
Genome-Wide Development of Polymorphic SNP Markers and Evaluation of Genetic Diversity of Litchi (Litchi chinensis Sonn.)
Abstract
:1. Introduction
2. Results
2.1. Development of Polymorphic SNP Markers for Litchi
2.2. Genetic Diversity Analysis
2.3. Genetic Identification of the Litchi Collection
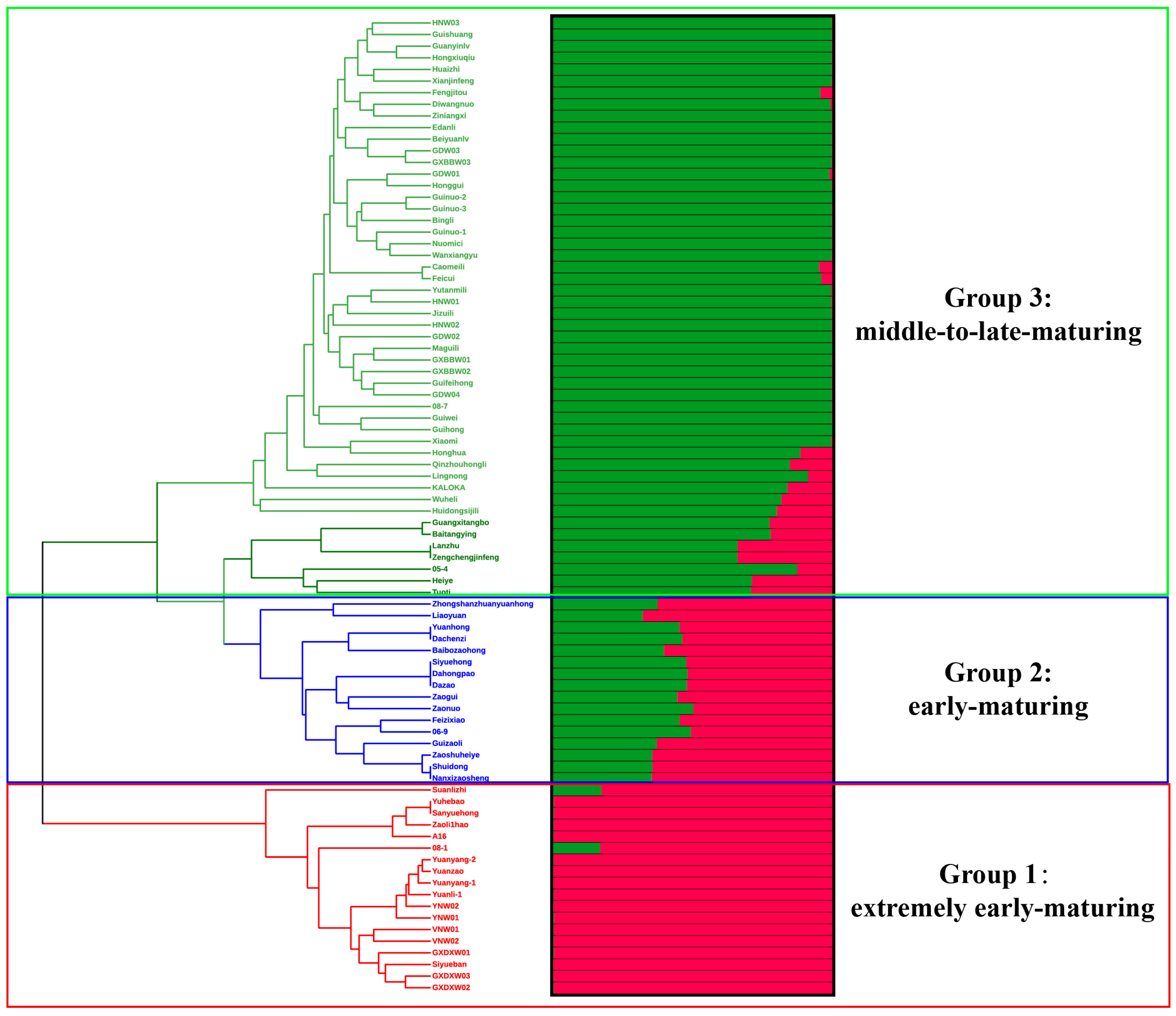
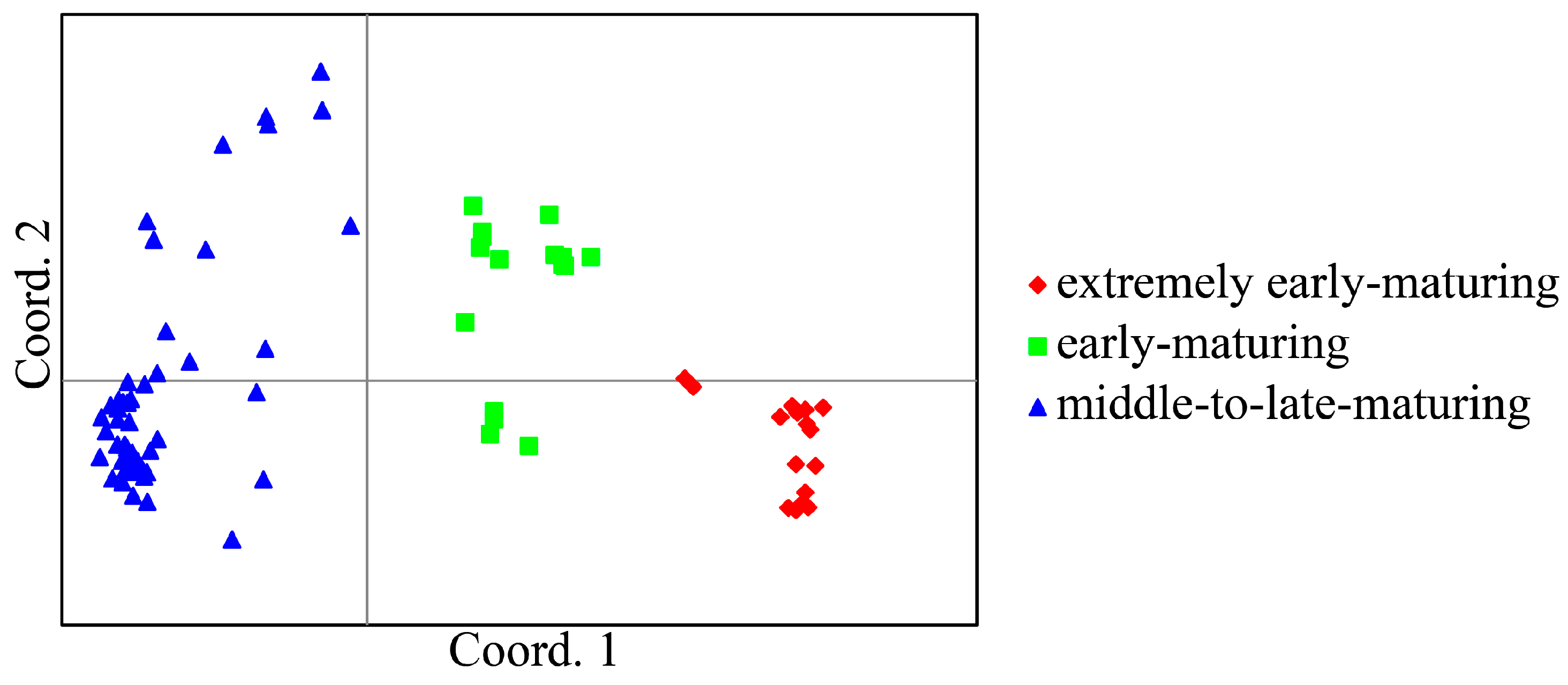
2.4. Phylogenetic and Population Structure Analyses of the Litchi Collection
3. Discussion
3.1. Genetic Diversity in the Studied Litchi Collection
3.2. Genetic Identification of the Studied Litchi Collection
3.3. Genetic Relationships and Population Structure among Litchi Accessions
4. Materials and Methods
4.1. Plant Material and DNA Extraction
4.2. SNP Selection and Genotyping
4.3. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Hu, G.B.; Feng, J.T.; Xiang, X.; Wang, J.B.; Salojärvi, J.; Liu, C.M.; Wu, Z.X.; Zhang, J.S.; Liang, X.M.; Jiang, Z.D.; et al. Two divergent haplotypes from a highly heterozygous lychee genome suggest independent domestication events for early and late-maturing cultivars. Nat. Genet. 2022, 54, 73–83. [Google Scholar] [CrossRef]
- Wu, S.X. Encyclopedia of China Fruits: Litchi; China Forestry Press: Beijing, China, 1998. [Google Scholar]
- Huang, X.M.; Subhadrabandhu, S.; Mitra, S.K.; Ben-Arie, R.; Stern, R.A. Origin, history, production and processing. In Litchi and Longan; Menzel, C.M., Waite, G.K., Eds.; Comwell Press: Trowbridge, UK, 2005; pp. 1–23. [Google Scholar]
- Qi, W.E.; Chen, H.B.; Li, J.X. Status, trend and countermeasures of development of litchi Industry in the mainland of China in 2022. Guangdong Agric. Sci. 2022, 50, 147–155. [Google Scholar]
- Wen, Y.J.; Ou, L.X.; Shi, F.C.; Yan, Q.; Cai, C.H.; Jiang, Y.H.; Liu, H.L.; Chen, J.Z. Conservation status and innovative utilization of litchi resources in the national litchi and banana germplasm repository (Guangzhou). J. Plant Genet. Resour. 2023, 24, 1205–1214. [Google Scholar]
- Li, J.G. The Litchi; China Agriculture Press: Beijing, China, 2008. [Google Scholar]
- Liu, W.; Xiao, Z.; Fan, C.; Jiang, N.; Meng, X.; Xiang, X. Cloning and characterization of a flavonol synthase gene from Litchi chinensis and its variation among litchi cultivars with different fruit maturation periods. Front. Plant Sci. 2018, 9, 567. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Wang, L.; Xiang, X.; Bi, F.; Zhang, Z. Morphological, chemical, and biosynthetic changes in pericarp waxes in response to the browning of litchi fruit during storage. Postharvest Biol. Technol. 2022, 191, 111968. [Google Scholar] [CrossRef]
