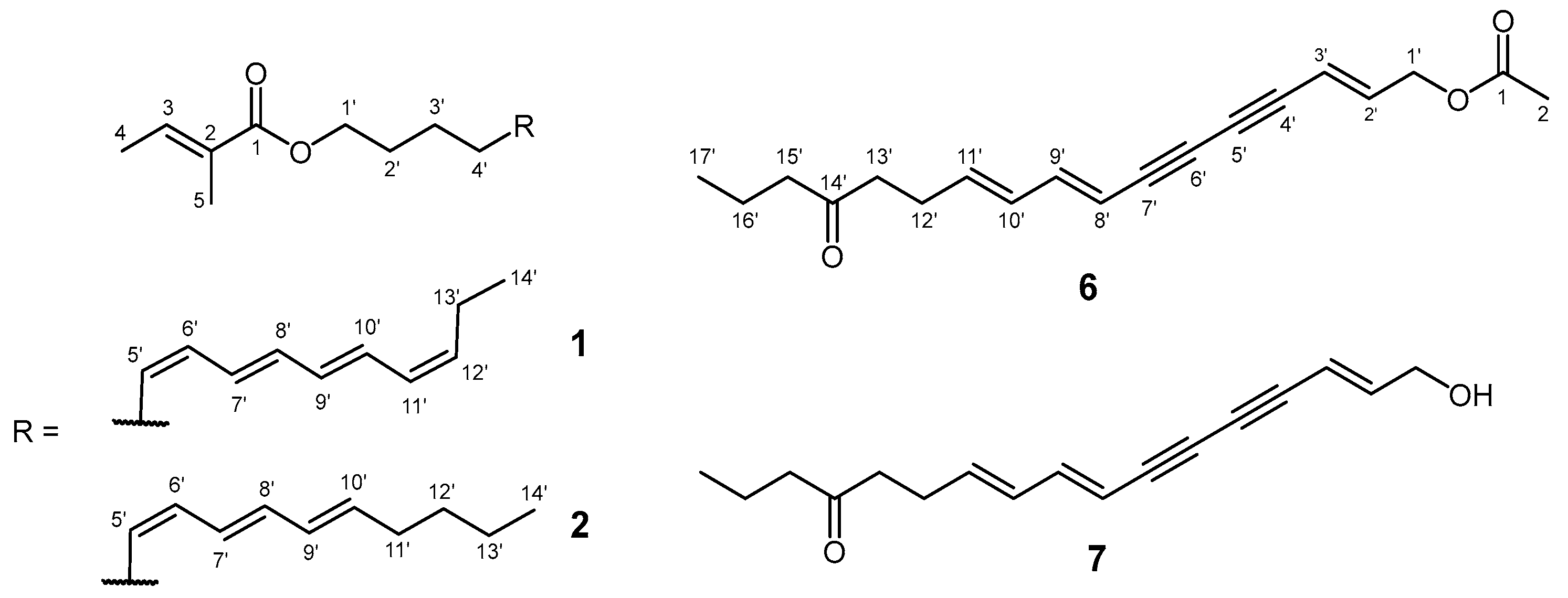
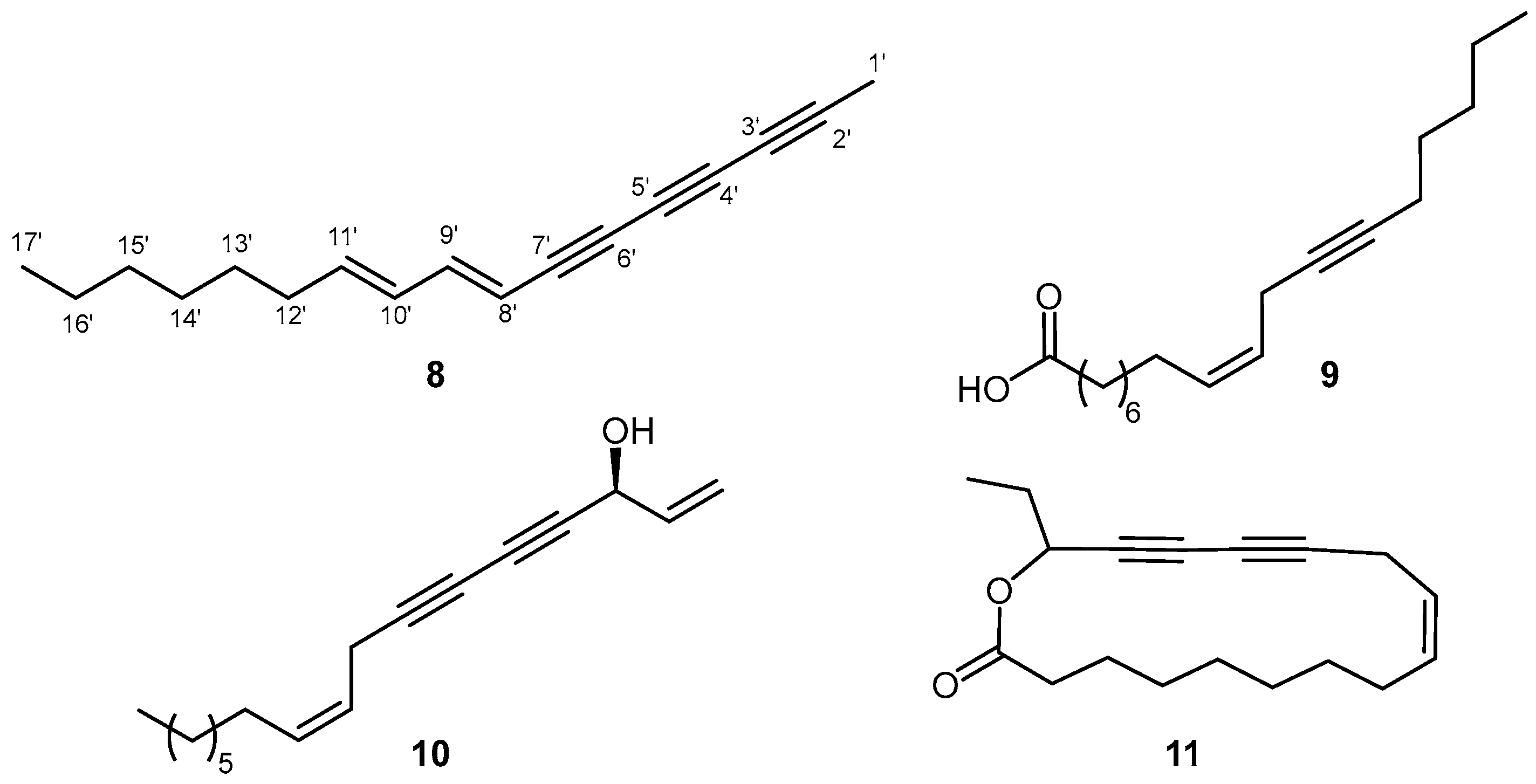
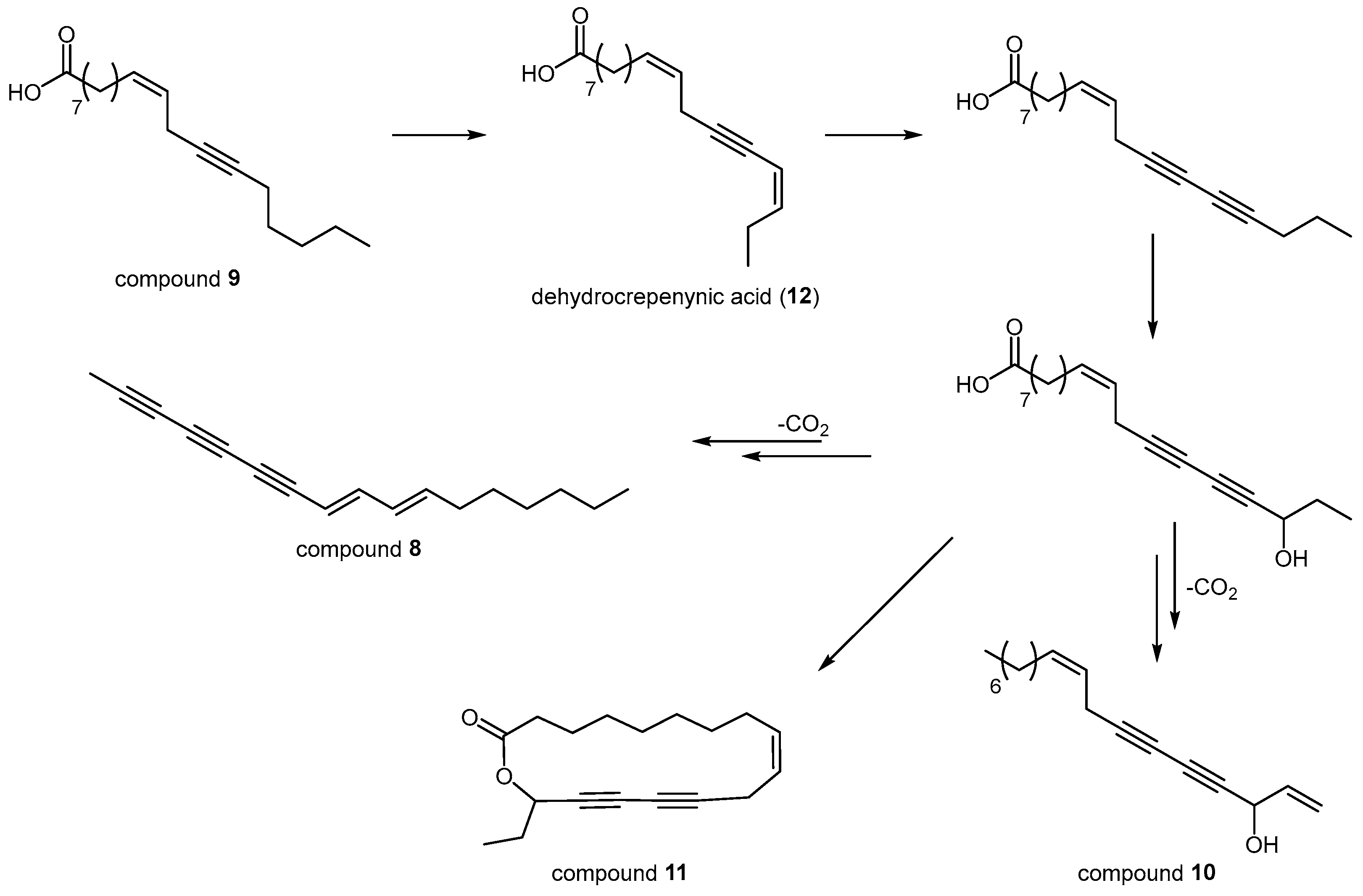
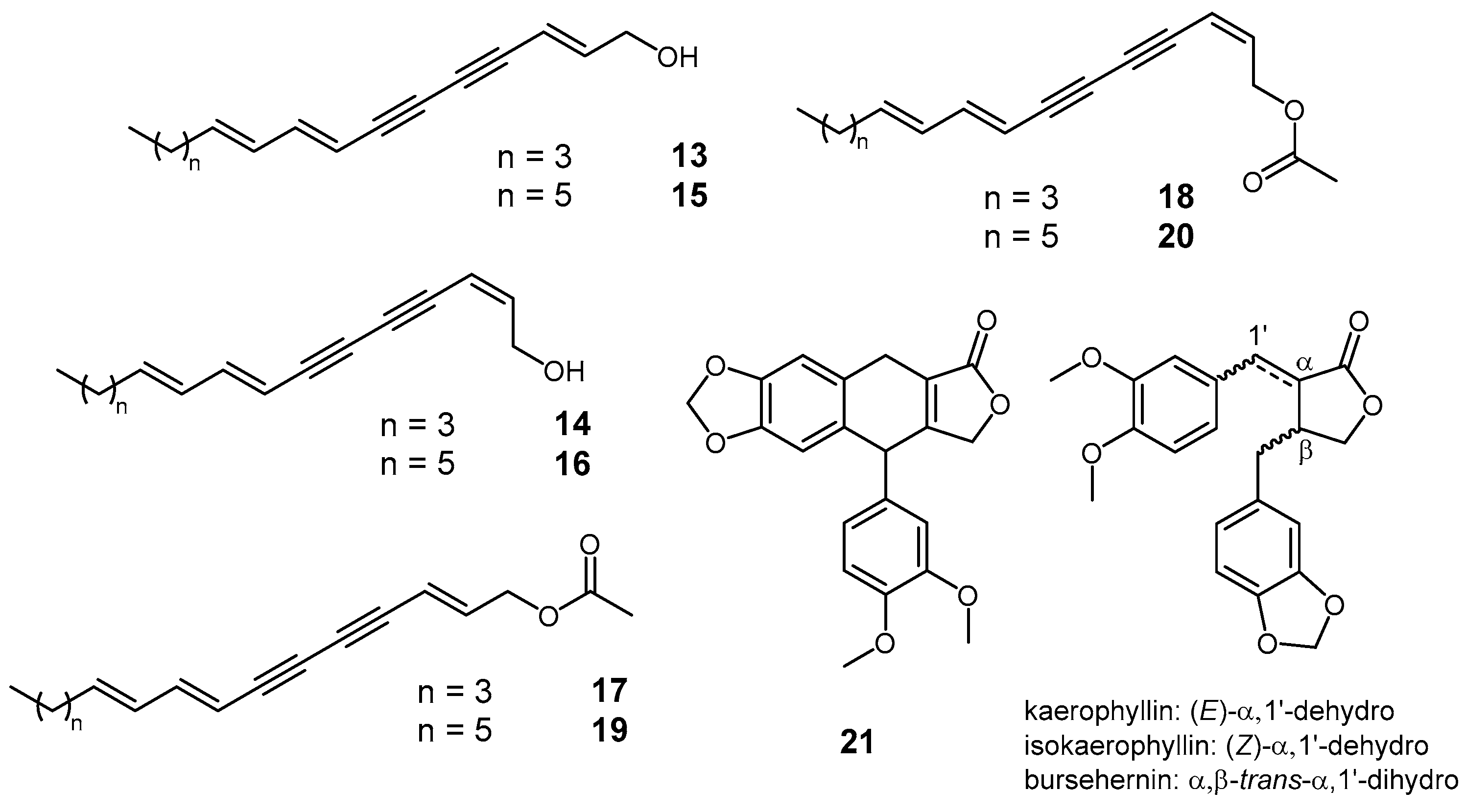
2. Results and Discussion
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Plant Material
3.3. Extraction and Isolation
3.4. Synthesis of Compounds 6 and 17–20
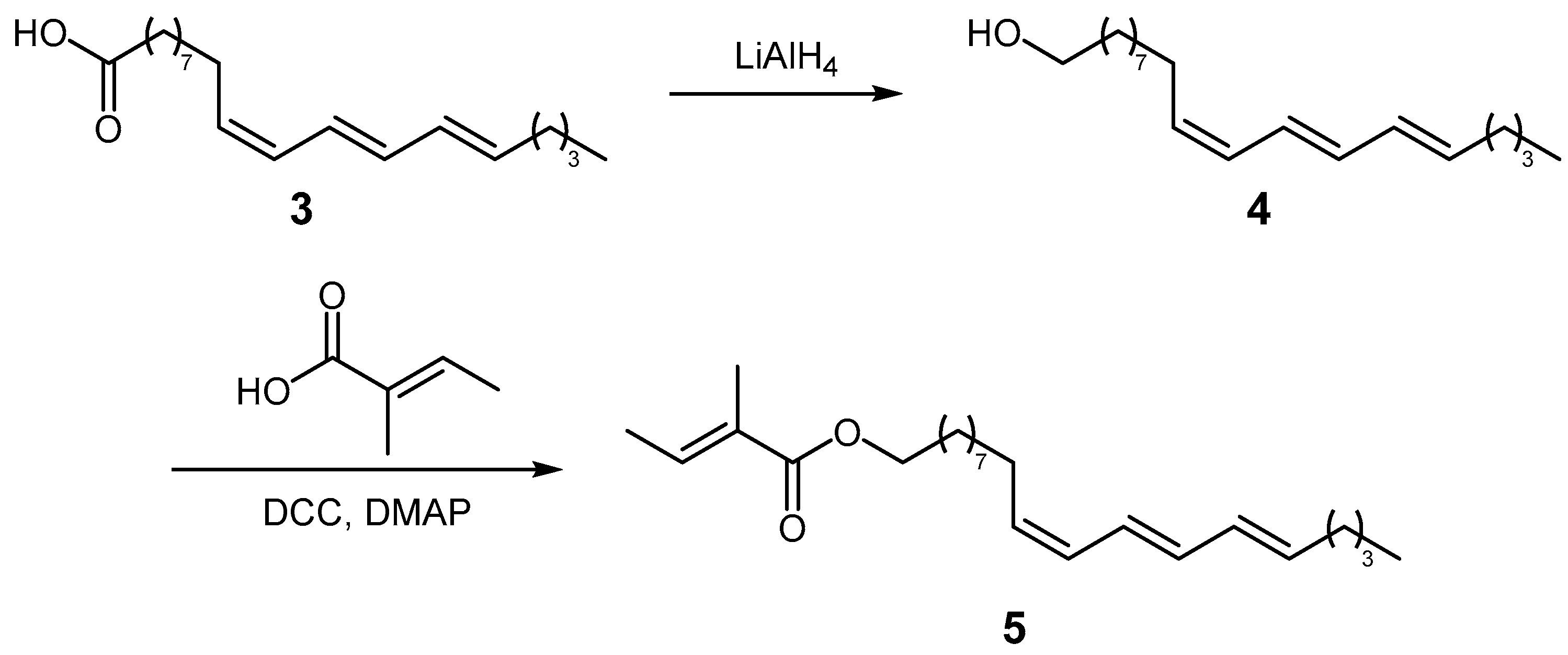
3.5. Reduction of Tung Oil
3.6. Synthesis of Compound 5
3.7. Experimental Spectral Data
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Teng, L.; Guo, X.; Ma, Y.; Xu, L.; Wei, J.; Xiao, P. A comprehensive review on traditional and modern research of the genus Bupleurum (Bupleurum L., Apiaceae) in recent 10 years. J. Ethnopharmacol. 2023, 306, 116129. [Google Scholar] [CrossRef]
- Yuan, B.; Yang, R.; Ma, Y.; Zhou, S.; Zhang, X.; Liu, Y. A systematic review of the active saikosaponins and extracts isolated from Radix Bupleuri and their applications. Pharm. Biol. 2016, 55, 620–635. [Google Scholar] [CrossRef] [PubMed]
- Nikolić, V. Flora SR Srbije V; Josifovic, M., Ed.; Serbian Academy of Sciences and Arts: Belgrade, Serbia, 1973; pp. 199–214. [Google Scholar]
- Micevski, K. The Flora of the Republic of Macedonia 1(6); Macedonian Academy of Sciences and Arts: Skopje, Republic of Macedonia, 2005; pp. 1433–1715. [Google Scholar]
- Tutin, T.G. Bupleurum L. In Flora Europaea; Tutin, T.G., Heywood, W.H., Burges, N.A., Moore, D.M., Valentine, D.H., Walters, S.M., Webb, D.A., Eds.; Cambridge University Press: London, UK, 1968; Volume 2, pp. 345–350. [Google Scholar]
- Yang, F.; Dong, X.; Yin, X.; Wang, W.; You, L.; Ni, J. Radix Bupleuri: A Review of Traditional Uses, Botany, Phytochemistry, Pharmacology, and Toxicology. BioMed Res. Int. 2017, 2017, 7597596. [Google Scholar] [CrossRef]
- Abolfazl, M.; Hossein, N.; Sohrab, I.; Hadi, A. Chemical composition and antifungal activity of essential oils from some medicinal plants of Iran. Int. J. Pharm. Sci. Res. 2014, 5, 1000–1006. [Google Scholar] [CrossRef]
- Abolfazl, M.; Frhad, M.; Ahmadreza, D.; Hossein, N. Chemical composition and antibacterial activity of the essential oils of some medicinal plants. Afr. J. Agric. Res. 2013, 8, 3151–3158. [Google Scholar] [CrossRef]
