
Identification of PgRg1-3 Gene for Ginsenoside Rg1 Biosynthesis as Revealed by Combining Genome-Wide Association Study and Gene Co-Expression Network Analysis of Jilin Ginseng Core Collection


 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Databases
2.2. Identification of Candidate Genes for Ginsenoside Rg1 Biosynthesis
2.2.1. Genome-Wide Association Study (GWAS)
Statistical Analysis of Ginsenoside Rg1 Content in Core Germplasm Resource Population
Genome-Wide Association Study Using Single-Locus Models
Multi-Locus Model Genome-Wide Association Study
Determination of Genes within Significantly Associated Intervals in Genome-Wide Association Study
2.2.2. Weighted Gene Co-Expression Network Analysis (WGCNA)
2.2.3. Study on the Effect of Gene Mutations on Rg1 Content through SNP-Rg1 Content Association Analysis
2.2.4. Gene Co-Expression Network Analysis
2.3. Functional Validation of Key Candidate Genes for Rg1
2.3.1. Validation of MeJA-Induced Regulation
2.3.2. Validation with RNA Interference (RNAi)
2.4. Exploration of the Molecular Mechanism of the PgRg1-3 Gene in Rg1 Biosynthesis
2.4.1. Functional Annotation of the PgRg1-3 Gene by GO and KEGG Pathway Localization, and Homology Analysis with Ginseng CYP Genes
2.4.2. Gene Expression Patterns at Different Times and in Different Cultivars
2.4.3. The Impacts of Gene Co-Expression Network Variations on Rg1 Biosynthesis
2.4.4. Genetic Effects of SNP Mutations in the PgRg1-3 Gene on Rg1 Biosynthesis
3. Results
3.1. Identification of Candidate Genes and Key Candidate Genes for Ginsenoside Rg1 Biosynthesis
3.1.1. Genome-Wide Association Study
Statistical Analysis of Ginsenoside Rg1 Content
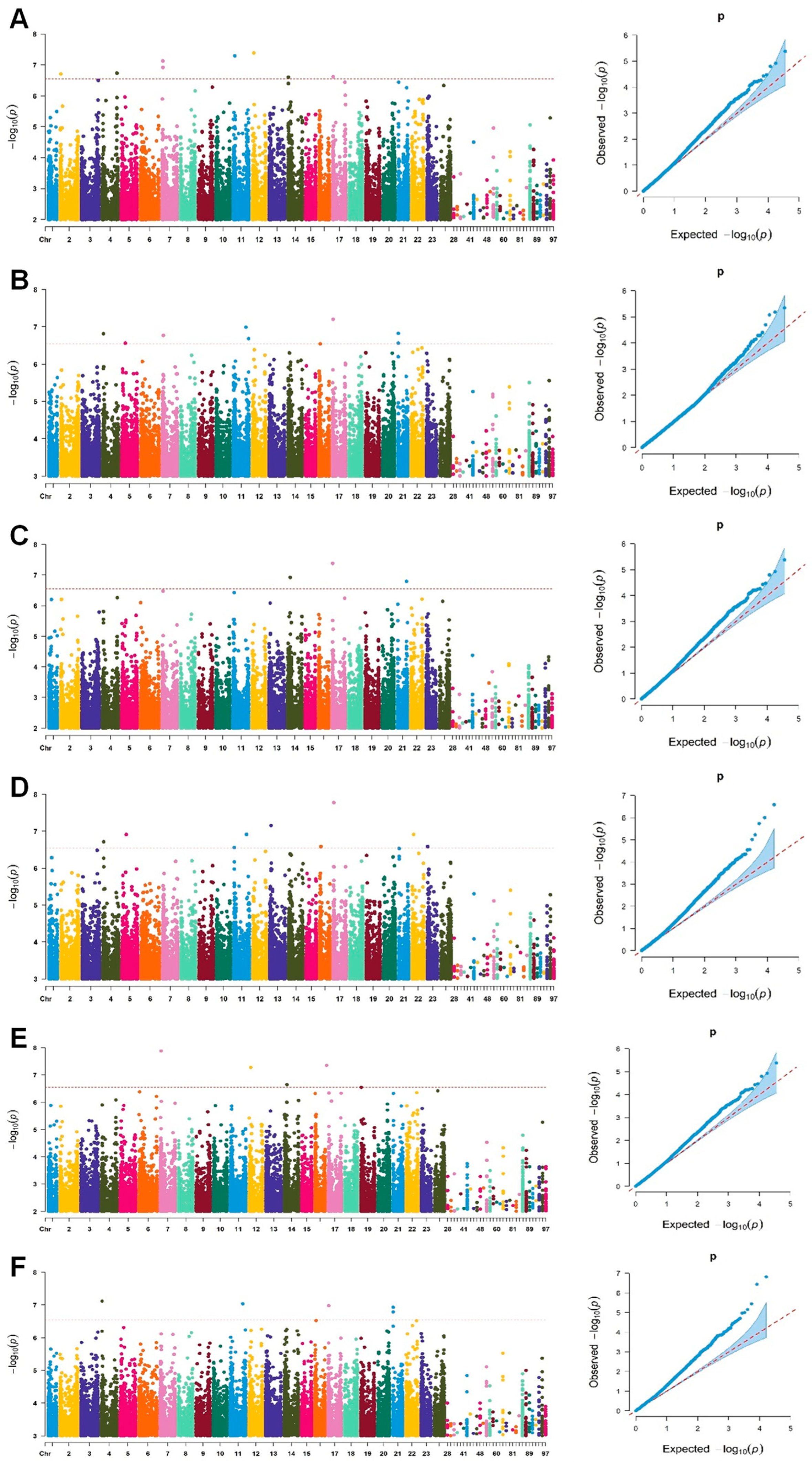
Genome-Wide Association Study of Ginsenoside Rg1 Content Using Single-Locus Models
Genome-Wide Association Study of Ginsenoside Rg1 Content Using Multi-Locus Models
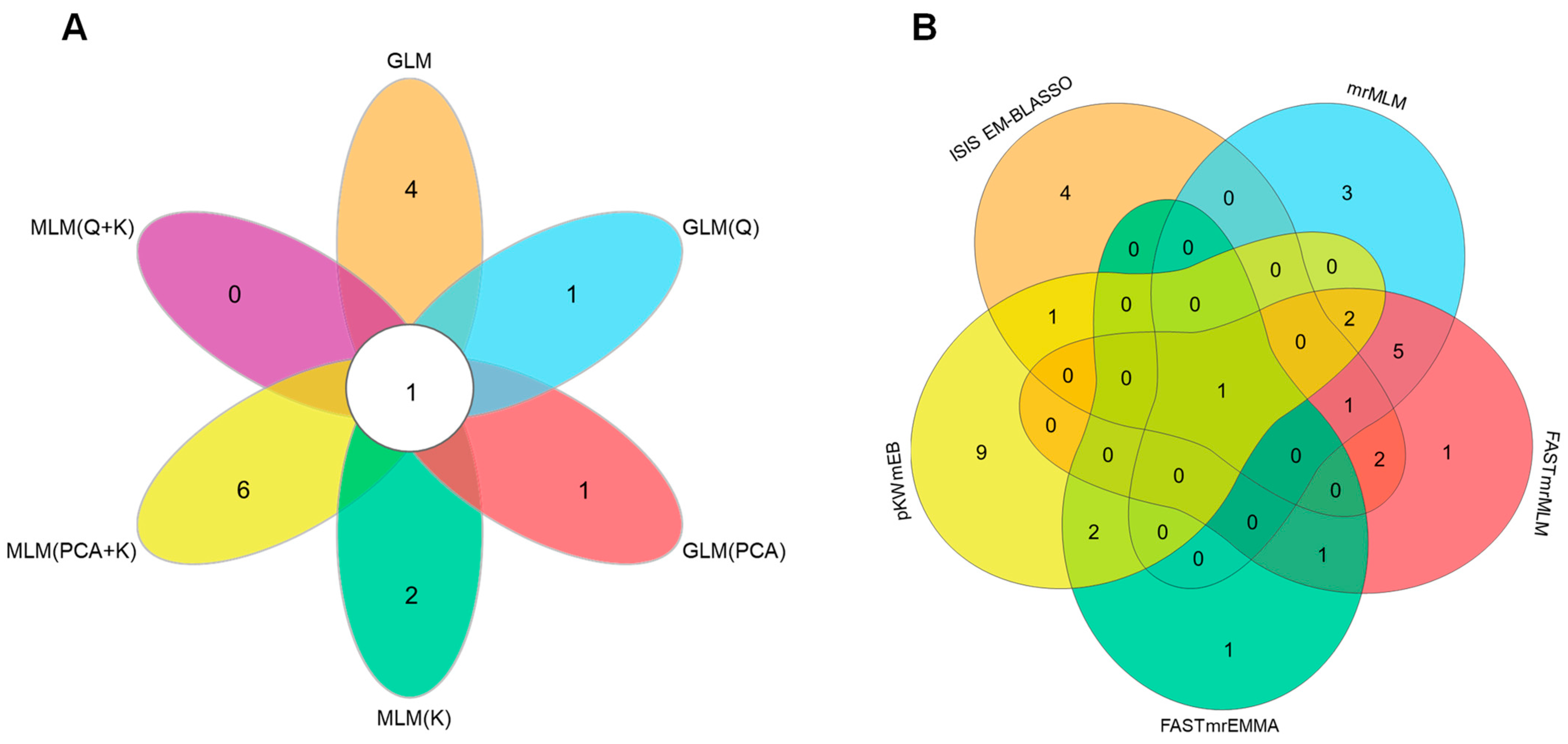
Identification of Genes in Significantly Associated Regions through Genome-Wide Association Study
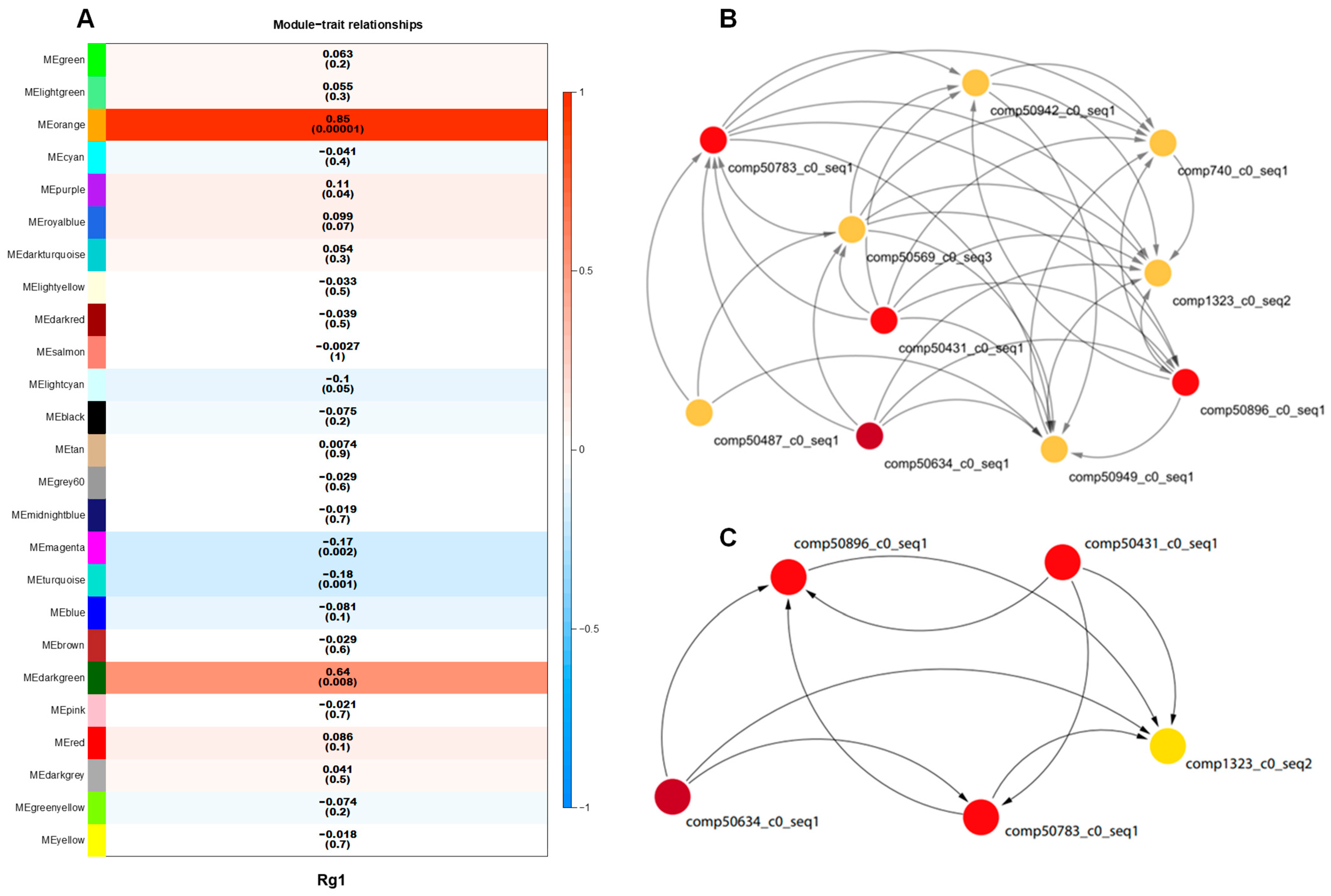
