
A Phylogenetic and Morphological Evolution Study of Ribes L. in China Using RAD-Seq


Abstract
1. Introduction
2. Results
2.1. RAD-Tag Sequencing and SNP Discovering
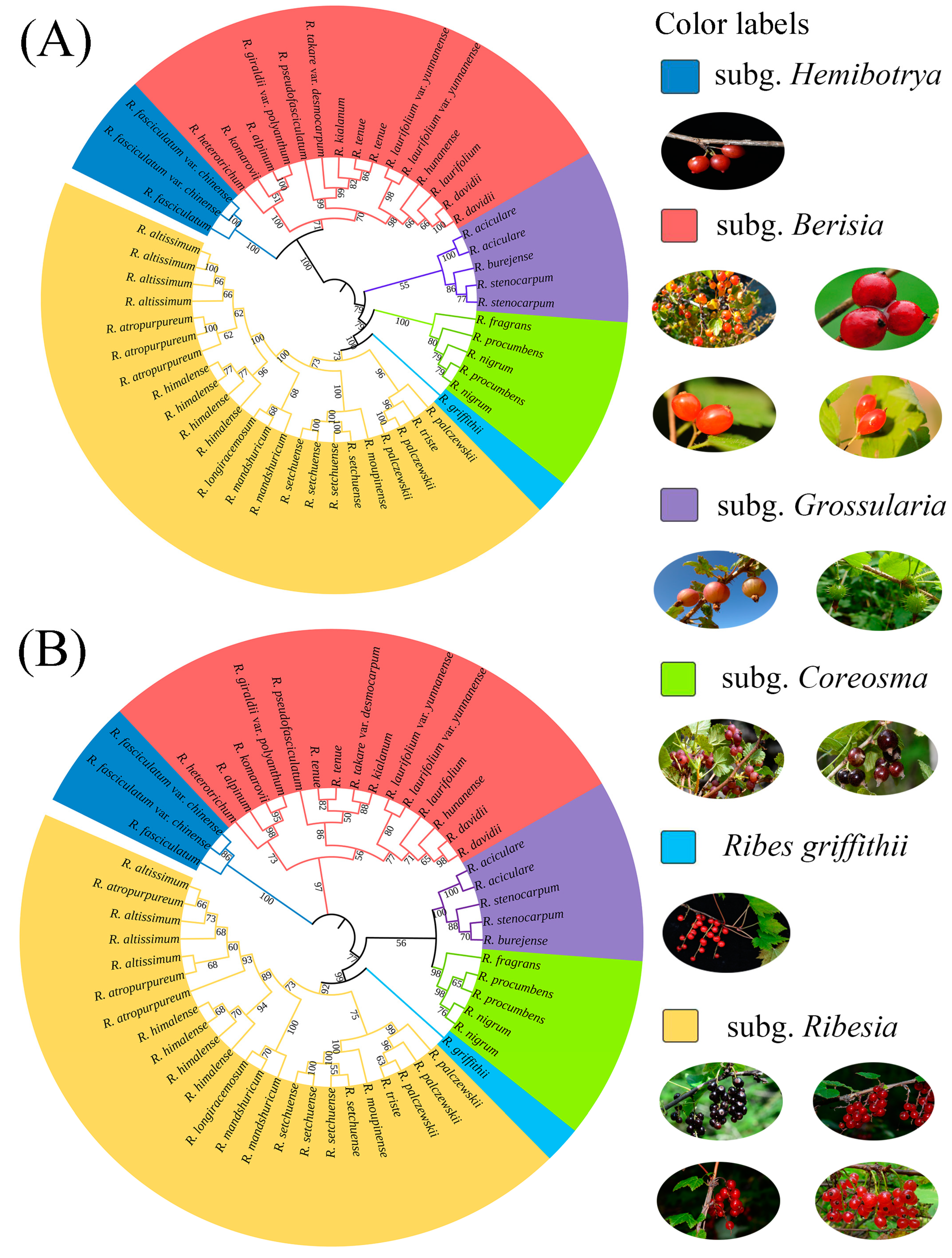
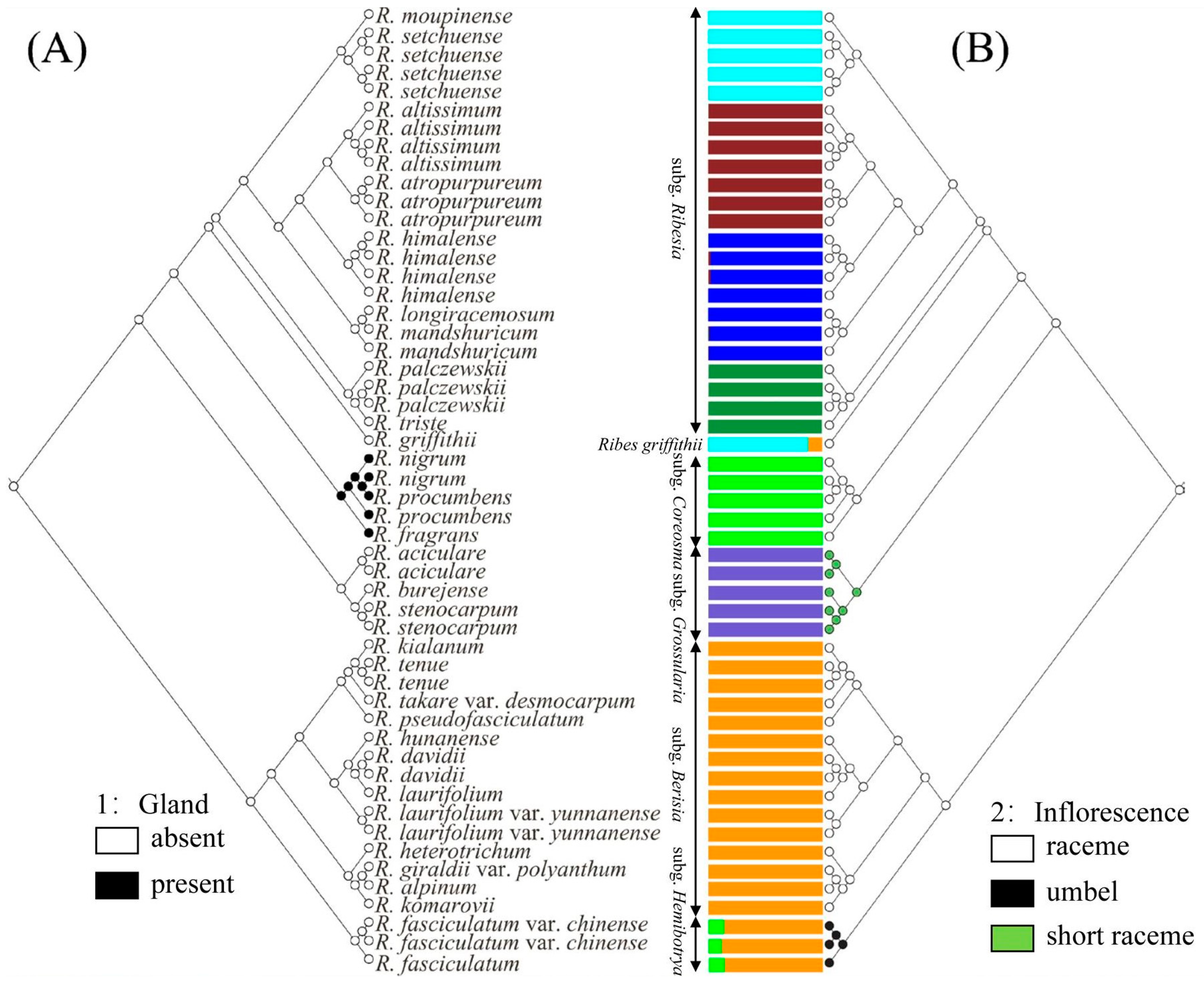
2.2. Phylogenetic Trees and Morphological Characteristics
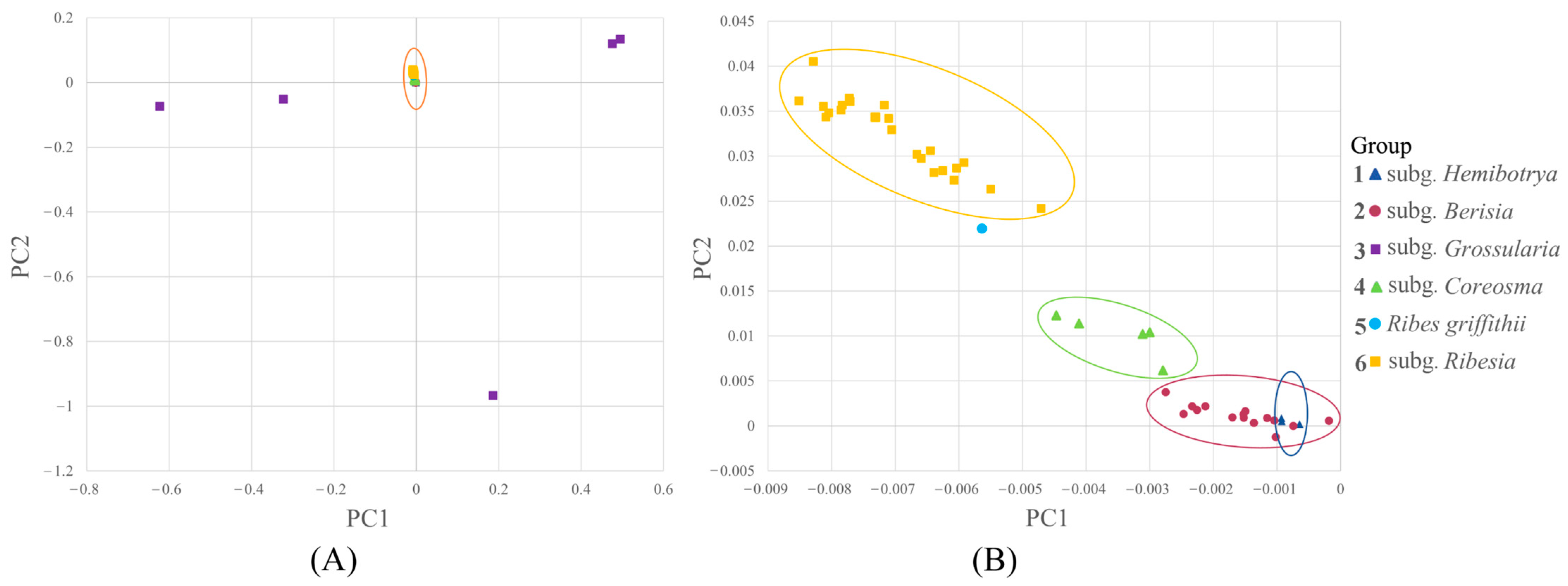
2.3. Principal Component Analysis of Ribes L.
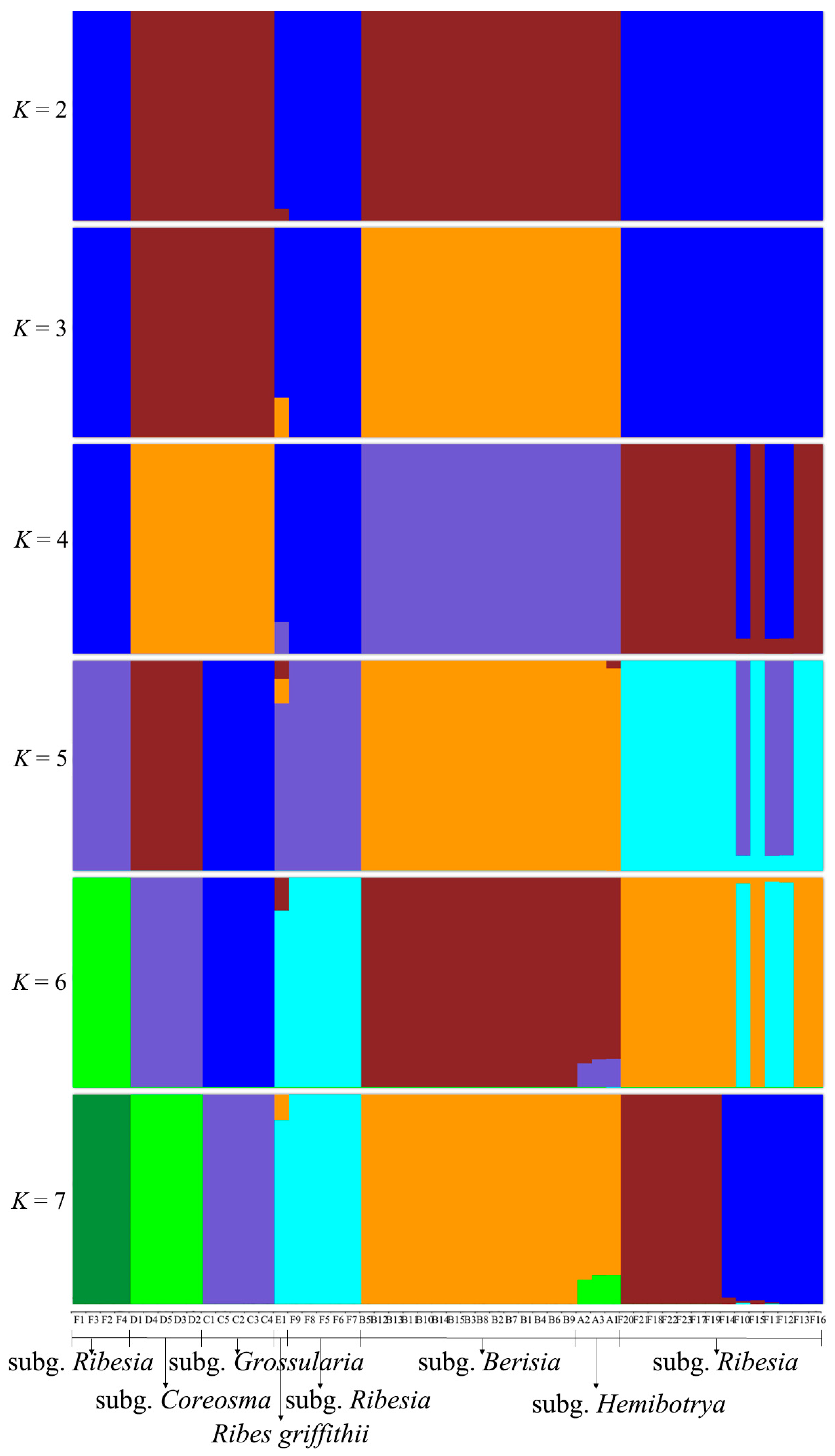
2.4. Population Genetic Structure of Ribes L.
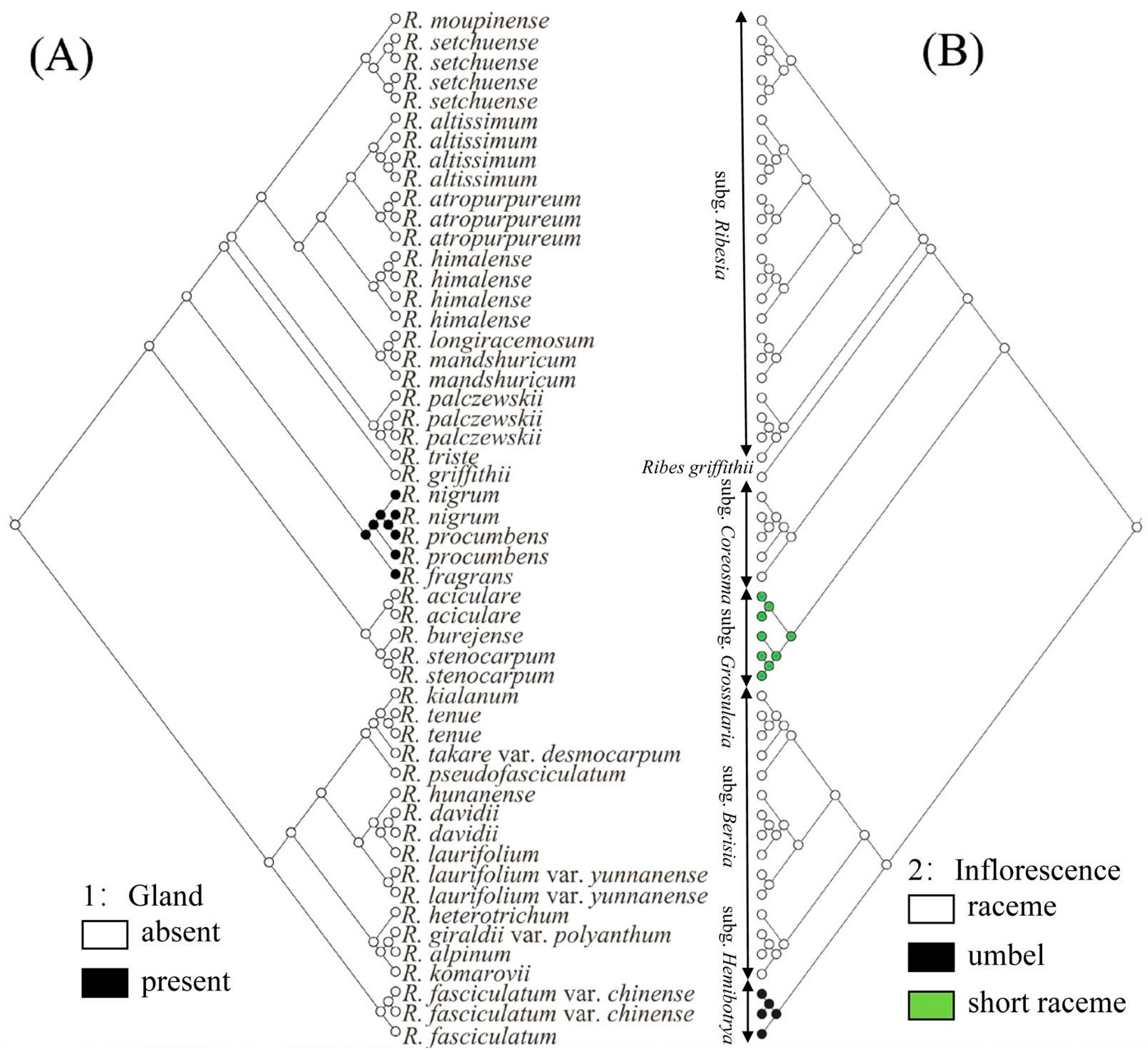
2.5. Ancestral Character Reconstruction
3. Discussion
3.1. Phylogenetic Relationships and Taxonomy of Ribes L.
3.2. Gene Exchange Analysis of Ribes L.
3.3. Revision of subg. Hemibotrya
4. Materials and Methods
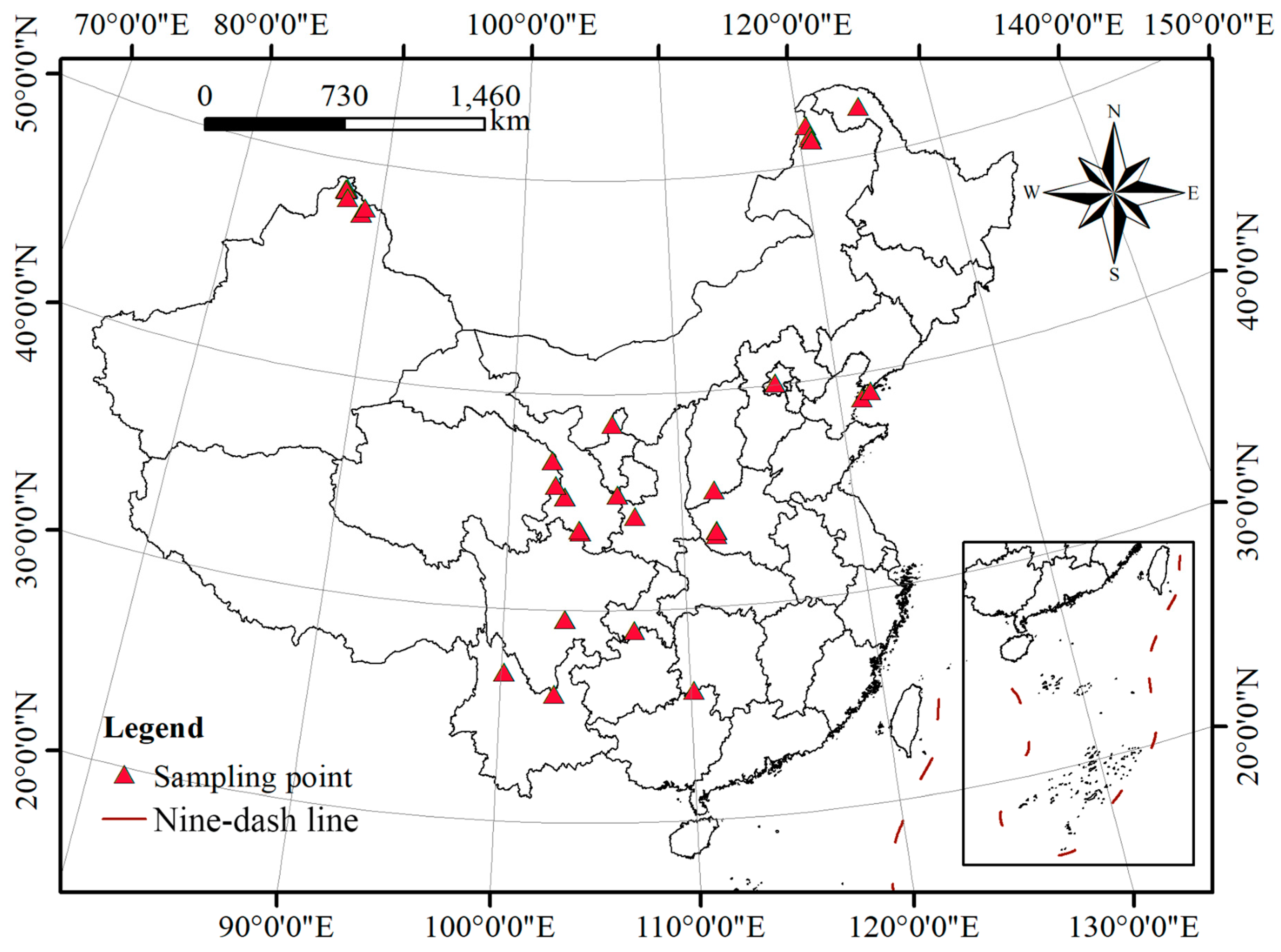
4.1. Plant Materials and DNA Isolation
4.2. RAD Library Preparation and Sequencing
4.3. Quality Filtering and SNP Discovery
4.4. Phylogenetic Analysis
4.5. Principle Component Analysis
4.6. Genetic Structure Analysis
4.7. Ancestral State Reconstruction of Morphological Characters
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Soltis, S.D.E. Phylogenetic Relationships in Ribes (Grossulariaceae) Inferred from ITS Sequence Data. Taxon 2003, 52, 51–66. [Google Scholar] [CrossRef]
- Lu, L.T. A Study on The Genus Ribes L. in China. Acta Phytotaxon. Sin. 1995, 33, 58–75. [Google Scholar]
- Hummer, K.E.; Dale, A. Horticulture of Ribes. For. Pathol. 2010, 40, 251–263. [Google Scholar] [CrossRef]
- Lyashenko, S.; Gonzalez-Fernandez, M.J.; Gomez-Mercado, F.; Yunusova, S.; Denisenko, O.; Guil-Guerrero, J.L. Ribes taxa: A promising source of gamma-linolenic acid-rich functional oils. Food Chem. 2019, 301, 125309. [Google Scholar] [CrossRef] [PubMed]
- Masierowska, M. Early Floral Resources for Urban Bees From Ornamental Shrubs Ribes Aureum, Ribes Sanguineum and Staphylea Pinnata. J. Apic. Sci. 2020, 64, 309–320. [Google Scholar] [CrossRef]
- de Janczewski, E. Monographie des groseilliers, Ribes L. Mémoires Société Phys. d’Hist. Nat. Genève 1907, 35, 199–517. [Google Scholar]
- Weigend, M. Grossulariaceae. In Flowering Plants. Eudicots; Springer: Berlin/Heidelberg, Germany, 2007; pp. 168–176. [Google Scholar]
- Berger, A. A taxonomic review of currants and gooseberries. Agric. Exp. Stn. 1924, 1, 35–38. [Google Scholar]
