

Mapping and Validation of Quantitative Trait Loci on Yield-Related Traits Using Bi-Parental Recombinant Inbred Lines and Reciprocal Single-Segment Substitution Lines in Rice (Oryza sativa L.)


Abstract
1. Introduction
2. Results
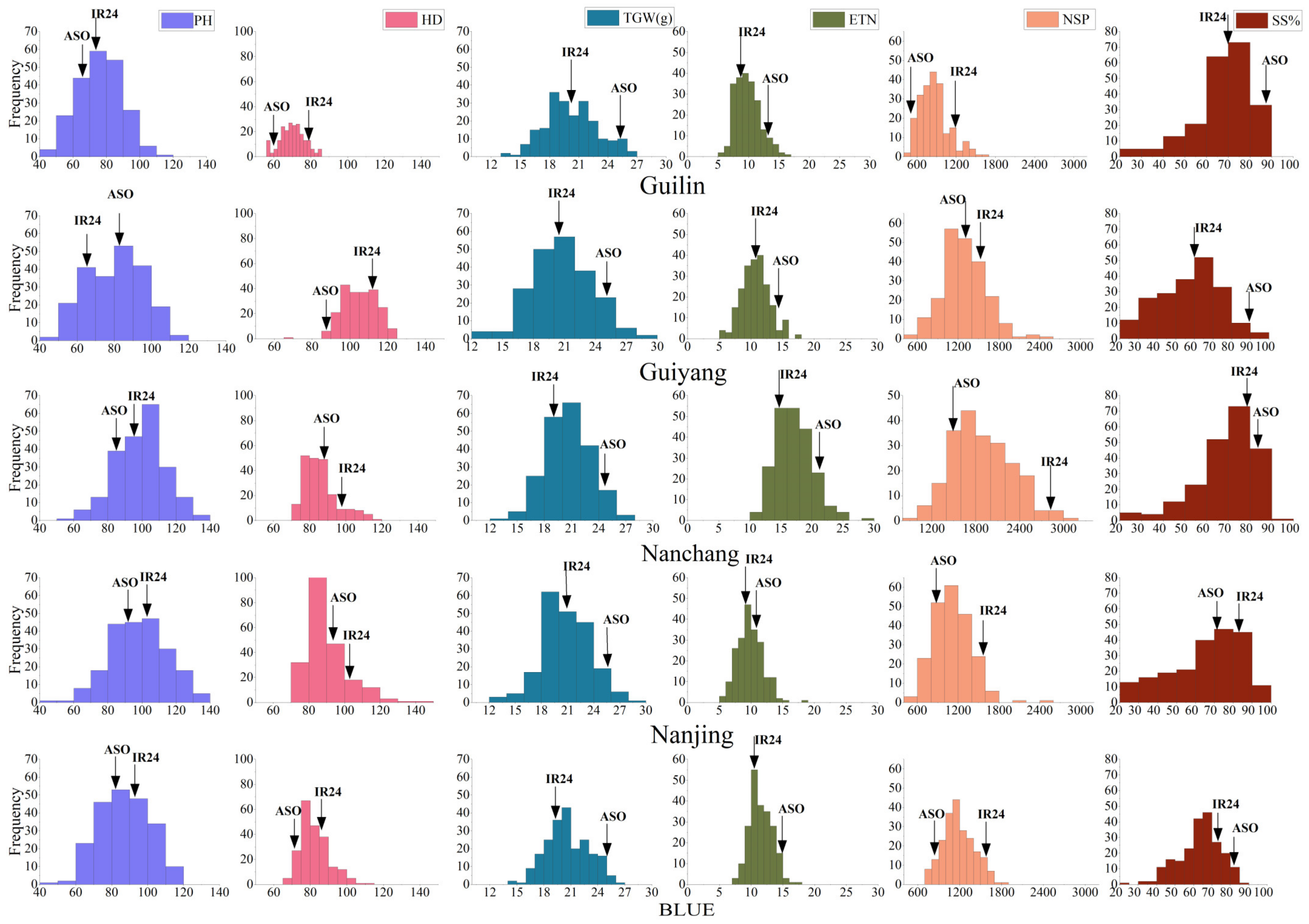
2.1. Phenotypic Distribution, Correlation, and Analysis of Variance (ANOVA)
2.2. QTLs Identified for Six Yield-Related Traits in the RIL Population
2.3. Evaluation of QTL-by-Environment Interactions in the RIL Population
2.4. Validation of the Stable QTLs on Yield-Related Traits by Reciprocal SSSL Populations
2.5. Validation of Two Linked QTLs for PH and HD by Reciprocal SSSLs
3. Discussion
4. Materials and Methods
4.1. Genetic Populations, Genotyping, and Linkage Map
4.2. Field Experiments and Trait Measurement
4.3. Phenotypic Data Analysis and the Estimation of Heritability
4.4. QTL Mapping Methods in the RIL and Reciprocal SSSL Populations
4.5. Physical Positions of SSR Markers and Genomic Visualization of Markers and QTLs
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jiao, Y.; Wang, Y.; Xue, D.; Wang, J.; Yan, M.; Liu, G.; Dong, G.; Zeng, D.; Lu, Z.; Zhu, X.; et al. Regulation of OsSPL14 by OsmiR156 defines ideal plant architecture in rice. Nat. Genet. 2010, 42, 541–545. [Google Scholar] [CrossRef] [PubMed]
- Marathi, B.; Guleria, S.; Mohapatra, T.; Parsad, R.; Mariappan, N.; Kurungara, V.K.; Atwal, S.S.; Prabhu, K.V.; Singh, N.K.; Singh, A.K. QTL analysis of novel genomic regions associated with yield and yield related traits in new plant type based recombinant inbred lines of rice (Oryza sativa L.). BMC Plant Biol. 2012, 12, 137. [Google Scholar] [CrossRef] [PubMed]
- Collard1, B.C.Y.; Jahufer, M.Z.Z.; Brouwer, J.B.; Pang, E.C.K. An introduction to markers, quantitative trait loci (QTL) mapping and marker-assisted selection for crop improvement: The basic concepts. Euphytica 2005, 142, 169–196. [Google Scholar] [CrossRef]
- Wang, J.; Wan, X.; Li, H.; Pfeiffer, W.H.; Crouch, J.; Wan, J. Application of identified QTL-marker associations in rice quality improvement through a design-breeding approach. Theor. Appl. Genet. 2007, 115, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yu, S.; Yan, S.; Wang, H.; Fang, W.; Chen, Y.; Ma, X.; Han, L. Progress in rice breeding based on genomic research. Genes 2024, 15, 564. [Google Scholar] [CrossRef]
- Mackay, T.F.; Stone, E.A.; Ayroles, J.F. The genetics of quantitative traits: Challenges and prospects. Nat. Rev. Genet. 2009, 10, 567. [Google Scholar] [CrossRef]
- Sasaki, A.; Ashikari, M.; Ueguchi-Tanaka, M.; Itoh, H.; Nishimura, A.; Swapan, D.; Ishiyama, K.; Saito, T.; Kobayashi, M.; Khush, G.S. A mutant gibberellin-synthesis gene in rice. Nature 2002, 416, 701–702. [Google Scholar] [CrossRef]