3.1.2. WGCNA Analysis
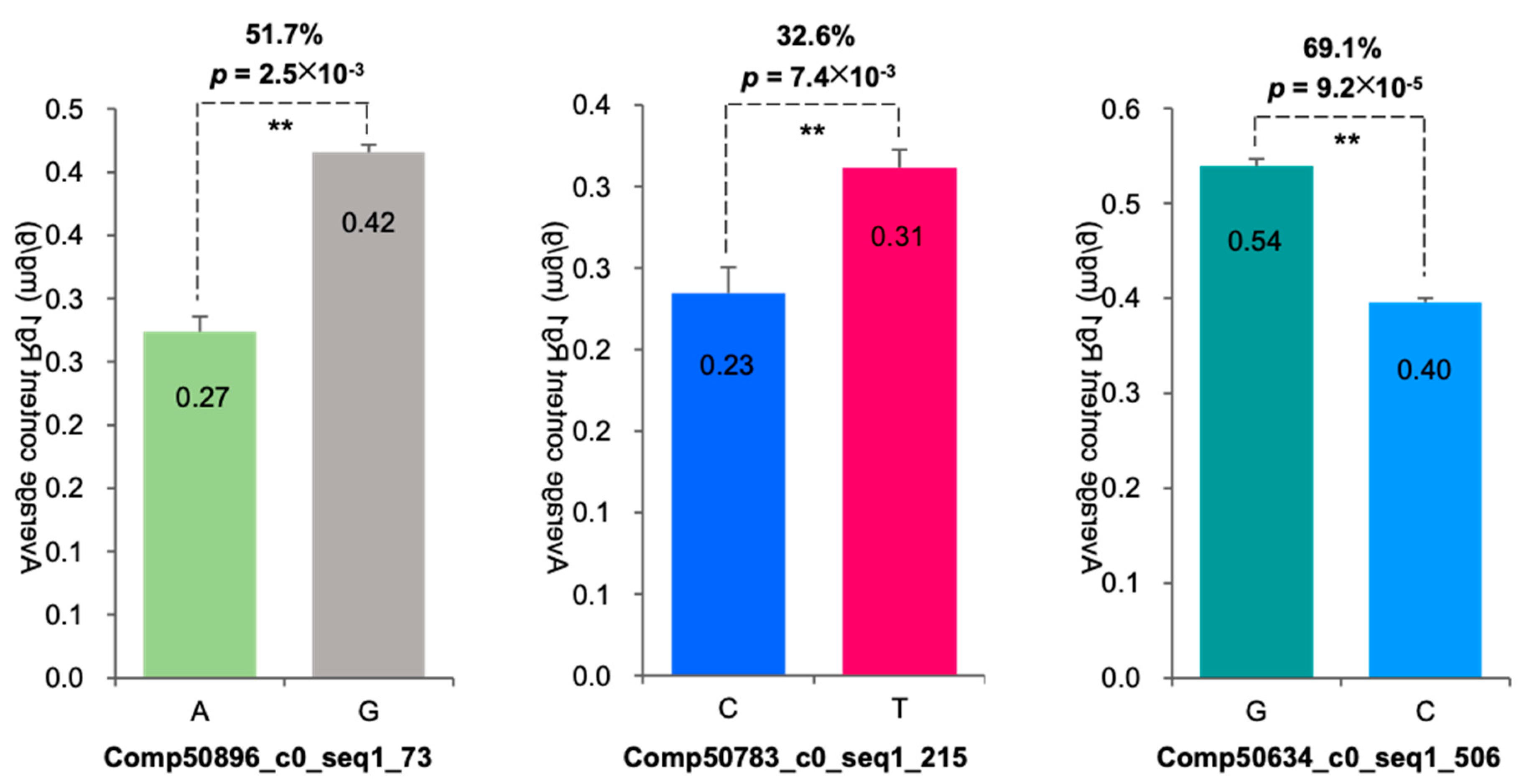
3.1.3. Association Analysis of SNPs with Rg1 Content Investigating the Impact of Gene Mutations on Rg1 Content
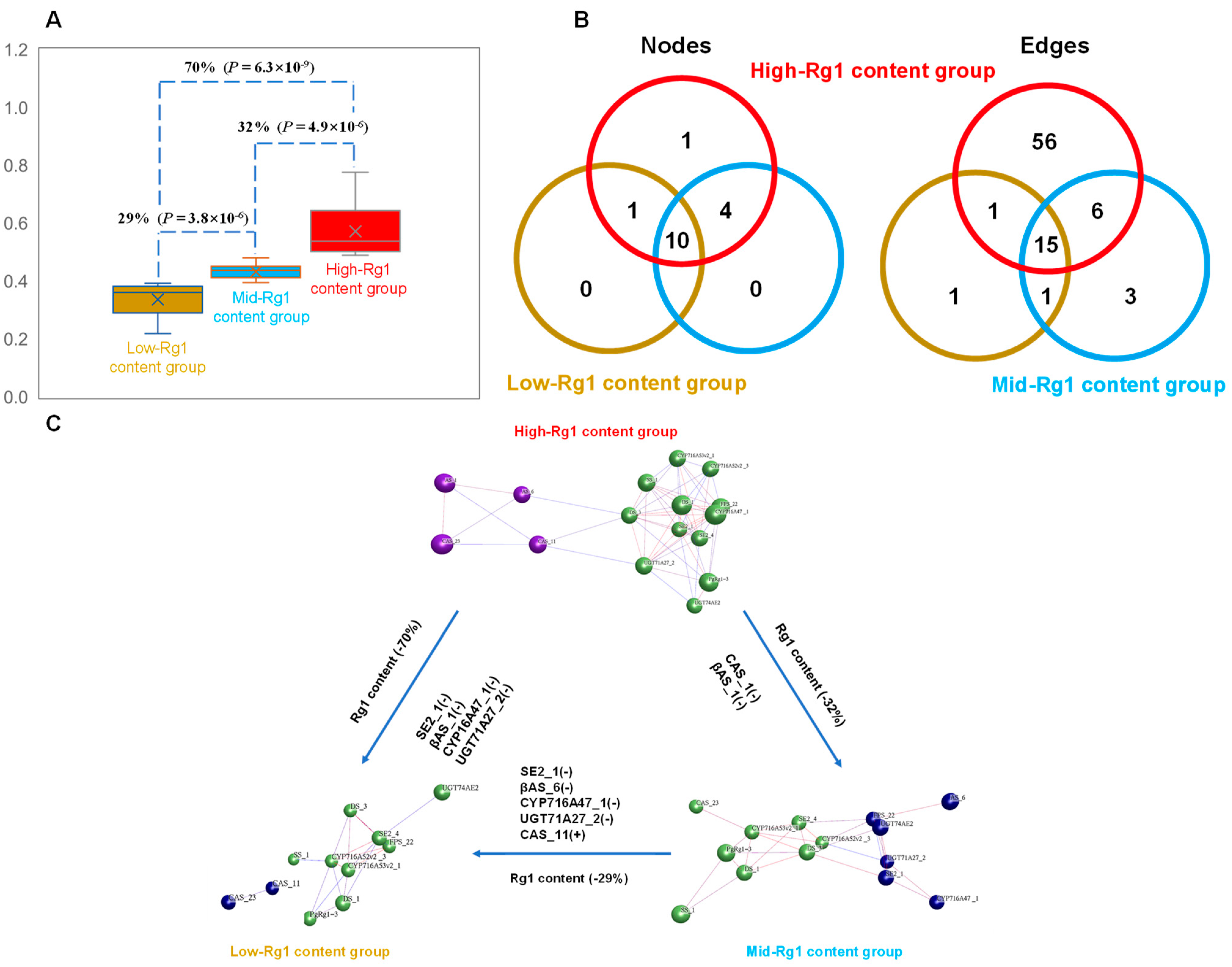
3.1.4. Network Analysis of Candidate Genes III for Rg1 Biosynthesis and 15 Key Enzyme Gene Transcripts Involved in Ginsenoside Biosynthesis
3.2. Functional Validation of the PgRg1-3 Key Candidate Gene for Rg1 Biosynthesis
3.2.1. MeJA-Induced Regulation
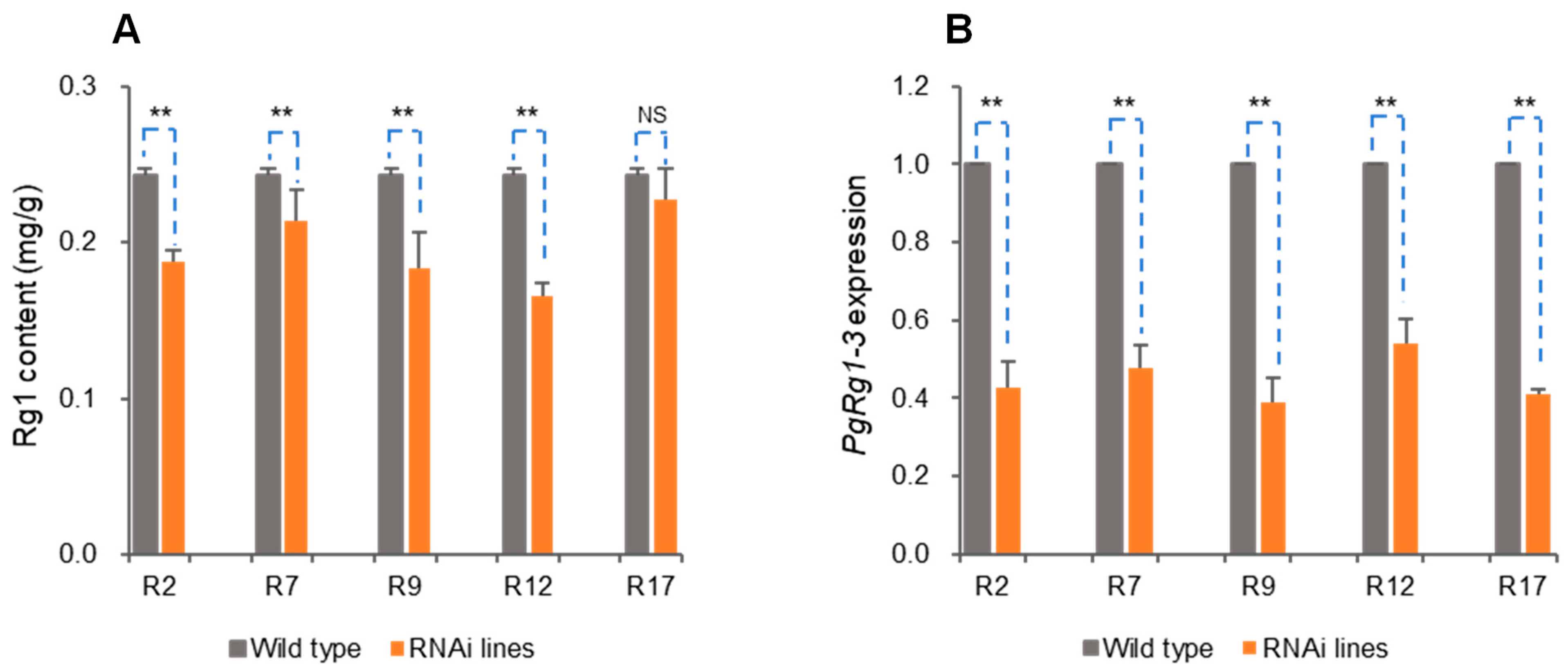
3.2.2. Genetic Transformation RNA Interference (RNAi)
3.3. Exploration of the Molecular Mechanism of the PgRg1-3 Gene in Rg1 Biosynthesis
3.3.1. Functional Annotation of PgRg1-3 Gene by GO, KEGG Localization, and Homology Comparison with Ginseng CYP Genes
3.3.2. Gene Expression Patterns at Different Spatiotemporal Points and in Different Cultivars
3.3.3. Impact of the Changes in Gene Co-Expression Networks on Rg1 Biosynthesis
3.3.4. Analysis of the Impact and Genetic Effects of SNP Mutations in the PgRg1-3 Gene on Rg1 Biosynthesis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Kim, K.H.; Lee, D.; Lee, H.L.; Kim, C.E.; Jung, K.; Kang, K.S. Beneficial effects of Panax ginseng for the treatment and prevention of neurodegenerative diseases: Past findings and future directions. J. Ginseng Res. 2018, 42, 239–247. [Google Scholar] [CrossRef]
- Mancuso, C.; Santangelo, R. Panax ginseng and Panax quinquefolius: From pharmacology to toxicology. Food Chem. Toxicol. 2017, 107, 362–372. [Google Scholar] [CrossRef]
- Taik-Koo, Y. Brief introduction of Panax ginseng C.A. Meyer. J. Korean Med. Sci. 2001, 16, S3–S5. [Google Scholar]
- Nam, M.H.; Kim, S.I.; Liu, J.R.; Yang, D.C.; Lim, Y.P.; Kwon, K.H.; Park, Y.M. Proteomic analysis of Korean ginseng (Panax ginseng C.A. Meyer). J. Chromatogr. B 2005, 815, 147–155. [Google Scholar] [CrossRef]
- Fuzzati, N. Analysis methods of ginsenosides. J. Chromatogr. B 2004, 812, 119–133. [Google Scholar] [CrossRef]