- Rustaiyan, A.; Masnabadi, N.; Masoudi, S.; Samadizadeh, M.; Firouznia, A.; Larijani, K. Composition of the Essential Oils of Bupleurum falcatum L. and Bupleurum gerardi All. from Iran. J. Essent. Oil Bear. Plants 2010, 6, 727–731. [Google Scholar] [CrossRef]
- Saraçoğlu, H.T.; Akin, M.; Demirci, B.; Baser, K.H.C. Chemical composition and antibacterial activity of essential oils from different parts of endemic Bupleurum L. species. Afr. J. Microbiol. Res. 2012, 6, 2899–2906. [Google Scholar] [CrossRef]
- Mohammadi, A.; Nazari, H.; Imani, S.; Amrollahi, H. Antifungal activities and chemical composition of some medicinal plants. J. Mycol. Med. 2014, 24, e1–e8. [Google Scholar] [CrossRef]
- Pistelli, L.; Cammilli, A.; Manunta, A.; Marsili, A.; Morelli, I. Triterpenoid saponins and flavonoid glycosides from Bupleurum falcatum subsp. Cernuum. Phytochem. 1993, 33, 1537–1539. [Google Scholar] [CrossRef]
- Radulović, N.; Stevanović, M.; Nešić, M.; Stojanović, N.; Ranđelović, P.; Ranđelović, V. Constituents of Bupleurum praealtum and Bupleurum veronense with potential immunomodulatory activity. J. Nat. Prod. 2020, 83, 2902–2914. [Google Scholar] [CrossRef]
- Nešić, M.D.; Nešić, M.S.; Dimitrijević, M.Ž.; Radulović, N.S. Essential Oil Composition of Bupleurum praealtum and Bupleurum affine: New Natural Constituents. Plants 2024, 13, 2076. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Zhang, W.; Su, J. Toxic polyacetylenes in the genus Bupleurum (Apiaceae)—Distribution, toxicity, molecular mechanism and analysis. J. Ethnopharmacol. 2016, 193, 566–573. [Google Scholar] [CrossRef] [PubMed]
- Madbouly, S.A.; Liu, K.; Xia, Y.; Kessler, M.R. Semi-interpenetrating polymer networks prepared from in situ cationic polymerization of bio-based tung oil with biodegradable polycaprolactone. RSC Adv. 2014, 4, 6710–6718. [Google Scholar] [CrossRef]
- Bohlmann, F.; Viehe, H.G. Polyacetylenverbindungen, X. Mitteil.: Synthese der Polyine aus Oenanthe crocata. Chem. Ber. 1955, 88, 1245–1251. [Google Scholar] [CrossRef]