- Lei, L.; Zheng, H.L.; Wang, J.G.; Liu, H.L.; Sun, J.; Zhao, H.W.; Yang, L.M.; Zou, D.T. Genetic dissection of rice (Oryza sativa L.) tiller, plant height, and grain yield based on QTL mapping and metaanalysis. Euphytica 2018, 214, 109. [Google Scholar] [CrossRef]
- Duan, M.; Sun, Z.; Shu, L.; Tan, Y.; Yu, D.; Sun, X.; Liu, R.; Li, Y.; Gong, S.; Yuan, D. Genetic analysis of an elite super-hybrid rice parent using high-density SNP markers. Rice 2013, 6, 21. [Google Scholar] [CrossRef]
- Yang, H.; Yang, Q.; Kang, Y.; Zhang, M.; Zhan, X.; Cao, L.; Cheng, S.; Wu, W.; Zhang, Y. Finding stable QTL for plant height in super hybrid rice. Agriculture 2022, 12, 165. [Google Scholar] [CrossRef]
- Yano, M.; Harushima, Y.; Nagamura, Y.; Kurata, N.; Minobe, Y.; Sasaki, T. Identification of quantitative trait loci controlling heading date in rice using a high-density linkage map. Theor. Appl. Genet. 1997, 95, 1025–1032. [Google Scholar] [CrossRef]
- Zhu, H.; Li, Y.; Liang, J.; Luan, X.; Xu, P.; Wang, S.; Zhang, G.; Liu, G. Analysis of QTLs on heading date based on single segment substitution lines in rice (Oryza Sativa L.). Sci. Rep. 2018, 8, 13232. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Liu, H.; Qi, F.; Zhang, Z.; Li, Q.; Han, Z.; Xing, Y. Genetic interactions among Ghd7, Ghd8, OsPRR37 and Hd1 contribute to large variation in heading date in rice. Rice 2019, 12, 48. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Chen, Z.; Wang, R.; Miao, Y.; Gao, H.; Tang, S.; Zhang, H.; Liu, Q. Characterization and fine-mapping of qHd2-1, a minor quantitative locus that affects heading date under long-day conditions in rice (Oryza sativa L.). Mol. Breed. 2020, 40, 27. [Google Scholar] [CrossRef]
- Sohail, A.; Shah, L.; Liu, L.; Islam, A.; Yang, Z.; Yang, Q.; Anis, G.B.; Xu, P.; Khan, R.M.; Li, J.; et al. Mapping and validation of qHD7b: Major heading-date QTL functions mainly under long-day conditions. Plants 2022, 11, 2288. [Google Scholar] [CrossRef]
- Li, S.; Luo, Y.; Wei, G.; Zong, W.; Zeng, W.; Xiao, D.; Zhang, H.; Song, Y.; Hao, Y.; Sun, K. Improving yield-related traits by editing the promoter of the heading date gene Ehd1 in rice. Theor. Appl. Genet. 2023, 136, 239. [Google Scholar] [CrossRef]
- Yan, W.; Liu, H.; Zhou, X.; Li, Q.; Zhang, J.; Lu, L.; Liu, T.; Liu, H.; Zhang, C.; Shen, G.; et al. Natural variation in Ghd7.1 plays an important role in grain yield and adaptation in rice. Cell Res. 2013, 23, 969–971. [Google Scholar] [CrossRef]