- Piao, X.; Zhang, H.; Kang, J.P.; Yang, D.U.; Li, Y.; Pang, S.; Jin, Y.; Yang, D.C.; Wang, Y. Advances in saponin diversity of Panax ginseng. Molecules 2020, 25, 3452. [Google Scholar] [CrossRef]
- Kim, N.H.; Jayakodi, M.; Lee, S.C.; Choi, B.S.; Jang, W.; Lee, J.; Kim, H.H.; Waminal, N.E.; Lakshmanan, M.; van Nguyen, B.; et al. Genome and evolution of the shade-requiring medicinal herb Panax ginseng. Plant Biotechnol. J. 2018, 16, 1904–1917. [Google Scholar] [CrossRef]
- Nam, M.H.; Kim, S.I.; Liu, J.R.; Yang, D.C.; Lim, Y.P.; Kwon, K.H.; Yoo, J.S.; Park, Y.M.; Cha, S.; Kim, M.Y. Ginsenosides are active ingredients in Panax ginseng with immunomodulatory properties from cellular to organismal levels. J. Ginseng Res. 2022, 46, 711–721. [Google Scholar]
- Wei, W.; Wang, P.; Wei, Y.; Liu, Q.; Yang, C.; Zhao, G.; Yue, J.; Yan, X.; Zhou, Z. Characterization of Panax ginseng UDP-glycosyltransferases catalyzing protopanaxatriol and biosyntheses of bioactive ginsenosides F1 and Rh1 in metabolically engineered yeasts. Mol. Plant 2015, 8, 1412–1424. [Google Scholar] [CrossRef]
- Jayakodi, M.; Choi, B.S.; Lee, S.C.; Kim, N.H.; Park, J.Y.; Jang, W.; Lakshmanan, M.; Mohan, S.V.G.; Lee, D.Y.; Yang, T.J. Ginseng Genome Database: An open-access platform for genomics of Panax ginseng. BMC Plant Biol. 2018, 18, s12870. [Google Scholar] [CrossRef]
- Lu, J.; Yao, L.; Li, J.X.; Liu, S.J.; Hu, Y.Y.; Wang, S.H.; Liang, W.X.; Huang, L.Q.; Dai, Y.J.; Wang, J.; et al. Characterization of UDP-glycosyltransferase involved in biosynthesis of ginsenosides Rg1 and Rb1 and identification of critical conserved amino acid residues for its function. J. Agric. Food Chem. 2018, 66, 9446–9455. [Google Scholar] [CrossRef]
- Zhao, J.N.; Wang, R.F.; Zhao, S.J.; Wang, Z.T. Advance in glycosyltransferases, the important bioparts for production of diversified ginsenosides. Chin. J. Nat. Med. 2020, 18, 643–658. [Google Scholar] [CrossRef]
- Zhou, C.; Gong, T.; Chen, J.-J.; Chen, T.-J.; Yang, J.-L.; Zhu, P. The glycosyltransferases involved in triterpenoid saponin biosynthesis: A review. Chin. J. Biotech. 2022, 38, 1004–1024. [Google Scholar]