- Sinnott, Q.P. A revision of Ribes L. subg. Grossularia (Mill.) Pers. Sect. Grossularia (Mill.) Nutt. (Grossulariaceae) in North America. Rhodora 1985, 87, 189–286. [Google Scholar]
- Rehder, A. Manual of Cultivated Trees and Shrubs; MacMillan: New York, NY, USA, 1951. [Google Scholar]
- Rehder, A. Manual of Cultivated Trees And Shrubs. Taxon 1927, 27, 424. [Google Scholar] [CrossRef]
- Zheng, B.J.; Li, G.X. Morphological observation of vessel elements in secondary xylem of Ribes. J. Beijing For. Univ. 2015, 37, 11. [Google Scholar] [CrossRef]
- Messinger, W.; Hummer, K.; Liston, A. Ribes (Grossulariaceae) phylogeny as indicated by restriction-site polymorphisms of PCR-amplified chloroplast DNA. Plant Syst. Evol. 1999, 217, 185–195. [Google Scholar] [CrossRef]
- Pikunova, A.V.; Martirosian, E.V.; Kniazev, S.D.; Ryzhova, N.N. Application of the RAPD-analysis for the study of genetic polymorphism and phylogenetic relationships in the Ribes L. genus. Russ. J. Genet. Appl. Res. 2012, 2, 141–151. [Google Scholar] [CrossRef]
- Weigend, M.; Mohr, O.; Motley, T.J. Phylogeny and classification of the genus Ribes (Grossulariaceae) based on 5S-NTS sequences and morphological and anatomical data. Bot. Jahrbücher 2002, 124, 163–182. [Google Scholar] [CrossRef]
- Schultheis, L.M.; Donoghue, M.J. Molecular phylogeny and biogeography of Ribes (Grossulariaceae), with an emphasis on gooseberries (subg. Grossularia). Syst. Bot. 2004, 29, 77–96. [Google Scholar] [CrossRef]
- Pan, L.; Zheng, B.J. Extraction of Genomic DNA from Ribes and Establishment of ITS-PCR Reaction Systems. J. Northeast For. Univ. 2012, 40, 41–43. [Google Scholar] [CrossRef]
- Liu, H.; Xue, Q.; Huang, W.; Tan, R. Comparative Analysis and Classification of Genus Ribes Based on the Secondary Structure of ITS2 Sequences. J. South-Cent. Univ. Natl. (Nat. Sci. Ed.) 2018, 37, 46–51. [Google Scholar]
- Huang, Y.; Liu, H.Y.; Xing, Y.Z. Next-Generation Sequencing Promoted the Release of Reference Genomes and Discovered Genome Evolution in Cereal Crops. Curr. Issues Mol. Biol. 2017, 27, 37–50. [Google Scholar] [CrossRef]
- Davey, J.W.; Blaxter, M.L. RADSeq: Next-generation population genetics. Brief. Funct. Genom. 2010, 9, 416–423. [Google Scholar] [CrossRef]