- Huang, H.; Liu, H.; Wang, L.; Xiang, X. Cuticular wax metabolism responses to atmospheric water stress on the exocarp surface of litchi fruit after harvest. Food Chem. 2023, 414, 135704. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Fu, D.; Jiang, Y.; Liu, H.; Shi, F.; Wen, Y.; Cai, C.; Chen, J.; Ou, L.; Yan, Q. Storability and linear regression models of pericarp browning and decay in fifty litchi (Litchi chinensis Sonn.) cultivars at room temperature storage. Foods 2023, 12, 1725. [Google Scholar] [CrossRef]
- Chen, H.; Ou, L.; Li, J.; Su, Z.; Yang, S.; Wu, Z.; Hu, Z. Fruit scientific research in New China in the past 70 years: Litchi. J. Fruit Sci. 2019, 36, 1399–1413. [Google Scholar]
- Andrijanić, Z.; Nazzicari, N.; Šarčević, H.; Sudarić, A.; Annicchiarico, P.; Pejić, I. Genetic diversity and population structure of European soybean germplasm revealed by single nucleotide polymorphism. Plants 2023, 12, 1837. [Google Scholar] [CrossRef]
- Guimarães, J.B.; Nunes, C.; Pereira, G.; Gomes, A.; Nhantumbo, N.; Cabrita, P.; Matos, J.; Simões, F.; Veloso, M.M. Genetic diversity and population structure of cowpea (Vigna unguiculata (L.) Walp.) landraces from portugal and mozambique. Plants 2023, 12, 846. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, Y.; Zong, X.; Teng, C.; Hou, W.; Li, P.; Du, D. Genetic diversity of global faba bean germplasm resources based on the 130K TNGS genotyping platform. Agronomy 2023, 13, 811. [Google Scholar] [CrossRef]
- Kimwemwe, P.K.; Bukomarhe, C.B.; Mamati, E.G.; Githiri, S.M.; Civava, R.M.; Mignouna, J.; Kimani, W.; Fofana, M. Population structure and genetic diversity of rice (Oryza sativa L.) germplasm from the Democratic Republic of Congo (DRC) using DArTseq-derived single nucleotide polymorphism (SNP). Agronomy 2023, 13, 1906. [Google Scholar] [CrossRef]
- Fang, J.; Zhu, X.; Wang, C.; Shangguan, L. Applications of DNA technologies in agriculture. Curr. Genom. 2016, 17, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, D.N.; Dillon, N.; Bally, I.; Groh, A.; Rahaman, J.; Warschefsky, E.; Freeman, B.; Innes, D.; Chambers, A.H. Estimation of genetic diversity and relatedness in a mango germplasm collection using SNP markers and a simplified visual analysis method. Sci. Hortic. 2019, 252, 156–168. [Google Scholar] [CrossRef]