- Wang, H.-J.; Li, Y.-X.; Tong, A.-Z.; Liu, W.; Hu, Y.-J. Research progress on the function of ginseng UGT gene family. Ginseng Res. 2022, 34, 54–57. [Google Scholar]
- Chen, Q.; Lei, J.; Liu, D.-Q.; Cui, X.-M.; Ge, F. Research progress on skeleton 597 modification of triterpenoid saponins of Panax. J. Chin. Med. Mater. 2020, 43, 2830–2836. [Google Scholar]
- Mohanan, P.; Yang, T.J.; Song, Y.H. Genes and regulatory mechanisms for ginsenoside biosynthesis. J. Plant Biol. 2023, 66, 87–97. [Google Scholar] [CrossRef]
- Xue, L.; He, Z.; Bi, X.; Xu, W.; Wei, T.; Wu, S.; Hu, S. Transcriptomic profiling reveals MEP pathway contributing to ginsenoside biosynthesis in Panax ginseng. BMC Genom. 2019, 20, 383. [Google Scholar] [CrossRef]
- Lu, J.; Li, J.; Wang, S.; Yao, L.; Liang, W.; Wang, J.; Gao, W. Advances in ginsenoside biosynthesis and metabolic regulation. Biotechnol. Appl. Biochem. 2018, 65, 514–522. [Google Scholar] [CrossRef]
- Huang, X.H.; Yang, S.H.; Gong, J.Y.; Zhao, Y.; Feng, Q.; Gong, H.; Li, W.; Zhan, Q.; Cheng, B.; Xia, J.; et al. Genomic analysis of hybrid rice varieties reveals numerous superior alleles that contribute to heterosis. Nat. Commun. 2015, 6, 6258. [Google Scholar] [CrossRef]
- Gong, Q.; Yang, Z.; Chen, E.; Sun, G.; He, S.; Butt, H.I.; Zhang, C.; Zhang, X.; Yang, Z.; Du, X.; et al. A Phi-class glutathione S-transferase gene for verticillium wilt resistance in Gossypium arboreum identified in a genome-wide association study. Plant Cell Physiol. 2018, 59, 275–289. [Google Scholar] [CrossRef]
- Wang, H.; Li, K.; Hu, X.; Liu, Z.; Wu, Y.; Huang, C. Genome-wide association analysis of forage quality in maize mature stalk. BMC Plant Biol. 2016, 16, 227. [Google Scholar] [CrossRef]
- Liu, S.; Jiang, Y.; Wang, Y.; Huo, H.; Cilkiz, M.; Chen, P.; Han, Y.; Li, L.; Wang, K.; Zhao, M.; et al. Genetic and molecular dissection of ginseng (Panax ginseng Mey.) germplasm using high-density genic SNP markers, secondary metabolites, and gene expressions. Front. Plant Sci. 2023, 14, 1165349. [Google Scholar] [CrossRef]