- Cruaud, A.; Gautier, M.; Galan, M.; Foucaud, J.; Sauné, L.; Genson, G.; Dubois, E.; Nidelet, S.; Deuve, T.; Rasplus, J.Y. Empirical assessment of RAD sequencing for interspecific phylogeny. Mol. Biol. Evol. 2014, 31, 1272–1274. [Google Scholar] [CrossRef]
- Zhu, J.Y. RAD-seq and its application in entomological research. Acta Entomol. Sin. 2016, 59, 767–774. [Google Scholar] [CrossRef]
- Miller, M.R.; Dunham, J.P.; Amores, A.; Cresko, W.A.; Johnson, E.A. Rapid and cost-effective polymorphism identification and genotyping using restriction site associated DNA (RAD) markers. Genome Res. 2007, 17, 240–248. [Google Scholar] [CrossRef]
- Wang, X.Q.; Ye, X.Y.; Zhao, L.; Li, D.Z.; Guo, Z.H.; Zhuang, H.F. Genome-wide RAD sequencing data provide unprecedented resolution of the phylogeny of temperate bamboos (Poaceae: Bambusoideae). Sci. Rep. 2017, 7, 11546. [Google Scholar] [CrossRef] [PubMed]
- Nagano, Y.; Mimura, T.; Kotoda, N.; Matsumoto, R.; Nagano, A.J.; Honjo, M.N.; Kudoh, H.; Yamamoto, M. Phylogenetic relationships of Aurantioideae (Rutaceae) based on RAD-Seq. Tree Genet. Genom. 2018, 14, 6. [Google Scholar] [CrossRef]
- Léveillé-Bourret, É.; Chen, B.H.; Garon-Labrecque, M.; Ford, B.A.; Starr, J.R. RAD sequencing resolves the phylogeny, taxonomy and biogeography of Trichophoreae despite a recent rapid radiation (Cyperaceae). Mol. Phylogent. Evol. 2020, 145, 106727. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.H. Pollen Morphology of Ribes L. and its Taxonomic Significance. Acta Phytotaxon. Sin. 1989, 27, 378–385. [Google Scholar]
- Yang, H.; Wei, C.L.; Liu, H.W.; Wu, J.L.; Li, Z.G.; Zhang, L.; Jian, J.B.; Li, Y.Y.; Tai, Y.L.; Zhang, J. Genetic divergence between Camellia sinensis and its wild relatives revealed via genome-wide SNPs from RAD sequencing. PLoS ONE 2016, 11, e0151424. [Google Scholar] [CrossRef]
- Wang, M.J.; Zhao, Q.L.; Li, X.Y.; Zheng, B.J. Leaf Epidermal Morphology and Taxonomic Significance of 12 Ribes L. Species. Bull. Bot. Res. 2018, 38, 490–496. [Google Scholar] [CrossRef]
- Wu, Z.Y.; Raven, P. Flora of China; Science Press: Beijing, China, 1994. [Google Scholar]
- Doyle, J. DNA protocols for plants. In Molecular Techniques in Taxonomy; Springer: Berlin/Heidelberg, Germany, 1991; pp. 283–293. [Google Scholar]