- Bohlmann, F.; Rode, K.M. Polyacetylenverbindungen, 147.: Die Polyine aus Oenanthe crocata L. Chem. Ber. 1968, 101, 1163–1175. [Google Scholar] [CrossRef]
- Sommerwerk, S.; Heller, L.; Siewert, B.; Csuk, R. Chemoenzymatic synthesis and cytotoxicity of oenanthotoxin and analogues. Bioorg. Med. Chem. 2015, 23, 5595–5602. [Google Scholar] [CrossRef]
- Zeisberg, R.; Bohlmann, F. Polyacetylenverbindungen, 229. 13C-NMR-Spektren von Polyinen. Chem. Ber. 1974, 107, 3800–3805. [Google Scholar] [CrossRef]
- Hearn, M.T.W.; Turner, J.L. The carbon-13 nuclear magnetic resonance spectra of the antibiotic polyacetylenic nitrile, diatretyne 2 (7-cyanohept-trans-2-ene-4,6-diynoic acid), and related compounds. J. Chem. Soc. Perkin Trans. 2 1976, 9, 1027–1029. [Google Scholar] [CrossRef]
- Bohlmann, F.; Brehm, M. Carbon-13 nuclear magnetic resonance spectroscopy of conjugated polyynes. Org. Magn. Reson. 1979, 12, 535–536. [Google Scholar] [CrossRef]
- Hansen, L.; Boll, P.M. Polyacetylenes in Araliaceae: Their Chemistry, Biosynthesis and Biological Significance. Phytochemistry 1986, 25, 285–293. [Google Scholar] [CrossRef]
- Dawid, C.; Dunemann, F.; Schwab, W.; Nothnagel, T.; Hofmann, T. Bioactive C17-Polyacetylenes in Carrots (Daucus carota L.): Current Knowledge and Future Perspectives. J. Agric. Food Chem. 2015, 63, 9211–9222. [Google Scholar] [CrossRef]
- Ahmad, T.; Cawood, M.; Iqbal, Q.; Ariño, A.; Batool, A.; Tariq, R.M.S.; Azam, M.; Akhtar, S. Phytochemicals in Daucus carota and Their Health Benefits—Review Article. Foods 2019, 8, 424. [Google Scholar] [CrossRef] [PubMed]
- Bohlmann, F.; Zdero, C. Über inhaltsstoffe der tribus mutisieae. Phytochemistry 1977, 16, 239–242. [Google Scholar] [CrossRef]
- Bohlmann, F.; Zdero, C.; Thefeld, W. Polyacetylenverbindungen, 200. Notiz über die Inhaltsstoffe von Bupleurum-Arten. Chem. Ber. 1971, 104, 2030–2032. [Google Scholar] [CrossRef]
- Huang, H.-Q.; Zhang, X.; Shen, Y.-H.; Su, J.; Liu, X.-H.; Tian, J.-M.; Lin, S.; Shan, L.; Zhang, W.-D. Polyacetylenes from Bupleurum longiradiatum. J. Nat. Prod. 2009, 72, 2153–2157. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.-Q.; Miao, Y.-H.; Gao, X.-X.; Chao, J.-B.; Zhang, X.; Qin, X.-M. Total syntheses of bupleurynol and its analog. Chin. Chem. Lett. 2017, 28, 1035–1038. [Google Scholar] [CrossRef]
