
Analysis of Wheat Virome in Korea Using Illumina and Oxford Nanopore Sequencing Platforms

Abstract
:1. Introduction
2. Results
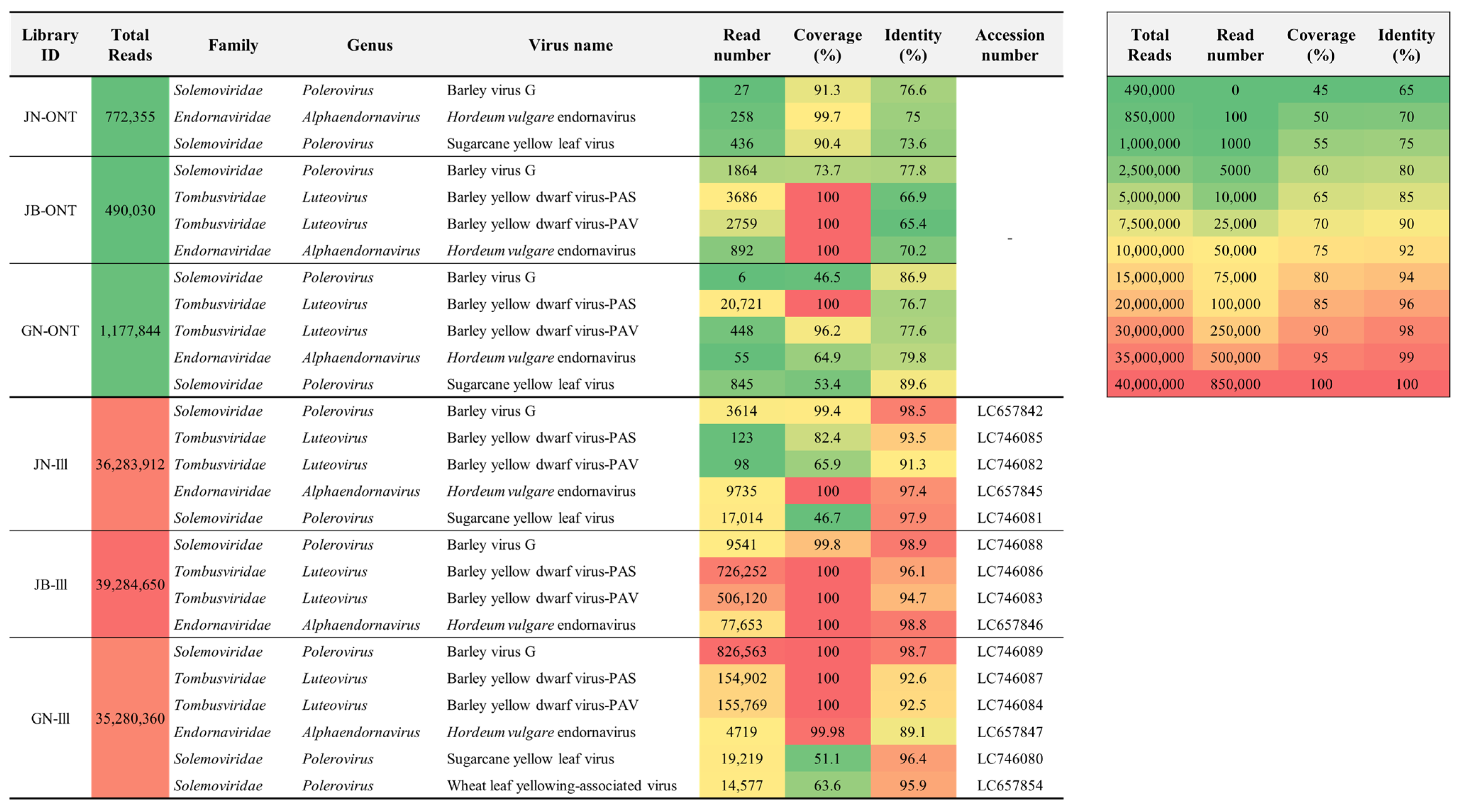
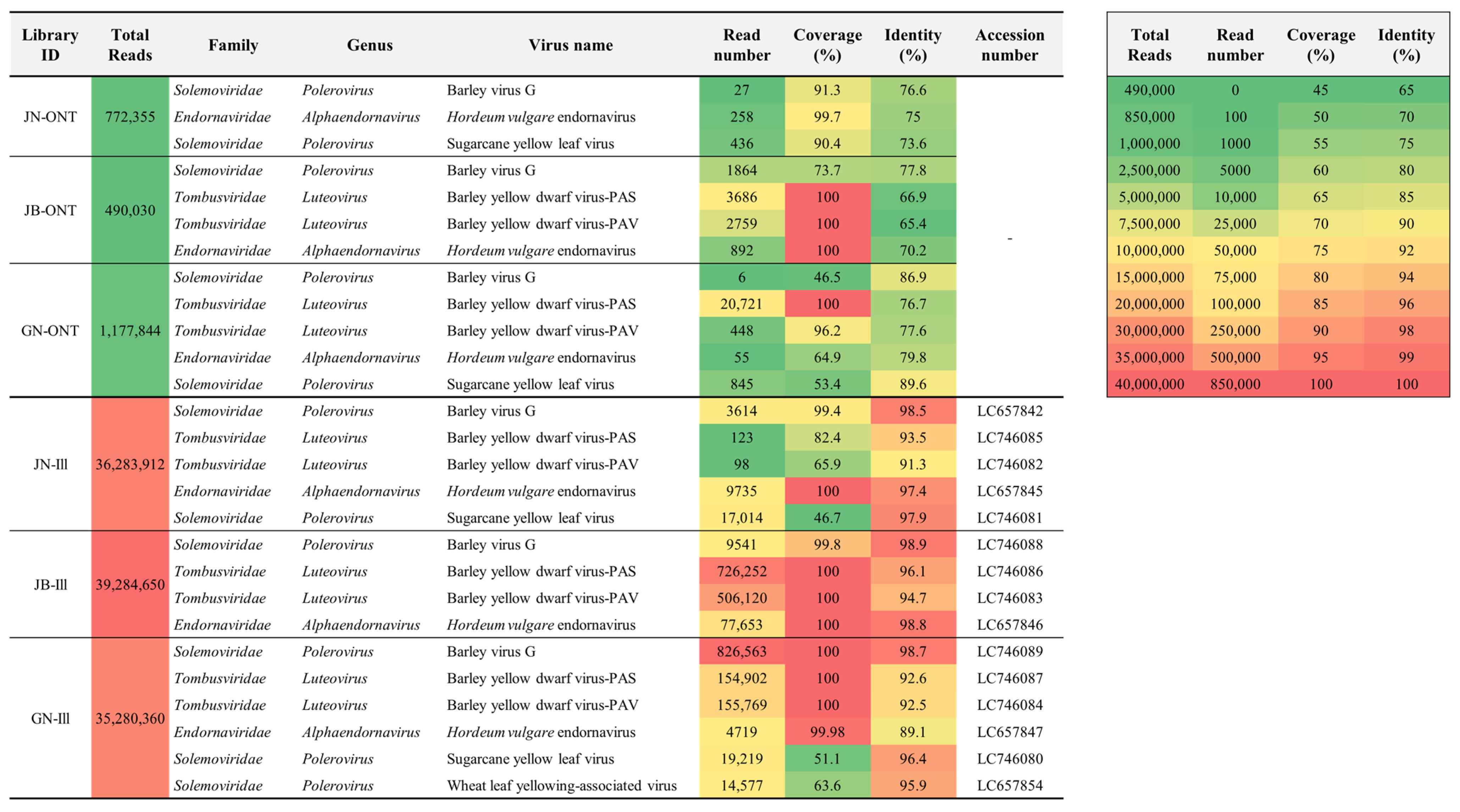
2.1. Virus Identification by ONT Sequencing
2.2. Virus Identification by Illumina Sequencing
2.3. Genome Assembly
2.4. Phylogenetic Analyses of Identified Viruses
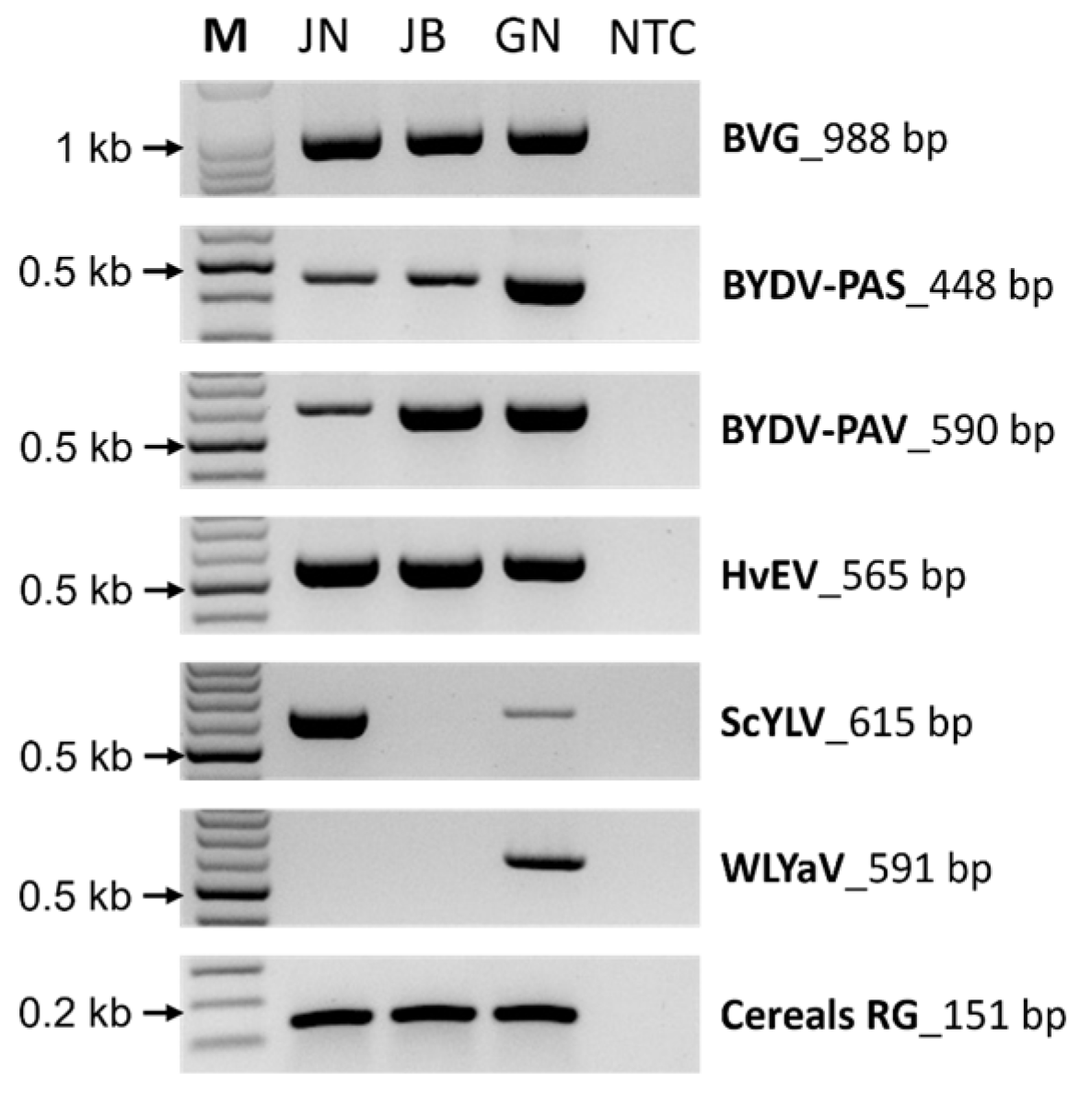
2.5. Validation of the Identified Virus by RT-PCR
3. Discussion
4. Materials and Methods
4.1. Sample Preparation and RNA Extraction
4.2. Library Preparation for ONT Platform and Virus Identification
4.3. Library Preparation for Illumina Platform and Virus Identification
4.4. Genome Mapping of Viral Sequences
4.5. Phylogenetic Analysis
4.6. Confirmation of the Identified Viruses by RT-PCR
4.7. Data Availability
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yang, W.; Liu, D.; Li, J.; Zhang, L.; Wei, H.; Hu, X.; Zheng, Y.; He, Z.; Zou, Y. Synthetic hexaploid wheat and its utilization for wheat genetic improvement in China. J. Genet. Genom. 2009, 36, 539–546. [Google Scholar] [CrossRef]
- Weaver, G.L. A miller’s perspective on the impact of health claims. Nutr. Today 2001, 36, 115–118. [Google Scholar] [CrossRef]
- Jo, Y.J.; You, T.Y.; Shin, T.W.; Sung, H.J.; Lee, H.W.; Kwak, J.E.; Kang, T.-S.; Lee, J.; Jeong, H.S. Quality characteristics of noodle made from domestic early maturity and high yield wheat cultivars. J. Korean Soc. Food Sci. Nutr. 2022, 51, 170–176. [Google Scholar] [CrossRef]