- Wang, K.; Jiang, S.; Sun, C.; Lin, Y.; Yin, R.; Wang, Y.; Zhang, M.P. The spatial and temporal transcriptomic landscapes of ginseng, Panax ginseng C.A. Meyer. Sci. Rep. 2015, 5, 18283. [Google Scholar] [CrossRef]
- Jiang, Y.; Liu, S.-Z.; Li, L.; Zang, K.-Y.; Wang, Y.-F.; Zhao, M.-Z.; Wang, K.-Y.; Zhu, L.; Chen, P.; Lei, J.; et al. Transcriptome and phenotype integrated analysis identifies genes controlling ginsenoside Rb1 biosynthesis and reveals their interactions in the process in Panax Ginseng. Int. J. Mol. Sci. 2022, 23, 14016. [Google Scholar] [CrossRef]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef]
- Wang, S.B.; Feng, J.Y.; Ren, W.L.; Huang, B.; Zhou, L.; Wen, Y.J.; Zhang, J.; Dunwell, J.M.; Xu, S.; Zhang, Y.M. Improving power and accuracy of genome-wide association studies via a multi-locus mixed linear model methodology. Sci. Rep. 2016, 6, 19444. [Google Scholar] [CrossRef]
- Tamba, C.L.; Zhang, Y.M. A fast mrMLM algorithm for multi-locus genome-wide association studies. bioRxiv 2018, 341784. [Google Scholar] [CrossRef]
- Wen, Y.J.; Zhang, H.; Ni, Y.L.; Huang, B.; Zhang, J.; Feng, J.Y.; Wang, S.B.; Dunwell, J.M.; Zhang, Y.M.; Wu, R. Methodological implementation of mixed linear models in multi-locus genome-wide association studies. Brief Bioinform. 2018, 19, 700–712. [Google Scholar] [CrossRef]
- Tamba, C.L.; Ni, Y.L.; Zhang, Y.M. Iterative sure independence screening EM-Bayesian LASSO algorithm for multi-locus genome-wide association studies. PLoS Comput. Biol. 2017, 13, e1005357. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Feng, J.Y.; Ni, Y.L.; Wen, Y.J.; Niu, Y.; Tamba, C.L.; Yue, C.; Song, Q.; Zhang, Y.M. pLARmEB: Integration of least angle regression with empirical Bayes for multi-locus genome-wide association studies. Heredity 2017, 118, 517–524. [Google Scholar] [CrossRef]