- López, H.; Valera, A.; Trujillo, J. Lignans from Bupleurum handiense. J. Nat. Prod. 1996, 59, 493–494. [Google Scholar] [CrossRef]
- Adams, R.P. Identification of Essential Oil Components by Gas Chromatography/Mass Spectrometry, 4th ed.; Allured Publishing: Carol Stream, IL, USA, 2007. [Google Scholar]
- National Institute of Standards and Technology. NIST 17; Mass Spectral Library (NIST/EPA/NIH); National Institute of Standards and Technology: Gaithersburg, MD, USA, 2017. [Google Scholar]
- McDoniel, P.B.; Cole, J.R. Antitumor Activity of Bursera schlechtendalii (Burseraceae): Isolation and Structure Determination of Two New Lignans. J. Pharm. Sci. 1972, 61, 1992–1994. [Google Scholar] [CrossRef]
- Mikaya, G.A.; Turabelidze, D.G.; Kemertelidze, E.P.; Wulfson, N.S. Kaerophyllin, A New Lignan from Chaerophyllum maculatum. Planta Med. 1981, 43, 378–380. [Google Scholar] [CrossRef]
- González, A.G.; Estévez-Reyes, R.; Mato, C.; Estévez-Braun, A.M. Isokaerophyllin, a butyrolactone from Bupleurum salicifolium. Phytochemistry 1990, 29, 675–678. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound 1 | Compound 2 | ||||
|---|---|---|---|---|---|
| Position | δH (m, J 1 (Hz), Integral) | δC (ppm) | Position | δH (m, J (Hz), Integral) | δC (ppm) |
| 1 | / | 168.32 | 1 | / | 168.33 |
| 2 | / | 128.87 | 2 | / | 128.85 |
| 3 | 6.85 (qq, 3J3,4 = 7.1, 4J3,5 = 1.4, 1 H) | 137.07 | 3 | 6.85 (qq, 3J3,4 = 7.0, 4J3,5 = 1.5, 1 H) | 137.30 |
| 4 | 1.78 (d, 3J3,4 = 7.1, 3 H) | 14.45 | 4 | 1.79 (dq, 3J3,4 = 7.0, 5J4,5 = 1.2, 3 H) | 14.39 |
| 5 | 1.83 (d, 4J3,5 = 1.4, 3 H) | 12.17 | 5 | 1.83 (dq, 4J3,5 = 1.5, 5J4,5 = 1.2, 3 H) | 11.78 |
| 1′a | 4.13 (dd, 3J1′a,2′b = 7.7, 3J1′a,2′a = 5.5, 1 H) | 64.39 | 1′a | 4.13 (dd, 3J1′a,2′b = 3J1′a,2′a = 7.0, 1 H) | 64.16 |
| 1′b | 4.13 (dd, 3J1′b,2′a = 7.7, 3J1′b,2′b = 5.5, 1 H) | 1′b | 4.13 (dd, 3J1′b,2′a = 3J1′b,2′b = 7.0, 1 H) | ||
| 2′a | 1.69 (dddd, 3J2′a,3′b = 9.8, 3J1′b,2′a = 7.7, 3J2′a,3′a = 5.6, 3J1′a,2′a = 5.5, 1 H) | 28.44 | 2′a | 1.69 (dddd, 3J2′a,3′b = 9.0, 3J1′b,2′a = 3J1′a,2′a = 7.0, 3J2′a,3′a = 5.0, 1 H) | 26.15 |