- Kim, M.; Jung, J.-K.; Shim, E.-J.; Chung, S.-M.; Park, Y.; Lee, G.P.; Sim, S.-C. Genome-wide SNP discovery and core marker sets for DNA barcoding and variety identification in commercial tomato cultivars. Sci. Hortic. 2021, 276, 109734. [Google Scholar] [CrossRef]
- Kuhn, D.N.; Livingstone, D.S.; Richards, J.H.; Manosalva, P.; Van den Berg, N.; Chambers, A.H. Application of genomic tools to avocado (Persea americana) breeding: SNP discovery for genotyping and germplasm characterization. Sci. Hortic. 2019, 246, 1–11. [Google Scholar] [CrossRef]
- Wang, D.; Zhou, Q.; Le, L.; Fu, F.; Wang, G.; Cao, F.; Yang, X. Molecular characterization and genetic diversity of ginkgo (Ginkgo biloba L.) based on insertions and deletions (InDel) Markers. Plants 2023, 12, 2567. [Google Scholar] [CrossRef]
- Akpertey, A.; Padi, F.K.; Meinhardt, L.; Zhang, D. Relationship between genetic distance based on single nucleotide polymorphism markers and hybrid performance in Robusta coffee (Coffea canephora). Plant Breed. 2022, 141, 286–300. [Google Scholar] [CrossRef]
- Catarcione, G.; Paolacci, A.R.; Alicandri, E.; Gramiccia, E.; Taviani, P.; Rea, R.; Costanza, M.T.; De Lorenzis, G.; Puccio, G.; Mercati, F.; et al. Genetic diversity and population structure of common bean (Phaseolus vulgaris L.) landraces in the Lazio region of Italy. Plants 2023, 12, 744. [Google Scholar] [CrossRef]
- Imai, A.; Kuniga, T. Genome-wide estimation of pedigree haplotypes offers genetic compositions and founder origins in Japanese citrus breeding materials. Sci. Hortic. 2021, 282, 110000. [Google Scholar] [CrossRef]
- Ding, X.D.; Lv, L.X.; Chen, X.J.; Guan, X. Identifying litchi cultivars and evaluating their genetic relationships by RAPD markers. J. Trop. Subtrop. Bot. 2000, 8, 49–54. [Google Scholar]
- Chen, Y.Y.; Deng, S.S.; Zhang, X.; Wei, S.X.; Gao, A.P.; Wang, J.B.; Zhou, B. RAPD analysis of genetic relationship among partial litchi germplasms in Hainan Island. Acta Hortic. Sin. 2004, 31, 224–226. [Google Scholar]
- Wang, J.B.; Deng, S.S.; Liu, Z.Y.; Liu, L.Z.; Du, Z.J.; Xu, B.Y.; Chen, Y.Y. RAPD analysis on main cultivars of litchi (Litchi chinensis Sonn.) in Hainan. J. Agric. Biotechnol. 2006, 14, 391–396. [Google Scholar]