- Zhang, Q.; Li, L.T.; VanBuren, R.; Liu, Y.L.; Yang, M.; Xu, L.M.; Bowers, J.E.; Zhong, C.H.; Han, Y.P.; Li, S.H. Optimization of linkage mapping strategy and construction of a high-density American lotus linkage map. BMC Genom. 2014, 15, 372. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Guo, J.B.; Zhou, X.J.; Wu, B.; Huang, L.; Luo, H.Y.; Chen, Y.N.; Chen, W.G.; Lei, Y.; Huang, Y. High-resolution mapping of a major and consensus quantitative trait locus for oil content to a ~0.8-Mb region on chromosome A08 in peanut (Arachis hypogaea L.). Theor. Appl. Genet. 2020, 133, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Yang, G.Q.; Chen, Y.M.; Li, D.Z.; Guo, Z.H. A comparison of different methods for preserving plant molecular materials and the effect of degraded DNA on ddRAD sequencing. Plant Divers. 2018, 40, 106–116. [Google Scholar] [CrossRef]
- Rochette, N.C.; Rivera-Colón, A.G.; Catchen, J.M. Stacks 2: Analytical methods for paired-end sequencing improve RADseq-based population genomics. Mol. Ecol. 2019, 28, 4737–4754. [Google Scholar] [CrossRef] [PubMed]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef] [PubMed]
- Hall, B.G. Building Phylogenetic Trees from Molecular Data with MEGA. Mol. Biol. Evol. 2013, 30, 1229–1235. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular Evolutionary Genetics Analysis Using Maximum Likelihood, Evolutionary Distance, and Maximum Parsimony Methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef]
- Patterson, N.; Price, A.L.; Reich, D. Population structure and eigenanalysis. PLoS Genet. 2006, 2, 2074–2093. [Google Scholar] [CrossRef]
- Blackburn, H.; Krehbiel, B.; Ericsson, S.; Wilson, C.; Caetano, A.; Paiva, S. A fine structure genetic analysis evaluating ecoregional adaptability of a Bos taurus breed (Hereford). PLoS ONE 2017, 12, e0176474. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef]
- Xia, Q.Y.; Guo, Y.R.; Zhang, Z.; Li, D.; Xuan, Z.L.; Li, Z.; Dai, F.Y.; Li, Y.R.; Cheng, D.J.; Li, R.Q.; et al. Complete Resequencing of 40 Genomes Reveals Domestication Events and Genes in Silkworm (Bombyx). Science 2009, 326, 433–436. [Google Scholar] [CrossRef]
- Derazmahalleh, S.M.; Mardi, M.; Zeinalabedini, M.; Kazemi, M.; Marashi, S.; Malekzadeh, S.; Khayam Nekoui, S.; Roodbar Shojaie, T.; Kolahi Zenoozi, S. The survey of genetic diversity and population structure analysis of iranian sweet pomegranate (Punica granatum L.) germplasm using SSR markers. Acta Hortic. 2013, 976, 271–276. [Google Scholar] [CrossRef]