- Choi, Y.-S.; Lee, J.-K.; Choi, Y.-H.; Kim, Y.-H.; Kang, C.-S.; Shin, M. Quality characteristics of wheat flours from new released Iksan370 with long spike and domestic wheat cultivars. Korean J. Food Cook. Sci. 2015, 31, 551–556. [Google Scholar] [CrossRef] [Green Version]
- Velandia, M.; Rejesus, R.M.; Jones, D.C.; Price, J.A.; Workneh, F.; Rush, C.M. Economic impact of Wheat streak mosaic virus in the Texas High Plains. Crop Prot. 2010, 29, 699–703. [Google Scholar] [CrossRef]
- Ordon, F.; Habekuss, A.; Kastirr, U.; Rabenstein, F.; Kühne, T. Virus resistance in cereals: Sources of resistance, genetics and breeding. J. Phytopathol. 2009, 157, 535–545. [Google Scholar] [CrossRef]
- Rotenberg, D.; Bockus, W.W.; Whitfield, A.E.; Hervey, K.; Baker, K.D.; Ou, Z.; Laney, A.G.; De Wolf, E.D.; Appel, J.A. Occurrence of viruses and associated grain yields of paired symptomatic and nonsymptomatic tillers in Kansas winter wheat fields. Phytopathology 2016, 106, 202–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleczewski, N.; Chapara, V.; Bradley, C.A. Occurrence of Viruses and Clavibacter michiganensis in Winter Wheat in Illinois, 2009 to 2011 and 2019 to 2020. Plant Health Prog. 2020, 21, 317–320. [Google Scholar] [CrossRef]
