



A Method for Electroporation of Cre Recombinase Protein into Intact Nicotiana tabacum Cells


{kind=link}
{kind=link}
Abstract
1. Introduction
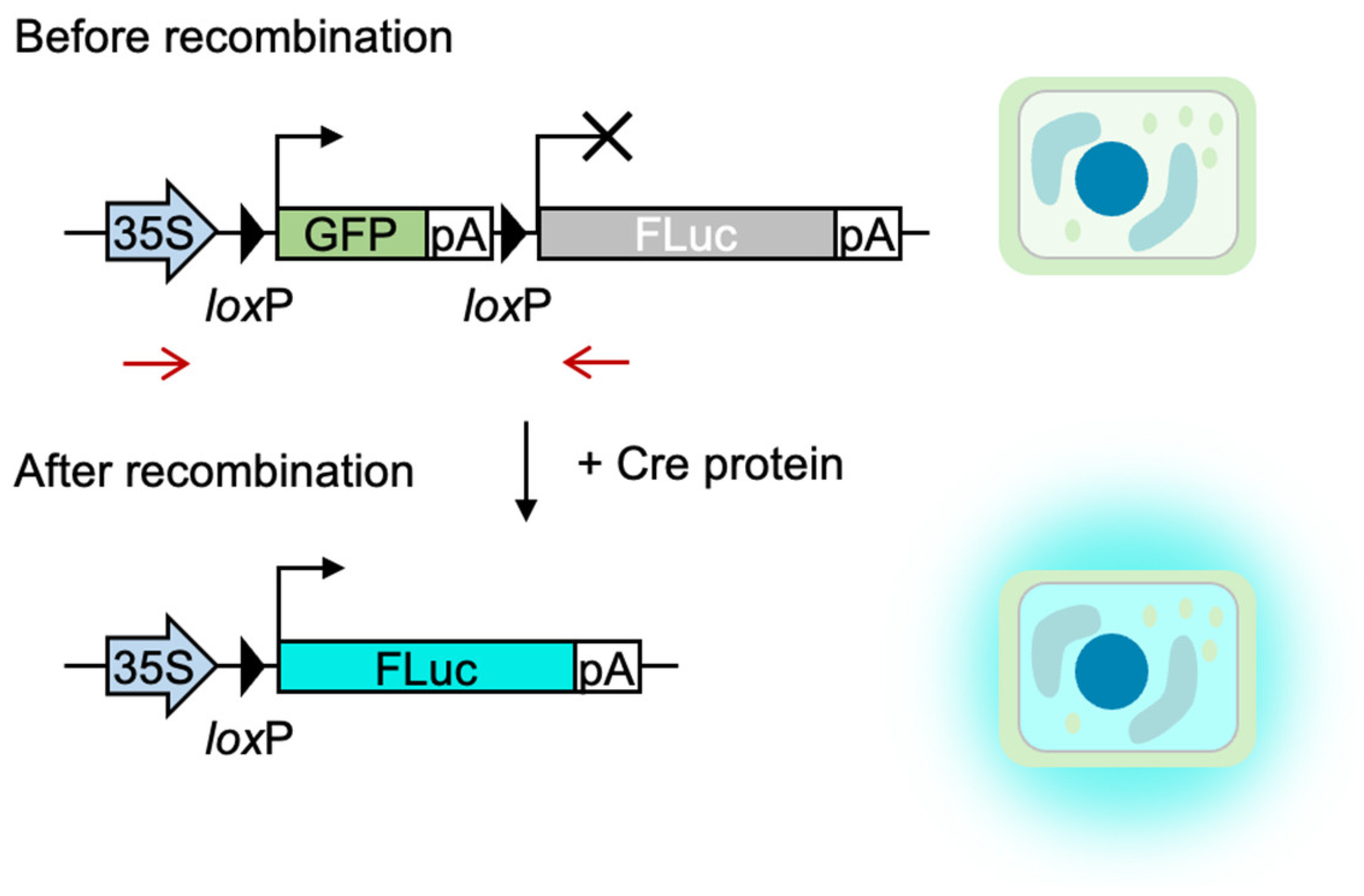
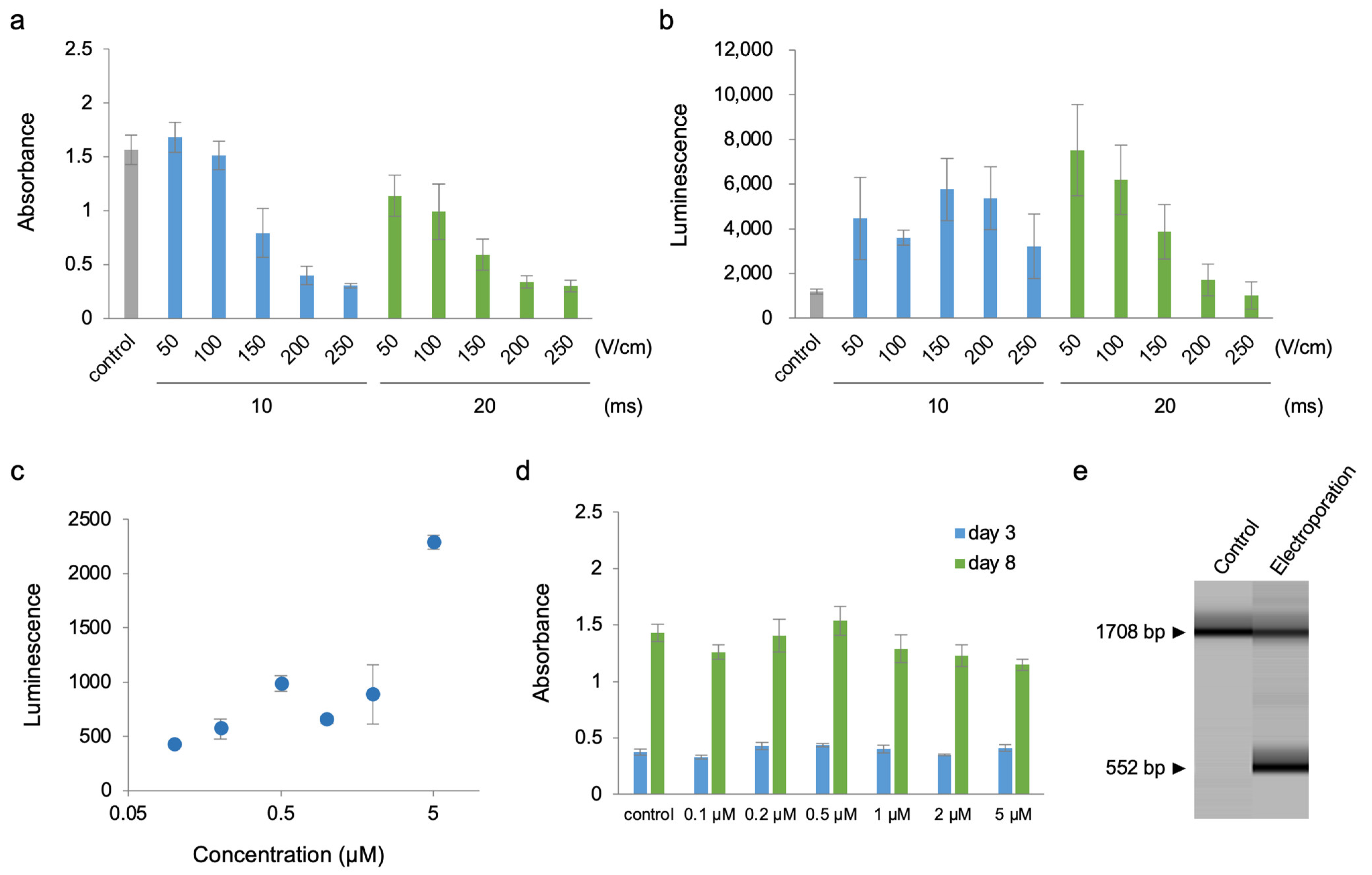
2. Results
3. Discussion
4. Methods
4.1. Plasmid Construct Preparation
4.2. Preparation of Cre Protein
4.3. BY-2 Cell Line and Agrobacterium Tumefaciens (Rhizobium radiobacter) Culture
4.4. Agrobacterium-Mediated Transformation of BY-2 Cells
4.5. Electroporation
4.6. Luminescence Quantification in Cells
4.7. Cell Counting Kit-8 (CCK8) Assay
4.8. Genomic PCR of Cre-Responsive Reporter Cassette
4.9. Quantification and Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Branda, C.S.; Dymecki, S.M. Talking about a revolution: The impact of site-specific recombinases on genetic analyses in mice. Dev. Cell 2004, 6, 7–28. [Google Scholar] [CrossRef] [PubMed]
- Nagy, A. Cre recombinase: The universal reagent for genome tailoring. Genesis 2000, 26, 99–109. [Google Scholar] [CrossRef]