- Zhang, H.; Zhu, Y.-J.; Zhu, A.-D.; Fan, Y.-Y.; Huang, T.-X.; Zhang, J.-F.; Xie, H.-A.; Zhuang, J.-Y. Fine-mapping of qTGW2, a quantitative trait locus for grain weight in rice (Oryza sativa L.). PeerJ 2020, 8, e8679. [Google Scholar] [CrossRef]
- Fan, C.; Xing, Y.; Mao, H.; Lu, T.; Han, B.; Xu, C.; Li, X.; Zhang, Q. GS3, a major QTL for grain length and weight and minor QTL for grain width and thickness in rice, encodes a putative transmembrane protein. Theor. Appl. Genet. 2006, 112, 1164–1171. [Google Scholar] [CrossRef]
- Song, X.-J.; Huang, W.; Shi, M.; Zhu, M.-Z.; Lin, H.-X. A QTL for rice grain width and weight encodes a previously unknown RING-type E3 ubiquitin ligase. Nat. Genet. 2007, 39, 6. [Google Scholar] [CrossRef]
- Hui, W.; Zhang, J.; Farkhanda, N.; Li, J.; Sun, S.; He, H.; Zhang, T.; Ling, Y.; Zhao, F. Identification of rice QTLs for important agronomic traits with long-kernel CSSL-Z741 and three SSSLs. Rice Sci. 2020, 27, 414–422. [Google Scholar] [CrossRef]
- Du, Z.; Huang, Z.; Li, J.; Bao, J.; Tu, H.; Zeng, C.; Wu, Z.; Fu, H.; Xu, J.; Zhou, D.; et al. qTGW12a, a naturally varying QTL, regulates grain weight in rice. Theor. Appl. Genet. 2021, 134, 2767–2776. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Qian, Q.; Fu, Z.; Wang, Y.; Xiong, G.; Zeng, D.; Wang, X.; Liu, X.; Teng, S.; Hiroshi, F. Control of tillering in rice. Nature 2003, 422, 618–621. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, K.; Maekawa, M.; Ujiie, S.; Satake, Y.; Furutani, I.; Okamoto, H.; Shimamoto, K.; Kyozuka, J. LAX and SPA: Major regulators of shoot branching in rice. Proc. Natl. Acad. Sci. USA 2003, 100, 11765–11770. [Google Scholar] [CrossRef]
- Ren, M.; Huang, M.; Qiu, H.; Chun, Y.; Li, L.; Kumar, A.; Fang, J.; Zhao, J.; He, H.; Li, X. Genome-wide association study of the genetic basis of effective tiller number in rice. Rice 2021, 14, 56. [Google Scholar] [CrossRef]
- Chen, H.; Fan, Y.; Li, D.; Sun, Z.; Ruan, Z.; Yang, H.; Kang, Y.; Wu, W.; Liu, Q.; Zhan, X.; et al. Genetic dissection of tiller number qTN4 in rice. Agriculture 2023, 13, 411. [Google Scholar] [CrossRef]
- Huang, X.; Qian, Q.; Liu, Z.; Sun, H.; He, S.; Luo, D.; Xia, G.; Chu, C.; Li, J.; Fu, X. Natural variation at the DEP1 locus enhances grain yield in rice. Nat. Genet. 2009, 41, 4. [Google Scholar] [CrossRef]
- Li, P.; Li, Q.; Lu, X.; Dai, L.; Yang, L.; Hong, Y.; Yan, T.; Shen, L.; Zhang, Q.; Ren, D.; et al. Genome-wide association study reveals novel QTLs and candidate genes for grain number in rice. Int. J. Mol. Sci. 2022, 23, 13617. [Google Scholar] [CrossRef]
- Lu, Y.; Chuan, M.; Wang, H.; Chen, R.; Tao, T.; Zhou, Y.; Xu, Y.; Li, P.; Yao, Y.; Xu, C.; et al. Genetic and molecular factors in determining grain number per panicle of rice. Front. Plant Sci. 2022, 13, 964246. [Google Scholar] [CrossRef]