| 2′b | 1.69 (dddd, 3J2′b,3′a = 9.8, 3J1′a,2′b = 7.7, 3J2′b,3′b = 5.6, 3J1′b,2′b = 5.5, 1 H) | 2′b | 1.69 (dddd, 3J2′b,3′a = 9.0, 3J1′a,2′b = 3J1′b,2′b = 7.0, 3J2′b,3′b = 5.0, 1 H) | ||
| 3′a | 1.49 (dddd, 3J2′b,3′a = 3J3′a,4′b = 9.8, 3J2′a,3′a = 3J3′a,4′a = 5.6, 1 H) | 26.23 | 3′a | 1.48 (dddd, 3J2′b,3′a = 3J3′a,4′b = 9.0, 3J2′a,3′a = 3J3′a,4′a = 5.0, 1 H) | 26.95 |
| 3′b | 1.49 (dddd, 3J2′a,3′b = 3J3′b,4′a = 9.8, 3J2′b,3′b = 3J3′b,4′b = 5.6, 1 H) | 3′b | 1.48 (dddd, 3J2′a,3′b = 3J3′b,4′a = 9.0, 3J2′b,3′b = 3J3′b,4′b = 5.0, 1 H) | ||
| 4′a | 2.23 (dddd, 3J3′b,4′a = 9.8, 3J4′a,5′ = 7.8, 3J3′a,4′a = 5.6, 4J4′a,6′ = 1.7, 1 H) | 27.61 | 4′a | 2.12 (dddd, 3J3′b,4′a = 9.0, 3J4′a,5′ = 7.3, 3J3′a,4′a = 5.0, 4J4′a,6′ = 1.3, 1 H) | 27.58 |
| 4′b | 2.23 (dddd, 3J3′a,4′b = 9.8, 3J4′b,5′ = 7.8, 3J3′b,4′b = 5.6, 4J4′b,6′ = 1.7, 1 H) | 4′b | 2.12 (dddd, 3J3′a,4′b = 9.0, 3J4′b,5′ = 7.3, 3J3′b,4′b = 5.0, 4J4′b,6′ = 1.3, 1 H) | ||
| 5′ | 5.46 (dddd, 3J5′,6′ = 11.0, 3J4′a,5′ = 3J4′b,5′ = 7.8, 4J5′,7′ = −1.7, 1 H) | 134.76 | 5′ | 5.38 (dddd, 3J5′,6′ = 10.8, 3J4′a,5′ = 3J4′b,5′ = 7.3, 4J5′,7′ = −0.8, 1 H) | 132.03 |
| 6′ | 6.06 (ddddd, 3J5′,6′ = 3J6′,7′ = 11.0, 4J4′a,6′ = 4J4′b,6′ = 1.7, 4J6′,8′ = −0.7, 1 H) | 129.39 | 6′ | 6.02 (ddddd, 3J6′,7′ = 11.4, 3J5′,6′ = 10.8, 4J4′a,6′ = 4J4′b,6′ = 1.3, 4J6′,8′ = −0.7, 1 H) | 129.34 |
| 7′ | 6.50 (ddddd, 3J7′,8′ = 15.2, 3J6′,7′ = 11.0, 4J5′,7′ = −1.7, 4J7′,9′ = −0.8, 5J7′,10′ = −0.6, 1 H) | 128.40 | 7′ | 6.36 (dddd, 3J7′,8′ = 14.8, 3J6′,7′ = 11.4, 4J5′,7′ = −0.8, 4J7′,9′ = −0.7, 1 H) | 125.86 |
| 8′ | 6.27 (dddd, 3J7′,8′ = 15.2, 3J8′,9′ = 12.0, 4J8′,10′ = −0.8, 4J6′,8′ = −0.7, 1 H) | 133.27 | 8′ | 6.17 (dddd, 3J7′,8′ = 14.8, 3J8′,9′ = 10.8, 4J8′,10′ = −0.7, 4J6′,8′ = −0.7, 1 H) | 132.03 |
| 9′ | 6.26 (dddd, 3J9′,10′ = 15.2, 3J8′,9′ = 12.0, 4J7′,9′ = −0.8, 4J9′,11′ = −0.7, 1 H) | 132.83 | 9′ | 6.0980 (ddddd, 3J9′,10′ = 15.0, 3J8′,9′ = 10.8, 4J7′,9′ = −0.7, 4J9′,11′a = 4J9′,11′b = 1.3, 1 H) | 130.62 |