- Zhang, M.; Liu, Y.H.; Xu, W.; Smith, C.W.; Murray, S.C.; Zhang, H.B. Analysis of the genes controlling three quantitative traits in three diverse plant species reveals the molecular basis of quantitative traits. Sci. Rep. 2020, 10, 10074. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Wang, Q.; Hu, Y.; Jia, Y.; Chen, J.; Liu, B.; Zhang, Z.; Guan, X.; Chen, S.; Zhou, B.; et al. Genomic analyses in cotton identify signatures of selection and loci associated with fiber quality and yield traits. Nat. Genet. 2017, 49, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.-X.; Zhong, J.-J. Role of jasmonic acid in alteration of ginsenoside heterogeneity in elicited cell cultures of Panax notoginseng. J. Biosci. Bioeng. 2007, 104, 513–516. [Google Scholar] [CrossRef] [PubMed]
- Kochan, E.; Balcerczak, E.; Lipert, A.; Szymańska, G.; Szymczyk, P. Methyl jasmonate as a control factor of the synthase squalene gene promoter and ginsenoside production in American ginseng hairy root cultured in shake flasks and a nutrient sprinkle bioreactor. Ind. Crops Prod. 2018, 115, 182–193. [Google Scholar] [CrossRef]
- Wu, X.H.; Fan, M.Z.; Li, X.F.; Piao, X.C.; Gao, R.; Lian, M.L. Involvement of putrescine, nitric oxide, and hydrogen peroxide in methyl jasmonate-induced ginsenoside synthesis in adventitious root cultures of Panax ginseng C.A. Meyer. J. Plant Growth Regul. 2021, 40, 1440–1449. [Google Scholar] [CrossRef]
- Lee, M.H.; Jeong, J.H.; Seo, J.W.; Shin, C.G.; Kim, Y.S.; In, J.G.; Yang, D.C.; Yi, J.S.; Choi, Y.E. Enhanced triterpene and phytosterol biosynthesis in Panax ginseng overexpressing squalene synthase gene. Plant Cell Physiol. 2004, 45, 976–984. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K.; Kim, Y.B.; Kim, J.K.; Kim, S.U.; Park, S.U. Molecular cloning and characterization of mevalonic acid (MVA) pathway genes and triterpene accumulation in Panax ginseng. J. Korean Soc. Appl. Biol. Chem. 2014, 57, 289–295. [Google Scholar] [CrossRef]
- Balusamy, S.R.D.; Rahimi, S.; Sukweenadhi, J.; Kim, Y.J.; Yang, D.C. Exogenous methyl jasmonate prevents necrosis caused by mechanical wounding and increases terpenoid biosynthesis in Panax ginseng. Plant Cell Tissue Organ Cult. 2015, 123, 341–348. [Google Scholar] [CrossRef]