- Wu, F.; Luo, X.; Wang, L.; Wei, Y.; Li, J.; Xie, H.; Zhang, J.; Xie, G. Genome-wide association study reveals the QTLs for seed storability in world rice core collections. Plants 2021, 10, 812. [Google Scholar] [CrossRef]
- Lee, C.-M.; Suh, J.-P.; Park, H.-S.; Baek, M.-K.; Jeong, O.-Y.; Yun, S.-J.; Cho, Y.-C.; Kim, S.-M. Identification of QTL combinations that cause spikelet sterility in rice derived from interspecific crosses. Rice 2021, 14, 99. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, H.; Zhang, L. Linkage Analysis and Gene Mapping; Science Press: Beijing, China, 2023. [Google Scholar]
- Xiao, J.; Li, J.; Yuan, L.; Tanksley, S.D. Dominance is the major genetic basis of heterosis in rice as revealed by QTL analysis using molecular markers. Genetics 1995, 140, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Hua, J.-P.; Xing, Y.-Z.; Xu, C.-G.; Sun, X.-L.; Yu, S.-B.; Zhang, Q. Genetic dissection of an elite rice hybrid revealed that heterozygotes are not always advantageous for performance. Genetics 2002, 162, 1885–1895. [Google Scholar] [CrossRef]
- Meng, L.; Zhao, X.; Ponce, K.; Ye, G.; Leung, H. QTL mapping for agronomic traits using multi-parent advanced generation inter-cross (MAGIC) populations derived from diverse elite indica rice lines. Field Crop. Res. 2016, 189, 19–42. [Google Scholar] [CrossRef]
- Qu, P.; Shi, J.; Chen, T.; Chen, K.; Shen, C.; Wang, J.; Zhao, X.; Ye, G.; Xu, J.; Zhang, L. Construction and integration of genetic linkage maps from three multi-parent advanced generation inter-cross populations in rice. Rice 2020, 13, 13. [Google Scholar] [CrossRef]
- Kubo, T.; Aida, Y.; Nakamura, K.; Tsunematsu, H.; Doi, K.; Yoshimura, A. Reciprocal chromosome segment substitution series derived from japonica and indica cross of rice (Oryza sativa L.). Breed Sci. 2002, 52, 319–325. [Google Scholar] [CrossRef]
- Zhang, B.; Shang, L.; Ruan, B.; Zhang, A.; Yang, S.; Jiang, H.; Liu, C.; Hong, K.; Lin, H.; Gao, Z. Development of three sets of high-throughput genotyped rice chromosome segment substitution lines and QTL mapping for eleven traits. Rice 2019, 12, 33. [Google Scholar] [CrossRef]
- Zhu, W.; Lin, J.; Yang, D.; Zhao, L.; Zhang, Y.; Zhu, Z.; Chen, T.; Wang, C. Development of chromosome segment substitution lines derived from backcross between two sequenced rice cultivars, indica recipient 93-11 and japonica donor Nipponbare. Plant Mol. Biol. Rep. 2009, 27, 126–131. [Google Scholar] [CrossRef]