- Liu, C.M.; Mei, M.T. Classification of lychee cultivars with RAPD analysis. Acta Hortic. 2005, 665, 149–159. [Google Scholar] [CrossRef]
- Yi, G.J.; Huo, H.Q.; Chen, D.C.; Huang, Z.R.; Cai, C.H.; Qiu, Y.P. Studies on genetic relationship among litchi varieties by using AFLP. Acta Hortic. Sin. 2003, 30, 399–403. [Google Scholar]
- Peng, H.X.; Cao, H.Q.; Zhu, J.H.; Liao, H.H.; Huang, F.Z.; Li, J.Z.; Liang, W. Studies on the application of AFLP molecular markers on genetic diversity and classification of good and rare litchi resources in Guangxi. Southwest China J. Agric. Sci. 2006, 19, 108–111. [Google Scholar]
- Zan, F.G.; Wu, Z.D.; Zeng, Q.; Zhang, H.Y.; Li, M.F.; Zheng, X.Q. Genetic diversity analysis of litchi germplasm by SRAP markers. Mol. Plant Breed. 2009, 7, 562–568. [Google Scholar]
- Wei, S.X.; Chen, Y.Y.; Xie, Z.S.; Zhu, M.; Luo, S.R. ISSR analysis of parts of lychee germplasm in Hainan province. Chin. J. Trop. Crops 2006, 27, 51–55. [Google Scholar]
- Yao, Q.R.; Zhao, C.Z.; Wang, W.Q. Analysis of genetic relationship of Hainan litchi germplasm resources by SSR marker. Bull. Bot. Res. 2009, 29, 628–632. [Google Scholar]
- Fu, J.X. Development and Application of Microsatellite Markers in Litchi, Longan and Selection of Excellent Individuals in Litchi Populations. Ph.D. Thesis, South China Agricultural University, Guangzhou, China, 2010. [Google Scholar]
- Xiang, X.; Ou, L.X.; Chen, H.B.; Sun, Q.M.; Chen, J.Z.; Cai, C.H.; Bai, L.J.; Zhao, J.S. EST-SSR analysis of genetic diversity in 96 litchi (Litchi chinensis Sonn.) germplasm resources in China. Genom. Appl. Biol. 2010, 29, 1082–1092. [Google Scholar]
- Rafalski, A. Applications of single nucleotide polymorphisms in crop genetics. Curr. Opin. Plant Biol. 2002, 5, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Brumfield, R.T.; Beerli, P.; Nickerson, D.A.; Edwards, S.V. The utility of single nucleotide polymorphisms in inferences of population history. Trends Ecol. Evol. 2003, 18, 249–256. [Google Scholar] [CrossRef]
- Cabezas, J.; Ibanez, J.; Lijavetzky, D.; Velez, D.; Bravo, G.; Rodriguez, V.; Carreno, I.; Jermakow, A.; Carreno, J.; Ruiz-Garcia, L.; et al. A 48 SNP set for grapevine cultivar identification. BMC Plant Biol. 2011, 11, 153. [Google Scholar] [CrossRef]