- Singh, K.; Wegulo, S.N.; Skoracka, A.; Kundu, J.K. Wheat streak mosaic virus: A century old virus with rising importance worldwide. Mol. Plant Pathol. 2018, 19, 2193–2206. [Google Scholar] [CrossRef] [Green Version]
- D’arcy, C.; Burnett, P. Barley Yellow Dwarf: 40 Years of Progress; Amer Phytopathological Society: Saint Paul, MN, USA, 1995. [Google Scholar]
- Clover, G.; Ratti, C.; Henry, C. Molecular characterization and detection of European isolates of Soil-borne wheat mosaic virus. Plant Pathol. 2001, 50, 761–767. [Google Scholar] [CrossRef]
- Almas, L.K.; Price, J.A.; Workneh, F.; Rush, C.M. Quantifying economic losses associated with levels of wheat streak mosaic incidence and severity in the Texas High Plains. Crop Prot. 2016, 88, 155–160. [Google Scholar] [CrossRef]
- Brakke, M. Virus diseases of wheat. Wheat Wheat Improv. 1987, 13, 585–624. [Google Scholar]
- Pichon, E.; Souquet, M.; Armand, T.; Jacquot, E. Wheat cultivars and natural-based substances: Impacts on epidemiological parameters of yellow dwarf disease. Plant Pathol. 2022, 71, 1293–1303. [Google Scholar] [CrossRef]
- King, A.M.; Lefkowitz, E.J.; Mushegian, A.R.; Adams, M.J.; Dutilh, B.E.; Gorbalenya, A.E.; Harrach, B.; Harrison, R.L.; Junglen, S.; Knowles, N.J. Changes to taxonomy and the International Code of Virus Classification and Nomenclature ratified by the International Committee on Taxonomy of Viruses (2018). Arch. Virol. 