- Sanchez-Quinto, F.; Botigue, L.R.; Civit, S.; Arenas, C.; Avila-Arcos, M.C.; Bustamante, C.D.; Comas, D.; Lalueza-Fox, C. North African Populations Carry the Signature of Admixture with Neandertals. PLoS ONE 2012, 7, e47765. [Google Scholar] [CrossRef]
- Maddison, W.P. Mesquite: A modular system for evolutionary analysis. Evolution 2008, 62, 1103–1118. [Google Scholar] [CrossRef]
- Lewis, P.O. A likelihood approach to inferring phylogeny from discrete morphological characters. Syst. Biol. 2001, 50, 913–925. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Code | Scientific Name | Clean Reads Number (M) | Q20 (%) | Homo SNPs | Hete SNPs | Total SNPs |
|---|---|---|---|---|---|---|
| A1 | Ribes fasciculatum | 7.68 | 98.06 | 18,169 | 264 | 18,433 |
| A2 | Ribes fasciculatum var. chinense | 10.56 | 98.08 | 23,189 | 322 | 23,511 |
| A3 | Ribes fasciculatum var. chinense | 14.26 | 98.01 | 25,609 | 577 | 26,186 |
| B1 | Ribes heterotrichum | 6.22 | 98 | 30,028 | 554 | 30,582 |
| B2 | Ribes komarovii | 17.2 | 97.89 | 33,558 | 797 | 34,355 |
| B3 | Ribes alpinum | 8.38 | 97.96 | 26,143 | 832 | 26,975 |
| B4 | Ribes giraldii var. polyanthum | 4.32 | 98.03 | 25,384 | 496 | 25,880 |
| B5 | Ribes pseudofasciculatum | 0.7 | 97.98 | 4982 | 66 | 50,48 |
| B6 | Ribes takare var. desmocarpum | 14.97 | 97.86 | 33,613 | 662 | 34,275 |
| B7 | Ribes kialanum | 6.95 | 98.18 | 29,730 | 627 | 30,357 |
| B8 | Ribes tenue | 9.34 | 97.82 | 25,164 | 523 | 25,687 |
| B9 | Ribes tenue | 12.47 | 98.1 | 33,332 | 843 | 34,175 |
| B10 | Ribes laurifolium var. yunnanense | 9.62 | 97.89 | 21,561 | 313 | 21,874 |
| B11 | Ribes laurifolium var. yunnanense | 10.65 | 98.02 | 23,175 | 368 | 23,543 |
| B12 | Ribes hunanense | 10.41 | 98.17 | 25,959 | 431 | 26,390 |
| B13 | Ribes laurifolium | 6.96 | 98.02 | 18,367 | 243 | 18,610 |
| B14 | Ribes davidii | 7.66 | 98.01 | 18,560 | 301 | 18,861 |
| B15 | Ribes davidii | 9.76 | 97.97 | 23,034 | 605 | 23,639 |
| C1 | Ribes aciculare | 10.83 | 97.84 | 127,633 | 14,111 | 141,744 |
| C2 | Ribes aciculare | 9.97 | 97.94 | 126,955 | 12,425 | 139,380 |
| C3 | Ribes burejense | 15.79 | 97.84 | 142,706 | 13,211 | 155,917 |
| C4 | Ribes stenocarpum | 2.07 | 98.01 | 141,709 | 10,065 | 151,774 |
| C5 | Ribes stenocarpum | 18.36 | 97.89 | 144,656 | 23,607 | 168,263 |
| D1 | Ribes fragrans | 11.8 | 98.12 | 45,922 | 1578 | 47,500 |