| 10′ | 6.48 (ddddd, 3J9′,10′ = 15.2, 3J10′,11′ = 11.0, 4J10′,12′ = 1.0, 4J8′,10′ = −0.8, 5J7′,10′ = −0.6, 1 H) | 128.04 | 10′ | 5.71 (dddd, 3J9′,10′ = 15.0, 3J10′,11′a = 3J10′,11′b = 6.9, 4J8′,10′ = −0.7, 1 H) | 135.60 |
| 11′ | 6.01 (ddddd, 3J10′,11′ = 3J11′,12′ = 11.0, 4J11′,13′a = 4J11′,13′b = 1.6, 4J9′,11′ = −0.7, 1 H) | 128.25 | 11′a | 2.10 (dddd, 3J10′,11′a = 6.9, 3J11′a,12′a = 3J11′a,12′b = 7.2, 4J9′,11′a = 1.3, 1 H) | 32.64 |
| 11′b | 2.10 (dddd, 3J10′,11′b = 6.9, 3 3J11′b,12′a = 3J11′b,12′b = 7.2, 4J9′,11′b = 1.3, 1 H) | ||||
| 12′ | 5.43 (dddd, 3J11′,12′ = 11.0, 3J12′,13′a = 3J12′,13′b = 7.8, 4J10′,12′ = 1.0, 1 H) | 132.10 | 12′a | 1.29 (dddd, 3J12a′,13′b = 9.0, 3J11′a,12′a = 3J11′b,12′a = 7.2, 3J12a′,13′a = 5.0, 1 H) | 31.58 |
| 12′b | 1.29 (dddd, 3J12b′,13′a = 9.0, 3J11′a,12′b = 3J11′b,12′b = 7.2, 3J12b′,13′b = 5.0, 1 H) | ||||
| 13′a | 2.24 (ddd, 3J12′,13′a = 7.8, 3J13′a,14′ = 7.5, 4J11′,13′a = 1.6, 1 H) | 21.39 | 13′a | 1.37 (ddd, 3J12b′,13′a = 9.0, 3J13′a, 14′ = 7.0, 3J12a′,13′a = 5.0, 1 H) | 22.55 |
| 13′b | 2.24 (ddd, 3J12′,13′b = 7.8, 3J13′b,14′ = 7.5, 4J11′,13′b = 1.6, 1 H) | 13′b | 1.37 (ddd, 3J12a′,13′b = 9.0, 3J13′b,14′ = 7.0, 3J12b′,13′b = 5.0, 1 H) | ||
| 14′ | 1.01 (t, 3J13′a, 14′ = 3J13′b,14′ = 7.5, 3 H) | 14.38 | 14′ | 0.90 (t, 3J13′a, 14′ = 3J13′b,14′ = 7.0, 3 H) | 14.07 |
| Position | δH (m, J 1 (Hz), Integral) | δC (ppm) |
|---|---|---|
| 1 | 1.99 (s, 3 H) | 4.86 |
| 2 | / 2 | 78.55 |
| 3 | / | 65.19 |
| 4 | / | 67.99 |
| 5 | / | 59.50 |
| 6 | / | 76.46 |
| 7 | / | 75.62 |
| 8 | 5.48 (ddd, 3J8,9 = 15.6, 4J8,10 = 0.7, 5J8,11 = 0.6, 1 H) | 106.75 |
| 9 | 6.74 (ddd, 3J8,9 = 15.6, 3J9,10 = 10.9, 4J9,11 = 0.6, 1 H) | 147.02 |
| 10 | 6.10 (ddtd, 3J10,11 = 15.1, 3J9,10 = 10.9, 4J10,12 = 1.3, 4J8′,10′ = 0.7, 1 H) | 129.55 |
| 11 | 5.89 (dtdd, 3J10,11 = 15.1, 3J11,12 = 7.2, 4J9,11 = 5J8′,11′ = 0.6, 1 H) | 141.07 |
| 12 | 2.12 (m, 3J11,12 = 7.2, 4J10,12 = 1.3, 2 H) | 33.06 |
| 13 3 | 1.37 (overlapped multiplets, 2 H) | 28.98 |
| 14 3 | 1.31 (overlapped multiplets, 2 H) | 29.85 |
| 15 3 | 1.25 (overlapped multiplets, 2 H) | 31.79 |
| 16 3 | 1.29 (m, 3J16,17 = 7.2, 2 H) | 22.71 |
| 17 | 0.88 (t, 3J16,17 = 7.2, 3 H) | 14.21 |
| RI 1 | RI 2 | Compound 3 | Relative Abundance 4 | |||