2018, 163, 2601–2631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nabi, S.U.; Baranwal, V.K.; Rao, G.P.; Mansoor, S.; Vladulescu, C.; Raja, W.H.; Jan, B.L.; Alansi, S. High-throughput RNA sequencing of mosaic infected and non-infected apple (Malus × domestica Borkh.) cultivars: From detection to the reconstruction of whole genome of viruses and viroid. Plants 2022, 11, 675. [Google Scholar] [CrossRef]
- Mehetre, G.T.; Leo, V.V.; Singh, G.; Sorokan, A.; Maksimov, I.; Yadav, M.K.; Upadhyaya, K.; Hashem, A.; Alsaleh, A.N.; Dawoud, T.M. Current developments and challenges in plant viral diagnostics: A systematic review. Viruses 2021, 13, 412. [Google Scholar] [CrossRef]
- Fajardo, T.V.M.; Bertocchi, A.A.; Nickel, O. Determination of the grapevine virome by high-throughput sequencing and grapevine viruses detection in Serra Gaúcha. Brazil. Rev. Ceres 2020, 67, 156–163. [Google Scholar] [CrossRef]
- Kreuze, J.F.; Perez, A.; Untiveros, M.; Quispe, D.; Fuentes, S.; Barker, I.; Simon, R. Complete viral genome sequence and discovery of novel viruses by deep sequencing of small RNAs: A generic method for diagnosis, discovery and sequencing of viruses. Virology 2009, 388, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalupowicz, L.; Dombrovsky, A.; Gaba, V.; Luria, N.; Reuven, M.; Beerman, A.; Lachman, O.; Dror, O.; Nissan, G.; Manulis-Sasson, S. Diagnosis of plant diseases using the Nanopore sequencing platform. Plant Pathol. 2019, 68, 229–238. [Google Scholar] [CrossRef]