| D2 | Ribes procumbens | 19.07 | 98.07 | 38,844 | 2554 | 41,398 |
| D3 | Ribes nigrum | 8.58 | 98.14 | 36,776 | 1009 | 37,785 |
| D4 | Ribes procumbens | 12.39 | 98.14 | 42,188 | 1394 | 43,582 |
| D5 | Ribes nigrum | 22.03 | 97.98 | 47,442 | 1601 | 49,043 |
| E1 | Ribes griffithii | 8.52 | 98.18 | 40,367 | 849 | 41,216 |
| F1 | Ribes palczewskii | 9.33 | 98.05 | 42,553 | 1036 | 43,589 |
| F2 | Ribes tenue | 11.72 | 98.14 | 43,108 | 1399 | 44,507 |
| F3 | Ribes palczewskii | 9.52 | 98.06 | 41,924 | 1114 | 43,038 |
| F4 | Ribes palczewskii | 21.29 | 97.94 | 45,801 | 1590 | 47,391 |
| F5 | Ribes moupinense | 7.57 | 98.16 | 40,291 | 805 | 41,096 |
| F6 | Ribes setchuense | 12.04 | 97.87 | 42,788 | 1120 | 43,908 |
| F7 | Ribes setchuense | 15.75 | 97.89 | 43,072 | 1100 | 44,172 |
| F8 | Ribes setchuense | 14.1 | 97.94 | 44,005 | 1365 | 45,370 |
| F9 | Ribes setchuense | 6.49 | 97.72 | 36,222 | 741 | 36,963 |
| F10 | Ribes mandshuricum | 14.35 | 97.85 | 44,773 | 1078 | 45,851 |
| F11 | Ribes mandshuricum | 15.01 | 98.16 | 45,074 | 988 | 46,062 |
| F12 | Ribes longiracemosum | 9.85 | 98.07 | 36,479 | 631 | 37,110 |
| F13 | Ribes himalense | 11.21 | 97.85 | 35,869 | 801 | 36,670 |
| F14 | Ribes himalense | 6.02 | 98.03 | 39,633 | 834 | 40,467 |
| F15 | Ribes himalense | 15.92 | 98.07 | 45,382 | 1185 | 46,567 |
| F16 | Ribes himalense | 15.03 | 98.06 | 37,404 | 947 | 38,351 |
| F17 | Ribes atropurpureum | 12.42 | 98 | 43,556 | 1244 | 44,800 |
| F18 | Ribes atropurpureum | 13.9 | 98.06 | 44,863 | 1240 | 46,103 |
| F19 | Ribes atropurpureum | 10.93 | 98.02 | 42,549 | 1254 | 43,803 |
| F20 | Ribes altissimum | 6.96 | 98.14 | 34,220 | 721 | 34,941 |
| F21 | Ribes altissimum | 14.78 | 97.73 | 42,454 | 1232 | 43,686 |
| F22 | Ribes altissimum | 14.17 | 98.06 | 44,704 | 1276 | 45,980 |
| F23 | Ribes altissimum | 14.48 | 98.07 | 43,845 | 1201 | 45,046 |
| Average | 11.35 | 98.00 | 45,314 | 2399 | 47,713 | |
| Total | 590.36 | 2,335,324 | 116,130 | 2,451,454 |
| Code | Scientific Name | Geographic Location | Altitude (Meters) | Latitude | Longitude |
|---|---|---|---|---|---|
| A1 | Ribes fasciculatum | Nanzhao, Henan | 613 | 33°33′27″ | 111°58′04″ |
| A2 | Ribes fasciculatum var. chinense | Beijing | 88 | 40°04′32″ | 116°13′58″ |
| A3 | Ribes fasciculatum var. chinense | Dalian, Liaoning | 352 | 38°50′21″ | 121°26′58″ |