|---|---|---|---|---|---|---|
| BFS | BFK | BFG | BS | |||
| 1100 | 1100 | Undecane 5 | 0.2 | - 6 | 0.3 | - |
| 1424 | 1419 | β-Ylangene | - | - | 0.1 | - |
| 1420 | 1417 | (E)-Caryophyllene 5 | 0.2 | - | - | - |
| 1489 | 1484 | Germacrene D 5 | 4.1 | - | tr 7 | tr |
| 1504 | 1500 | Bicyclogermacrene | 0.2 | - | - | - |
| 1512 | 1505 | β-Bisabolene | - | - | 0.1 | - |
| 1550 | 1542 | (E)-α-Bisabolene | - | - | 0.1 | - |
| 1844 | 1841 | Neophytadiene (Isomer 1) | - | - | 0.2 | - |
| 2116 | / 8 | Compound 14 | 2.2 | 4.5 | - | - |
| 2110 | 2104 | (E)-Phytol 5 | - | - | 0.8 | - |
| 2196 | / | Compound 13 | 10.1 | 17.6 | - | - |
| 2210 | / | Compound 8 | - | - | 5.1 | - |
| 2219 | / | Compound 17 stereoisomer 9 | - | 0.3 | - | - |
| 2229 | / | Praealtaester B | 0.6 | - | - | - |
| 2232 | / | Praealtaester B stereoisomer 1 | 1.5 | 0.1 | - | 0.7 |
| 2236 | / | Praealtaester B stereoisomer 2 | - | 0.3 | - | 0.7 |
| 2240 | / | Praealtaester B stereoisomer 3 | - | 1.0 | - | 4.5 |
| 2245 | / | Compound 18 | 0.3 | - | - | - |
| 2253 | / | Compound 2 | 0.6 | 0.6 | - | 1.4 |
| 2270 | / | Praealtaester B stereoisomer 4 | - | 0.6 | - | 1.6 |
| 2274 | / | Praealtaester B stereoisomer 5 | - | 0.5 | - | 2.6 |
| 2300 | 2300 | Tricosane 5 | - | 0.1 | 0.1 | 0.4 |
| 2310 | / | Compound 17 | 2.2 | 10.5 | - | - |
| 2331 | / | Compound 1 | 10.3 | 1.0 | - | 1.4 |
| 2338 | / | Compound 16 | tr | 0.7 | 1.8 | - |
| 2341 | / | Compound 1 stereoisomer 1 | 3.4 | 4.7 | - | 6.1 |
| 2358 | / | Compound 15 stereoisomer 1 | - | - | - | 4.9 |
| 2371 | / | Compound 1 stereoisomer 2 | 0.3 | 1.2 | - | 1.3 |
| 2378 | / | Compound 1 stereoisomer 3 | tr | 1.9 | - | 2.8 |
| 2416 | / | Compound 15 | 4.6 | 6.3 | 0.8 | - |
| 2432 | 2432 | Docosanal 5 | - | - | 0.1 | - |
| 2434 | / | Compound 15 stereoisomer 2 | - | - | - | 5.2 |
| 2500 | 2500 | Pentacosane 5 | 0.4 | 0.1 | 0.5 | 0.8 |
| 2532 | / | Compound 19 | 0.5 | 4.8 | 0.1 | - |
| 2595 | 2595 | 1-Hexacosene | - | - | 0.1 | - |
| 2600 | 2600 | Hexacosane 5 | - | - | 0.1 | - |
| 2639 | 2632 | Tetracosanal 5 | 0.2 | 0.4 | 0.2 | 0.4 |
| 2700 | 2700 | Heptacosane 5 | 0.9 | 0.2 | 1.5 | 1.1 |