- Ghaffari, S.; Hasnaoui, N.; Zinelabidine, L.; Ferchichi, A.; Martínez-Zapater, J.; Ibáñez, J. Genetic diversity and parentage of Tunisian wild and cultivated grapevines (Vitis vinifera L.) as revealed by single nucleotide polymorphism (SNP) markers. Tree Genet. Genomes 2014, 10, 1103–1112. [Google Scholar] [CrossRef]
- Wu, B.; Zhong, G.Y.; Yue, J.Q.; Yang, R.T.; Li, C.; Li, Y.J.; Zhong, Y.; Wang, X.; Jiang, B.; Zeng, J.W.; et al. Identification of pummelo cultivars by using a panel of 25 selected SNPs and 12 DNA segments. PLoS ONE 2014, 9, e94506. [Google Scholar] [CrossRef] [PubMed]
- Ophir, R.; Sherman, A.; Rubinstein, M.; Eshed, R.; Sharabi Schwager, M.; Harel-Beja, R.; Bar-Ya’akov, I.; Holland, D. Single nucleotide polymorphism markers from de-novo assembly of the pomegranate transcriptome reveal germplasm genetic diversity. PLoS ONE 2014, 9, e88998. [Google Scholar] [CrossRef] [PubMed]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—An update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef]
- Tieman, D.; Zhu, G.; Resende, M.F.R.; Lin, T.; Nguyen, C.; Bies, D.; Rambla, J.L.; Beltran, K.S.O.; Taylor, M.; Zhang, B.; et al. A chemical genetic roadmap to improved tomato flavor. Science 2017, 355, 391–394. [Google Scholar] [CrossRef]
- Li, N.; He, Q.; Wang, J.; Wang, B.; Zhao, J.; Huang, S.; Yang, T.; Tang, Y.; Yang, S.; Aisimutuola, P.; et al. Super-pangenome analyses highlight genomic diversity and structural variation across wild and cultivated tomato species. Nat. Genet. 2023, 55, 852–860. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]
- Liu, W.; Xiao, Z.; Bao, X.; Yang, X.; Fang, J.; Xiang, X. Identifying litchi (Litchi chinensis Sonn.) cultivars and their genetic relationships using single nucleotide polymorphism (SNP) markers. PLoS ONE 2015, 10, e0135390. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, E.; Ferreira, C.; da Silva Santos, V.; de Jesus, O.; Oliveira, G.; da Silva, M. Potential of SNP markers for the characterization of Brazilian cassava germplasm. Theor. Appl. Genet. 2014, 127, 1423–1440. [Google Scholar] [CrossRef] [PubMed]
- Park, G.; Sim, S.-C.; Jung, J.-K.; Shim, E.-J.; Chung, S.-M.; Lee, G.P.; Park, Y. Development of genome-wide single nucleotide polymorphism markers for variety identification of F1 hybrids in cucumber (Cucumis sativus L.). Sci. Hortic. 2021, 285, 110173. [Google Scholar] [CrossRef]