| B1 | Ribes heterotrichum | Fuhai, Xinjiang | 916 | 47°34′59″ | 88°44′36″ |
| B2 | Ribes komarovii | Lishan, Shanxi | 2358 | 35°24′58″ | 111°59′12″ |
| B3 | Ribes alpinum | Beijing | 88 | 40°04′32″ | 116°13′58″ |
| B4 | Ribes giraldii var. polyanthum | Dalian, Liaoning | 105 | 39°06′05″ | 122°00′03″ |
| B5 | Ribes pseudofasciculatum | Xining, Qinghai | 2332 | 36°56′37″ | 102°27′49″ |
| B6 | Ribes takare var. desmocarpum | Zhouqu, Gansu | 2578 | 33°43′50″ | 104°06′12″ |
| B7 | Ribes kialanum | Lijiang, Yunnan | 3812 | 27°01′34″ | 100°10′43″ |
| B8 | Ribes tenue | Hezheng, Gansu | 2684 | 35°15′14″ | 103°14′27″ |
| B9 | Ribes tenue | Xining, Qinghai | 2518 | 35°47′37″ | 102°40′37″ |
| B10 | Ribes laurifolium var. yunnanense | Leshan, Sichuan | 3099 | 29°34′26″ | 103°21′23″ |
| B11 | Ribes laurifolium var. yunnanense | Leshan, Sichuan | 3099 | 29°34′26″ | 103°21′23″ |
| B12 | Ribes hunanense | Chengbu, Hunan | 1669 | 26°09′39″ | 110°11′34″ |
| B13 | Ribes laurifolium | Leshan, Sichuan | 3099 | 29°34′26″ | 103°21′23″ |
| B14 | Ribes davidii | Nanchuan, Chongqing | 726 | 29°03′19″ | 107°08′04″ |
| B15 | Ribes davidii | Nanchuan, Chongqing | 726 | 29°03′19″ | 107°08′04″ |
| C1 | Ribes aciculare | Burqin, Xinjiang | 1240 | 48°12′29″ | 87°37′10″ |
| C2 | Ribes aciculare | Fuhai, Xinjiang | 916 | 47°34′59″ | 88°44′36″ |
| C3 | Ribes burejense | Nanzhao, Henan | 1349 | 33°32′04″ | 111°57′28″ |
| C4 | Ribes stenocarpum | Xining, Qinghai | 2332 | 36°56′37″ | 102°27′49″ |
| C5 | Ribes stenocarpum | Guyuan, Ningxia | 2928 | 35°21′51″ | 106°19′54″ |
| D1 | Ribes fragrans | Moerdaoga, Inner Mongolia | 1256 | 51°22′43″ | 120°49′48″ |
| D2 | Ribes procumbens | Daxing’anling, Heilongjiang | 1001 | 52°20′28″ | 124°42′12″ |
| D3 | Ribes nigrum | Hemu, Xinjiang | 1389 | 48°34′27″ | 87°21′32″ |
| D4 | Ribes procumbens | Xiniuerhe, Inner Mongolia | 707 | 51°15′51″ | 120°47′29″ |
| D5 | Ribes nigrum | Ergun, Inner Mongolia | 1404 | 51°09′19″ | 120°54′37″ |
| E1 | Ribes griffithii | Lijiang, Yunnan | 3812 | 27°01′34″ | 100°10′43″ |
| F1 | Ribes palczewskii | Hemu, Xinjiang | 1099 | 48°34′13″ | 87°25′43″ |
| F2 | Ribes tenue | Moerdaoga, Inner Mongolia | 869 | 51°09′19″ | 120°54′37″ |
| F3 | Ribes palczewskii | Xiniuerhe, Inner Mongolia | 533 | 51°52′41″ | 120°37′56″ |