- Zhang, B.; Ma, L.; Wu, B.; Xing, Y.; Qiu, X. Introgression lines: Valuable resources for functional genomics research and breeding in rice (Oryza sativa L.). Front. Plant Sci. 2022, 13, 863789. [Google Scholar] [CrossRef]
- Takahashi, Y.; Shomura, A.; Sasaki, T.; Yano, M. Hd6, a rice quantitative trait locus involved in photoperiod sensitivity, encodes the α subunit of protein kinase CK2. Proc. Natl. Acad. Sci. USA 2001, 98, 7922–7927. [Google Scholar] [CrossRef]
- Li, H.; Ye, G.; Wang, J. A modified algorithm for the improvement of composite interval mapping. Genetics 2007, 175, 361–374. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Meyer, E.; McKay, J.K.; Matz, M.V. 2b-RAD: A simple and flexible method for genome-wide genotyping. Nat. Methods 2012, 9, 808–810. [Google Scholar] [CrossRef] [PubMed]
- Yin, C.; Li, H.; Li, S.; Xu, L.; Zhao, Z.; Wang, J. Genetic dissection on rice grain shape by the two-dimensional image analysis in one japonica× indica population consisting of recombinant inbred lines. Theor. Appl. Genet. 2015, 128, 1969–1986. [Google Scholar] [CrossRef] [PubMed]
- Yin, C.; Li, H.; Zhao, Z.; Wang, Z.; Liu, S.; Chen, L.; Liu, X.; Tian, Y.; Ma, J.; Xu, L. Genetic dissection of top three leaf traits in rice using progenies from a japonica× indica cross. J. Integr. Plant Biol. 2017, 59, 866–880. [Google Scholar] [CrossRef]
- Meng, L.; Li, H.; Zhang, L.; Wang, J. QTL IciMapping: Integrated software for genetic linkage map construction and quantitative trait locus mapping in biparental populations. Crop J. 2015, 121, 269–283. [Google Scholar] [CrossRef]
- Li, S.; Wang, J.; Zhang, L. Inclusive composite interval mapping of QTL by environment interactions in biparental populations. PLoS ONE 2015, 10, e0132414. [Google Scholar] [CrossRef]
- Youens-Clark, K.; Buckler, E.; Casstevens, T.; Chen, C.; DeClerck, G.; Derwent, P.; Dharmawardhana, P.; Jaiswal, P.; Kersey, P.; Karthikeyan, A.S.; et al. Gramene database in 2010: Updates and extensions. Nucleic Acids Res. 2010, 39, D1085–D1094. [Google Scholar] [CrossRef]
- Wang, Y.; Jia, L.; Tian, G.; Dong, Y.; Zhang, X.; Zhou, Z.; Luo, X.; Li, Y.; Yao, W. ShinyCircos-V2.0: Leveraging the creation of circos plot with enhanced usability and advanced features. iMeta 2023, 2, e109. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Population | Trait | Trait | ||||
|---|---|---|---|---|---|---|
| PH | HD | NSP | SS | TGW | ||
| RIL | HD | 0.0269 ns | ||||
| NSP | 0.1253 ns | −0.0786 ns | ||||
| SS | 0.2108 ** | −0.0278 ns | 0.0381 ns | |||
| TGW | 0.2404 *** | −0.1788 ** | −0.2968 *** | 0.2519 *** | ||
| ETN | −0.2524 *** | −0.2821 *** | 0.1355 * | 0.0088 ns | −0.1234 ns | |
| AIS | HD | 0.5006 *** | ||||
| NSP | 0.3245 ** | 0.2382 ns | ||||
| SS | 0.1209 ns | 0.0846 ns | −0.0422 ns | |||