- Chen, P.; Liu, J.; Wang, L.; Liu, X.; Guo, L.; Li, F.; Li, D.; Chai, L.; Xu, Q.; Li, X.; et al. Genetic background of the citrus landrace ‘Huarongdao Zhoupigan’ revealed by simple sequence repeat marker and genomic analyses. Sci. Hortic. 2021, 289, 110456. [Google Scholar] [CrossRef]
- Geukens, E.; Haegeman, A.; Van Meulder, J.; Van Laere, K.; Smolders, E.; Ruttink, T.; Leus, L. Exploring genetic diversity in an Ilex crenata breeding germplasm. Horticulturae 2023, 9, 485. [Google Scholar] [CrossRef]
- Sun, Q.; Ma, S.; Ma, W.; Zhao, J.; Fang, J.; Yang, X.; Xiang, X. Identification and genetic diversity analysis of two F1 hybrid populations of litchi (Litchi chinensis Sonn.) using EST-SSR markers. Mol. Plant Breed. 2014, 12, 87–95. [Google Scholar]
- Murray, M.G.; Thompson, W.F. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 1980, 8, 4321–4326. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef]
- Rosenberg, N.A. Distruct: A program for the graphical display of population structure. Mol. Ecol. Notes 2004, 4, 137–138. [Google Scholar] [CrossRef]
- Excoffier, L.; Laval, G.; Schneider, S. Arlequin (version 3.0): An integrated software package for population genetics data analysis. Evol. Bioinform. 2005, 1, 47. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number of Accessions | He | Ho | |
|---|---|---|---|
| Overall | 84 | 0.364 | 0.267 |
| Cultivars | 55 | 0.358 | 0.278 |
| (1) Old cultivars | 37 | 0.371 | 0.322 |
| (2) Modern cultivars | 18 | 0.259 | 0.187 |
| Hybrids | 12 | 0.348 | 0.275 |
| Wild accessions | 17 | 0.365 | 0.127 |
| Source of Variation | Origin | Fruit Maturation Period | ||||
|---|---|---|---|---|---|---|
| df | SS | % | df | SS | % | |
| Among Pops | 3 | 169.666 | 5% | 2 | 1118.304 | 51% |
| Among Indiv | 80 | 1807.197 | 33% | 81 | 858.559 | 0% |
| Within Indiv | 84 | 930.500 | 63% | 84 | 930.500 | 49% |
| Total | 167 | 2907.363 | 100% | 167 | 2907.363 | 100% |
| Origin | Old Cultivars | Modern Cultivars | Hybrids | Wild Accessions |
|---|---|---|---|---|
| Old cultivars | - | |||
| Modern cultivars | 0.118 | - | ||
| Hybrids | 0.035 | 0.031 | - | |
| Wild accessions | 0.106 | 0.004 | 0.027 | - |
| Fruit maturation period | EEM group | EM group | MLM group | |
| EEM group | - | |||
| EM group | 0.283 | - | ||
| MLM group | 0.691 | 0.348 | - |