- Ankrum, J.A.; Dastidar, R.G.; Ong, J.F.; Levy, O.; Karp, J.M. Performance-enhanced mesenchymal stem cells via intracellular delivery of steroids. Sci. Rep. 2014, 4, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Livet, J.; Weissman, T.A.; Kang, H.; Draft, R.W.; Lu, J.; Bennis, R.A.; Sanes, J.R.; Lichtman, J.W. Transgenic strategies for combinatorial expression of fluorescent proteins in the nervous system. Nature 2007, 450, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Furuhata, Y.; Sakai, A.; Murakami, T.; Morikawa, M.; Nakamura, C.; Yoshizumi, T.; Fujikura, U.; Nishida, K.; Kato, Y. A method using electroporation for the protein delivery of Cre recombinase into cultured Arabidopsis cells with an intact cell wall. Sci. Rep. 2019, 9, 2163. [Google Scholar] [CrossRef] [PubMed]
- Furuhata, Y.; Sakai, A.; Kato, Y. Protein electroporation of Cre recombinase into cultured Arabidopsis cells with an intact cell wall. Protoc. Exch. 2019, 1–9. [Google Scholar] [CrossRef]
- Doran, P.M. Therapeutically important proteins from in vitro plant tissue culture systems. Curr. Med. Chem. 2013, 20, 1047–1055. [Google Scholar]
- Leifert, J.A.; Harkins, S.; Whitton, J.L. Full-length proteins attached to the HIV tat protein transduction domain are neither transduced between cells, nor exhibit enhanced immunogenicity. Gene Ther. 2002, 9, 1422–1428. [Google Scholar] [CrossRef]
- Cedeño, C.; Pauwels, K.; Tompa, P. Protein delivery into plant cells: Toward in vivo structural biology. Front. Plant Sci. 2017, 8, 519. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Furuhata, Y.; Egi, E.; Murakami, T.; Kato, Y. A Method for Electroporation of Cre Recombinase Protein into Intact Nicotiana tabacum Cells. Plants 2023, 12, 1631. https://doi.org/10.3390/plants12081631
Furuhata Y, Egi E, Murakami T, Kato Y. A Method for Electroporation of Cre Recombinase Protein into Intact Nicotiana tabacum Cells. Plants. 2023; 12(8):1631. https://doi.org/10.3390/plants12081631
Chicago/Turabian StyleFuruhata, Yuichi, Emiko Egi, Tomi Murakami, and Yoshio Kato. 2023. "A Method for Electroporation of Cre Recombinase Protein into Intact Nicotiana tabacum Cells" Plants 12, no. 8: 1631. https://doi.org/10.3390/plants12081631
APA StyleFuruhata, Y., Egi, E., Murakami, T., & Kato, Y. (2023). A Method for Electroporation of Cre Recombinase Protein into Intact Nicotiana tabacum Cells. Plants, 12(8), 1631. https://doi.org/10.3390/plants12081631