| TGW | 0.0080 ns | −0.1884 ns | −0.3176 ** | 0.5028 *** | ||
| ETN | −0.3906 ** | −0.2123 ns | 0.4161 *** | −0.1006 ns | −0.2667 * | |
| IAS | HD | 0.3301 * | ||||
| NSP | −0.1597 ns | −0.1537 ns | ||||
| SS | 0.1907 ns | −0.1937 ns | −0.1716 ns | |||
| TGW | −0.1635 ns | −0.1528 ns | −0.5439 *** | 0.0444 ns | ||
| ETN | −0.4406 *** | −0.1418 ns | 0.4891 *** | 0.2823 * | −0.1543 ns | |
| Population | Trait | Variance Components | Heritability | ||||
|---|---|---|---|---|---|---|---|
| Genotype | Environment | GE Interaction | Random Error | Plot Level | Genotypic Mean Level | ||
| RIL | PH | 189.17 *** | 140.66 *** | 35.57 *** | 21.21 | 0.77 | 0.94 |
| HD | 62.45 *** | 206.86 *** | 26.02 *** | 6.07 | 0.66 | 0.90 | |
| TGW | 5.39 *** | 0.09 *** | 1.78 *** | 0.95 | 0.66 | 0.90 | |
| ETN | 2.60 *** | 12.22 *** | 1.01 *** | 4.04 | 0.34 | 0.78 | |
| NSP | 48,804.04 *** | 192,351.84 *** | 23,251.20 *** | 60,185.55 | 0.37 | 0.78 | |
| SS | 114.45 *** | 41.54 *** | 6.05 *** | 197.71 | 0.31 | 0.67 | |
| AIS | PH | 28.28 *** | 97.99 *** | 7.78 ns | 16.16 | 0.54 | 0.88 |
| HD | 4.03 *** | 196.51 *** | 4.08 *** | 1.77 | 0.41 | 0.76 | |
| TGW | 1.30 *** | 0.24 *** | 0.22 ns | 0.54 | 0.63 | 0.91 | |
| ETN | 0.60 ns | 16.54 *** | 0.00 ns | 10.01 | 0.06 | 0.32 | |
| NSP | 2617.29 ns | 126,986.19 *** | 6615.26 ns | 48,633.88 | 0.05 | 0.25 | |
| SS | 7.63 *** | 19.05 *** | 19.37 *** | 26.19 | 0.14 | 0.48 | |
| IAS | PH | 59.96 *** | 265.84 *** | 4.06 ns | 11.75 | 0.79 | 0.96 |
| HD | 6.28 *** | 213.46 *** | 1.79 *** | 1.57 | 0.65 | 0.91 | |
| TGW | 1.35 *** | 0.52 *** | 0.42 *** | 0.65 | 0.56 | 0.88 | |
| ETN | 0.60 ns | 14.31 *** | 0.42 ns | 2.76 | 0.16 | 0.57 | |
| NSP | 10,910.70 ns | 378,504.78 *** | 378,504.78 ns | 79,878.49 | 0.11 | 0.46 | |
| SS | 11.78 *** | 130.52 *** | 16.94 *** | 17.17 | 0.26 | 0.65 | |
| QTL Name a | Chr. | Environment | Genetic Position (cM) b | Left Marker c1 | Right Marker c2 | Physical Position (bp) d | e LOD | f PVE (%) | g ADD |
|---|---|---|---|---|---|---|---|---|---|
| qPH1 | 1 | GY, NC | 97~97 | RM1003 | RM486 | 33477070~34956587 | 2.51~3.77 | 2.66~4.3 | −4.13~−3.15 |
| qPH2 | 2 | NJ, GL, GY, BL | 129~134 | RM6 | RM425 | 29585867~32303944 | 3.10~8.52 | 10.66~17.4 | 8.37~9.76 |
| qPH3-1 | 3 | NC, NJ, BL | 54~57 | RM251 | RM5748 | 9949856~12327987 | 3.53~4.57 | 3.73~7.21 | 4.46~6.15 |
| qPH3-2 | 3 | NC, NJ, BL | 110~114 | RM411 | RM7097 | 21430854~26881168 | 2.67~5.02 | 4.06~10.87 | 4.71~4.78 |
| qHD3-1 | 3 | GL, GY, NC, NJ, BL | 0~2 | RM523 | RM5474 | 1320598~3804270 | 3.18~16.4 | 3.55~15.47 | 1.68~3.9 |
| qHD3-2 | 3 | GL, GY, NC, NJ, BL | 163~166 | RM8269 | RM448 | 31339287~31399595 | 4.79~8.80 | 4.68~11.56 | 2.22~3.10 |