| No. | Accession Name | Status | Geographic Origin | Maturation Period | No. | Accession Name | Status | Geographic Origin | Maturation Period |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Sanyuehong | old cultivar | GD | EEM | 43 | Guanyinlv | modern cultivar | GD | MLM |
| 2 | Dazao | old cultivar | GD | EM | 44 | Beiyuanlv | modern cultivar | GD | MLM |
| 3 | Zhongshanzhuanyuanhong | old cultivar | GD | EM | 45 | Bingli | modern cultivar | GD | MLM |
| 4 | Shuidong | old cultivar | GD | EM | 46 | Lingnong | modern cultivar | GD | MLM |
| 5 | Feizixiao | old cultivar | GD | EM | 47 | Guishuang | modern cultivar | GD | MLM |
| 6 | Guiwei | old cultivar | GD | MLM | 48 | Maguili | modern cultivar | GD | MLM |
| 7 | Nuomici | old cultivar | GD | MLM | 49 | Hongxiuqiu | modern cultivar | GD | MLM |
| 8 | Huaizhi | old cultivar | GD | MLM | 50 | Fengjitou | modern cultivar | GD | MLM |
| 9 | Heiye | old cultivar | GD | MLM | 51 | Wanxiangyu | modern cultivar | GD | MLM |
| 10 | Zengchengjinfeng | old cultivar | GD | MLM | 52 | Diwangnuo | modern cultivar | GD | MLM |
| 11 | Huidongsijili | old cultivar | GD | MLM | 53 | Caomeili | modern cultivar | GX | MLM |
| 12 | Baitangying | old cultivar | GD | MLM | 54 | Guifeihong | modern cultivar | GX | MLM |
| 13 | Yuhebao | old cultivar | GX | EEM | 55 | Yutanmili | modern cultivar | HN | MLM |
| 14 | Siyueban | old cultivar | GX | EEM | 56 | A16 | hybrid | - | EEM |
| 15 | Siyuehong | old cultivar | GX | EM | 57 | 08-1 | hybrid | - | EEM |
| 16 | Zaoshuheiye | old cultivar | GX | EM | 58 | 06-9 | hybrid | - | EM |
| 17 | Guangxitangbo | old cultivar | GX | MLM | 59 | Zaogui | hybrid | - | EM |
| 18 | Qinzhouhongli | old cultivar | GX | MLM | 60 | Zaonuo | hybrid | - | EM |
| 19 | Jizuili | old cultivar | GX | MLM | 61 | Guinuo-1 | hybrid | - | MLM |
| 20 | Yuanhong | old cultivar | FJ | EM | 62 | Guinuo-2 | hybrid | - | MLM |
| 21 | Baibozaohong | old cultivar | FJ | EM | 63 | Guinuo-3 | hybrid | - | MLM |
| 22 | Dachenzi | old cultivar | FJ | EM | 64 | Honggui | hybrid | - | MLM |
| 23 | Lanzhu | old cultivar | FJ | MLM | 65 | Guihong | hybrid | - | MLM |
| 24 | Edanli | old cultivar | HN | MLM | 66 | 05-4 | hybrid | - | MLM |
| 25 | Wuheli | old cultivar | HN | MLM | 67 | 08-7 | hybrid | - | MLM |
| 26 | Ziniangxi | old cultivar | HN | MLM | 68 | YNW01 | wild accession | YN | EEM |
| 27 | Suanlizhi | old cultivar | SC | EEM | 69 | YNW02 | wild accession | YN | EEM |