- Lee, Y.S.; Park, H.S.; Lee, D.K.; Jayakodi, M.; Kim, N.H.; Koo, H.J.; Lee, S.C.; Kim, Y.J.; Kwon, S.W.; Yang, T.J. Integrated transcriptomic and metabolomic analysis of five Panax ginseng cultivars reveals the dynamics of ginsenoside biosynthesis. Front. Plant Sci. 2017, 8, 1048. [Google Scholar] [CrossRef]
- Kang, K.B.; Jayakodi, M.; Lee, Y.S.; Nguyen, V.B.; Park, H.S.; Koo, H.J.; Choi, I.Y.; Kim, D.H.; Chung, Y.J.; Ryu, B.; et al. Identification of candidate UDP-glycosyltransferases involved in protopanaxadiol-type ginsenoside biosynthesis in Panax ginseng. Sci. Rep. 2018, 8, 11744. [Google Scholar] [CrossRef]
- Zeng, X.; Luo, T.; Li, J.; Li, G.; Zhou, D.; Liu, T.; Zou, X.; Pandey, A.; Luo, Z. Transcriptomics-based identification and characterization of 11 CYP450 genes of Panax ginseng responsive to MeJA. Acta Biochim. Biophys. Sin. 2018, 50, 1094–1103. [Google Scholar] [CrossRef]
- Liu, T.; Luo, T.; Guo, X.; Zou, X.; Zhou, D.; Afrin, S.; Li, G.; Zhang, Y.; Zhang, R.; Luo, Z. PgMYB2, a MeJA-responsive transcription factor, positively regulates the dammarenediol synthase gene expression in Panax Ginseng. Int. J. Mol. Sci. 2019, 20, 2219. [Google Scholar] [CrossRef] [PubMed]
- Han, J.Y.; In, J.G.; Kwon, Y.S.; Choi, Y.E. Regulation of ginsenoside and phytosterol biosynthesis by RNA interferences of squalene epoxidase gene in Panax ginseng. Phytochemistry 2010, 71, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Han, J.Y.; Kwon, Y.S.; Yang, D.C.; Jung, Y.R.; Choi, Y.E. Expression and RNA interference-induced silencing of the dammarenediol synthase gene in Panax ginseng. Plant Cell Physiol. 2006, 47, 1653–1662. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhao, S.J.; Liang, Y.L.; Wang, L.; Cao, H.J. Regulation and differential expression of protopanaxadiol synthase in Asian and American ginseng ginsenoside biosynthesis by RNA interferences. Plant Growth Regul. 2013, 71, 207–217. [Google Scholar] [CrossRef]
- Lu, C.; Zhao, S.; Wei, G.; Zhao, H.; Qu, Q. Functional regulation of ginsenoside biosynthesis by RNA interferences of a UDP-glycosyltransferase gene in Panax ginseng and Panax quinquefolius. Plant Physiol. Biochem. 2017, 111, 67–76. [Google Scholar] [CrossRef]