| F4 | Ribes palczewskii | Ergun, Inner Mongolia | 1404 | 51°09′19″ | 120°54′37″ |
| F5 | Ribes moupinense | Kunming, Yunnan | 3419 | 26°04′46″ | 102°50′05″ |
| F6 | Ribes setchuense | Zhouqu, Gansu | 2705 | 33°39′45″ | 104°09′41″ |
| F7 | Ribes setchuense | Zhouqu, Gansu | 2705 | 33°39′45″ | 104°09′41″ |
| F8 | Ribes setchuense | Hezheng, Gansu | 2684 | 35°15′14″ | 103°14′27″ |
| F9 | Ribes setchuense | Hezheng, Gansu | 2684 | 35°15′14″ | 103°14′27″ |
| F10 | Ribes mandshuricum | Nanzhao, Henan | 1372 | 33°21′11″ | 111°57′17″ |
| F11 | Ribes mandshuricum | Guyuan, Ningxia | 2928 | 35°21′51″ | 106°19′54″ |
| F12 | Ribes longiracemosum | Baoji, Shaanxi | 568 | 34°21′22″ | 107°18′21″ |
| F13 | Ribes himalense | Hezheng, Gansu | 2684 | 35°15′14″ | 103°14′27″ |
| F14 | Ribes himalense | Xining, Qinghai | 2332 | 36°56′37″ | 102°27′49″ |
| F15 | Ribes himalense | Xining, Qinghai | 2229 | 35°48′10″ | 102°40′53″ |
| F16 | Ribes himalense | Yinchuan, Ningxia | 3556 | 38°37′54″ | 106°03′35″ |
| F17 | Ribes atropurpureum | Burqin, Xinjiang | 1245 | 48°37′05″ | 87°30′03″ |
| F18 | Ribes atropurpureum | Burqin, Xinjiang | 1245 | 48°37′05″ | 87°30′03″ |
| F19 | Ribes atropurpureum | Fuhai, Xinjiang | 1489 | 47°52′05″ | 88°56′56″ |
| F20 | Ribes altissimum | Hemu, Xinjiang | 1446 | 48°33′07″ | 87°28′24″ |
| F21 | Ribes altissimum | Hemu, Xinjiang | 1446 | 48°33′07″ | 87°28′24″ |
| F22 | Ribes altissimum | Hemu, Xinjiang | 1099 | 48°34′13″ | 87°25′43″ |
| F23 | Ribes altissimum | Hemu, Xinjiang | 1099 | 48°34′13″ | 87°25′43″ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, B.; Yu, Z.; Xu, Z.; Zheng, B. A Phylogenetic and Morphological Evolution Study of Ribes L. in China Using RAD-Seq. Plants 2023, 12, 829. https://doi.org/10.3390/plants12040829
Zhang B, Yu Z, Xu Z, Zheng B. A Phylogenetic and Morphological Evolution Study of Ribes L. in China Using RAD-Seq. Plants. 2023; 12(4):829. https://doi.org/10.3390/plants12040829
Chicago/Turabian StyleZhang, Baoshan, Ziyang Yu, Zhichao Xu, and Baojiang Zheng. 2023. "A Phylogenetic and Morphological Evolution Study of Ribes L. in China Using RAD-Seq" Plants 12, no. 4: 829. https://doi.org/10.3390/plants12040829
APA StyleZhang, B., Yu, Z., Xu, Z., & Zheng, B. (2023). A Phylogenetic and Morphological Evolution Study of Ribes L. in China Using RAD-Seq. Plants, 12(4), 829. https://doi.org/10.3390/plants12040829