| 2742 | 2738 | Pentacosanal | tr | 0.1 | - | tr |
| 2800 | 2800 | Octacosane 5 | tr | - | - | - |
| 2831 | 2835 | (E,E,E,E)-Squalene 5 | - | - | 0.2 | - |
| 2845 | 2833 | Hexacosanal 5 | 6.0 | 9.6 | 6.0 | 7.1 |
| 2900 | 2900 | Nonacosane 5 | - | 0.3 | 1.1 | 1.0 |
| 2908 | 2906 | 1-Hexacosanol 5 | 9.8 | - | 3.0 | 0.9 |
| 2946 | 2944 | Heptacosanal | 0.3 | 0.1 | 0.3 | - |
| 3049 | 3040 | Octacosanal 5 | 4.1 | 3.9 | 3.3 | 4.0 |
| 3097 | 3090 | 10-Nonacosanone | 15.3 | 8.8 | 0.6 | 40.1 |
| 3100 | 3100 | Hentriacontane 5 | - | 0.1 | 0.4 | - |
| 3111 | 3111 | 1-Octacosanol 5 | 6.7 | 4.8 | - | - |
| 3112 | 3111 | 10-Nonacosanol | - | - | 9.5 | - |
| 3120 | / | Bursehernin 10 | - | - | 21.3 | - |
| 3148 | 3149 | α-Tocopherol 5 | 1.6 | - | 3.6 | 2.8 |
| 3175 | / | Kaerophyllin 11 | - | - | 12.6 | - |
| 3186 | / | Isokaerophyllin 12 | - | - | 2.7 | - |
| 3251 | 3250 | Triacontanal | - | - | 0.4 | 0.8 |
| Total identified: | 86.6 | 85.1 | 77.0 | 92.6 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nešić, M.D.; Nešić, M.S.; Raca, I.L.; Bukleski, M.; Radulović, N.S. Unmasking the Aliphatic Repertoire: New Polyunsaturated Metabolites in Bupleurum falcatum sensu lato Provide Chemotaxonomic Insights. Plants 2025, 14, 1432. https://doi.org/10.3390/plants14101432
Nešić MD, Nešić MS, Raca IL, Bukleski M, Radulović NS. Unmasking the Aliphatic Repertoire: New Polyunsaturated Metabolites in Bupleurum falcatum sensu lato Provide Chemotaxonomic Insights. Plants. 2025; 14(10):1432. https://doi.org/10.3390/plants14101432
Chicago/Turabian StyleNešić, Milica D., Milan S. Nešić, Irena Lj. Raca, Miha Bukleski, and Niko S. Radulović. 2025. "Unmasking the Aliphatic Repertoire: New Polyunsaturated Metabolites in Bupleurum falcatum sensu lato Provide Chemotaxonomic Insights" Plants 14, no. 10: 1432. https://doi.org/10.3390/plants14101432
APA StyleNešić, M. D., Nešić, M. S., Raca, I. L., Bukleski, M., & Radulović, N. S. (2025). Unmasking the Aliphatic Repertoire: New Polyunsaturated Metabolites in Bupleurum falcatum sensu lato Provide Chemotaxonomic Insights. Plants, 14(10), 1432. https://doi.org/10.3390/plants14101432