- Park, S.B.; Chun, J.H.; Ban, Y.W.; Han, J.Y.; Choi, Y.E. Alteration of Panax ginseng saponin composition by overexpression and RNA interference of the protopanaxadiol 6-hydroxylase gene (CYP716A53v2). J. Ginseng Res. 2016, 40, 47–54. [Google Scholar] [CrossRef]
- Krieger, U.; Lippman, Z.B.; Zamir, D. The flowering gene SINGLEFLOWER TRUSS drives heterosis for yield in tomato. Nat. Genet. 2010, 42, 459–463. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, S.; Chen, X.; Zhao, T.; Yu, J.; Chen, P.; Wang, Y.; Wang, K.; Zhao, M.; Jiang, Y.; Wang, Y.; et al. Identification of PgRg1-3 Gene for Ginsenoside Rg1 Biosynthesis as Revealed by Combining Genome-Wide Association Study and Gene Co-Expression Network Analysis of Jilin Ginseng Core Collection. Plants 2024, 13, 1784. https://doi.org/10.3390/plants13131784
Liu S, Chen X, Zhao T, Yu J, Chen P, Wang Y, Wang K, Zhao M, Jiang Y, Wang Y, et al. Identification of PgRg1-3 Gene for Ginsenoside Rg1 Biosynthesis as Revealed by Combining Genome-Wide Association Study and Gene Co-Expression Network Analysis of Jilin Ginseng Core Collection. Plants. 2024; 13(13):1784. https://doi.org/10.3390/plants13131784
Chicago/Turabian StyleLiu, Sizhang, Xiaxia Chen, Tianqi Zhao, Jinghui Yu, Ping Chen, Yanfang Wang, Kangyu Wang, Mingzhu Zhao, Yue Jiang, Yi Wang, and et al. 2024. "Identification of PgRg1-3 Gene for Ginsenoside Rg1 Biosynthesis as Revealed by Combining Genome-Wide Association Study and Gene Co-Expression Network Analysis of Jilin Ginseng Core Collection" Plants 13, no. 13: 1784. https://doi.org/10.3390/plants13131784
APA StyleLiu, S., Chen, X., Zhao, T., Yu, J., Chen, P., Wang, Y., Wang, K., Zhao, M., Jiang, Y., Wang, Y., & Zhang, M. (2024). Identification of PgRg1-3 Gene for Ginsenoside Rg1 Biosynthesis as Revealed by Combining Genome-Wide Association Study and Gene Co-Expression Network Analysis of Jilin Ginseng Core Collection. Plants, 13(13), 1784. https://doi.org/10.3390/plants13131784