| qHD5 | 5 | GL, GY | 1~16 | RM13 | RM413 | 2011198~2212829 | 3.02~3.72 | 2.92~3.99 | 1.31~1.70 |
| qHD6 | 6 | NC, NJ, GY, GL, BL | 48~55 | RM557 | RM136 | 8552551~8752551 | 3.73~6.28 | 6.82~11.61 | −1.98~−4.93 |
| qHD8 | 8 | NC, NJ, GY, GL, BL | 31~37 | RM6863 | RM72 | 2012432~2212432 | 7.06~15.3 | 9.21~20.00 | 6.70~−1.97 |
| qHD12 | 12 | GL, GY, NC, BL | 27~32 | RM6296 | RM7102 | 3201718~12952364 | 2.63~5.33 | 5.18~7.08 | 1.54~2.78 |
| qTGW2-1 | 2 | GL, BL | 22~26 | RM211 | RM71 | 8560434~8760434 | 2.50~3.08 | 7.63~8.22 | −0.97~−0.95 |
| qTGW2-2 | 2 | GL, GY | 133~136 | RM6 | RM425 | 29585849~32303935 | 4.17~7.20 | 5.20~7.20 | −0.87~−0.75 |
| qTGW3-1 | 3 | NC, GY, BL | 112~117 | RM411 | RM7097 | 21430825~26881139 | 3.98~6.02 | 4.83~9.86 | 0.77~0.97 |
| qTGW3-2 | 3 | GL, NC, BL | 183~187 | RM448 | RM570 | 31399595~35595760 | 4.35~5.44 | 8.97~10.72 | −1.09~−0.92 |
| qTGW5 | 5 | GL, GY, NC, BL | 15~16 | RM405 | RM430 | 3073440~18753934 | 5.91~6.53 | 5.47~6.63 | -096~−0.79 |
| qETN11 | 11 | NJ, BL | 16~19 | RM4 | RM6288 | 932168~2166293 | 3.36~3.53 | 3.10~3.62 | −0.57~−0.46 |
| qNSP6 | 6 | NC, NJ, GL, BL | 61~79 | RM6818 | RM541 | 16582523~19514548 | 3.73~6.28 | 6.82~11.61 | 65.62~144.85 |
| qSS1 | 1 | GY, BL | 137~150 | RM14 | RM5410 | 35477070~41094758 | 3.17~4.10 | 9.03~7.22 | 4.11~−8.26 |
| QTL | Position | Left Marker | Right Marker | a1 LOD | a2 LOD(AbyE) | b1 PVE | b2 PVE(AbyE) | c Add | d1 LeftCI | d2 RightCI |
|---|---|---|---|---|---|---|---|---|---|---|
| qPH1 | 97 | RM1003 | RM486 | 10.56 | 0.55 | 3.07 | 0.18 | −2.84 | 96.5 | 97.5 |
| qPH2 | 138 | RM6 | RM425 | 3.26 | 3.10 | 0.73 | 0.66 | 0.44 | 132.5 | 151.5 |
| qPH3-1 | 55 | RM251 | RM5748 | 17.33 | 0.21 | 5.70 | 0.17 | 4.34 | 52.5 | 58.5 |
| qPH3-2 | 111 | RM411 | RM7097 | 18.42 | 0.99 | 5.81 | 0.06 | 3.99 | 105.5 | 115.5 |
| qHD3-1 | 1 | RM523 | RM5474 | 36.84 | 6.47 | 7.99 | 0.50 | 2.60 | 0 | 2.5 |
| qHD3-2 | 164 | RM8269 | RM448 | 36.64 | 4.53 | 7.92 | 0.08 | 2.64 | 163.5 | 164.5 |
| qHD5 | 1 | RM13 | RM413 | 5.16 | 1.79 | 0.87 | 0.1 | 0.83 | 0 | 4.5 |
| qHD6 | 49 | RM557 | RM136 | 31.36 | 13.79 | 7.76 | 3.96 | −1.95 | 47.5 | 50.5 |
| qHD8 | 35 | RM6863 | RM72 | 56.01 | 2.47 | 15.38 | 1.43 | −3.51 | 32.5 | 36.5 |
| qHD12 | 29 | RM6296 | RM7102 | 15.70 | 1.09 | 3.66 | 0.13 | 1.76 | 21.5 | 35.5 |
| qTGW2-1 | 34 | RM211 | RM71 | 7.28 | 0.72 | 2.61 | 0.06 | −0.41 | 21.5 | 43.5 |
| qTGW2-2 | 133 | RM6 | RM425 | 9.87 | 2.74 | 3.43 | 0.87 | −0.41 | 126.5 | 137.5 |
| qTGW3-1 | 113 | RM411 | RM7097 | 15.42 | 1.32 | 5.76 | 0.26 | 0.60 | 107.5 | 117.5 |
| qTGW3-2 | 187 | RM448 | RM570 | 15.05 | 1.39 | 5.60 | 0.07 | −0.61 | 180.5 | 192.5 |