| 28 | Dahongpao | old cultivar | SC | EM | 70 | GXDXW01 | wild accession | DX-GX | EEM |
| 29 | Tuoti | old cultivar | SC | MLM | 71 | GXDXW02 | wild accession | DX-GX | EEM |
| 30 | Yuanli-1 | old cultivar | YN | EEM | 72 | GXDXW03 | wild accession | DX-GX | EEM |
| 31 | Yuanzao | old cultivar | YN | EEM | 73 | GXBBW01 | wild accession | BB-GX | MLM |
| 32 | Yuanyang-1 | old cultivar | YN | EEM | 74 | GXBBW02 | wild accession | BB-GX | MLM |
| 33 | Yuanyang-2 | old cultivar | YN | EEM | 75 | GXBBW03 | wild accession | BB-GX | MLM |
| 34 | Nanxizaosheng | old cultivar | TW | EM | 76 | GDW01 | wild accession | GD | MLM |
| 38 | Xiaomi | old cultivar | VN | MLM | 77 | GDW02 | wild accession | GD | MLM |
| 39 | Honghua | old cultivar | VN | MLM | 78 | GDW03 | wild accession | GD | MLM |
| 40 | KALOKA | old cultivar | TL | MLM | 79 | GDW4 | wild accession | GD | MLM |
| 35 | Zaoli1hao | modern cultivar | GD | EEM | 80 | HNW01 | wild accession | HN | MLM |
| 36 | Guizaoli | modern cultivar | GX | EM | 81 | HNW02 | wild accession | HN | MLM |
| 37 | Liaoyuan | modern cultivar | YN | EM | 82 | HNW03 | wild accession | HN | MLM |
| 41 | Xinjinfeng | modern cultivar | GD | MLM | 83 | VNW01 | wild accession | VN | EEM |
| 42 | Feicui | modern cultivar | GD | MLM | 84 | VNW02 | wild accession | VN | EEM |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, W.; Xiao, Z.; Jiang, N.; Fan, C.; Xiang, X. Genome-Wide Development of Polymorphic SNP Markers and Evaluation of Genetic Diversity of Litchi (Litchi chinensis Sonn.). Plants 2023, 12, 3949. https://doi.org/10.3390/plants12233949
Liu W, Xiao Z, Jiang N, Fan C, Xiang X. Genome-Wide Development of Polymorphic SNP Markers and Evaluation of Genetic Diversity of Litchi (Litchi chinensis Sonn.). Plants. 2023; 12(23):3949. https://doi.org/10.3390/plants12233949
Chicago/Turabian StyleLiu, Wei, Zhidan Xiao, Nonghui Jiang, Chao Fan, and Xu Xiang. 2023. "Genome-Wide Development of Polymorphic SNP Markers and Evaluation of Genetic Diversity of Litchi (Litchi chinensis Sonn.)" Plants 12, no. 23: 3949. https://doi.org/10.3390/plants12233949
APA StyleLiu, W., Xiao, Z., Jiang, N., Fan, C., & Xiang, X. (2023). Genome-Wide Development of Polymorphic SNP Markers and Evaluation of Genetic Diversity of Litchi (Litchi chinensis Sonn.). Plants, 12(23), 3949. https://doi.org/10.3390/plants12233949