| qTGW5 | 16 | RM405 | RM430 | 26.18 | 2.37 | 9.17 | 0.32 | −0.77 | 14.5 | 18.5 |
| qETN11 | 1 | RM286 | RM4 | 7.38 | 1.56 | 2.47 | 0.45 | −0.33 | 0 | 5.5 |
| qNSP6 | 61 | RM6818 | RM541 | 17.23 | 3.69 | 7.78 | 2.12 | 67.1 | 60.5 | 65.5 |
| qSS1 | 150 | RM14 | RM5410 | 3.31 | 2.12 | 2.73 | 1.93 | −1.14 | 139.5 | 151 |
| QTL | Environment | Marker Name | Chr. | Pos. (cM) | LOD a | PVE (%) b | ADD c | SSSL Population | Previously Published QTLs/Genes |
|---|---|---|---|---|---|---|---|---|---|
| qPH3-1 | NC, NJ | RM251 | 3 | 51.33 | 2.69~3.24 | 14.54~17.01 | 4.89~5.35 | AIS | qPH3.1, qPH3b [9,10] |
| qHD3-1 | GY, NC, NJ, BL | RM523 | 3 | 0 | 6.07~13.50 | 9.66~30.10 | 2.91~4.87 | AIS, IAS | qHd3-1 [12] |
| qHD6 | NC, NJ, BL | RM557 | 6 | 42.23 | 3.47~16.20 | 7.34~38.45 | 5.10~1.35 | AIS | qHD6a, qHD6b, HD6 [15,41] |
| qHD8 | NC, NJ, BL | RM72 | 8 | 37.45 | 8.21~11.38 | 17.68~22.23 | −6.26~3.58 | AIS | qHD8a [15] |
| qTGW2-2 | GY, NC | RM425 | 2 | 143.95 | 2.99~2.64 | 19.40~15.30 | 1.97~−1.73 | AIS | qTGW2 [18] |
| qETN11 | GY, BL | RM6288 | 11 | 19.35 | 2.83~3.03 | 20.44~21.72 | −2.02~−1.23 | IAS | qTN11-1 [8] |
| qNSP6 | GL, GY | RM6818 | 6 | 59.92 | 3.92~3.83 | 24.59~20.33 | 4.66~192.07 | AIS | GN1a [27] |
| qSS1 | GY, BL | RM5410 | 1 | 151.42 | 4.00~2.70 | 21.48~13.20 | −9.89~−4.66 | AIS, IAS | qSS1.1 [30] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manzoor, G.A.; Yin, C.; Zhang, L.; Wang, J. Mapping and Validation of Quantitative Trait Loci on Yield-Related Traits Using Bi-Parental Recombinant Inbred Lines and Reciprocal Single-Segment Substitution Lines in Rice (Oryza sativa L.). Plants 2025, 14, 43. https://doi.org/10.3390/plants14010043
Manzoor GA, Yin C, Zhang L, Wang J. Mapping and Validation of Quantitative Trait Loci on Yield-Related Traits Using Bi-Parental Recombinant Inbred Lines and Reciprocal Single-Segment Substitution Lines in Rice (Oryza sativa L.). Plants. 2025; 14(1):43. https://doi.org/10.3390/plants14010043
Chicago/Turabian StyleManzoor, Ghulam Ali, Changbin Yin, Luyan Zhang, and Jiankang Wang. 2025. "Mapping and Validation of Quantitative Trait Loci on Yield-Related Traits Using Bi-Parental Recombinant Inbred Lines and Reciprocal Single-Segment Substitution Lines in Rice (Oryza sativa L.)" Plants 14, no. 1: 43. https://doi.org/10.3390/plants14010043
APA StyleManzoor, G. A., Yin, C., Zhang, L., & Wang, J. (2025). Mapping and Validation of Quantitative Trait Loci on Yield-Related Traits Using Bi-Parental Recombinant Inbred Lines and Reciprocal Single-Segment Substitution Lines in Rice (Oryza sativa L.). Plants, 14(1), 43. https://doi.org/10.3390/plants14010043