- Dong, Z.-X.; Lin, C.-C.; Chen, Y.-K.; Chou, C.-C.; Chen, T.-C. Identification of an emerging cucumber virus in Taiwan using Oxford nanopore sequencing technology. Plant Methods 2022, 18, 143. [Google Scholar] [CrossRef]
- Šašić Zorić, L.; Janjušević, L.; Djisalov, M.; Knežić, T.; Vunduk, J.; Milenković, I.; Gadjanski, I. Molecular Approaches for Detection of Trichoderma Green Mold Disease in Edible Mushroom Production. Biology 2023, 12, 299. [Google Scholar] [CrossRef]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef]
- Barba, M.; Czosnek, H.; Hadidi, A. Historical perspective, development and applications of next-generation sequencing in plant virology. Viruses 2014, 6, 106–136. [Google Scholar] [CrossRef] [Green Version]
- Pecman, A.; Adams, I.; Gutiérrez-Aguirre, I.; Fox, A.; Boonham, N.; Ravnikar, M.; Kutnjak, D. Systematic Comparison of Nanopore and Illumina Sequencing for the Detection of Plant Viruses and Viroids Using Total RNA Sequencing Approach. Front. Microbiol. 2022, 13, 1424. [Google Scholar] [CrossRef]
- Boykin, L.M.; Sseruwagi, P.; Alicai, T.; Ateka, E.; Mohammed, I.U.; Stanton, J.-A.L.; Kayuki, C.; Mark, D.; Fute, T.; Erasto, J. Tree lab: Portable genomics for early detection of plant viruses and pests in sub-saharan africa. Genes 2019, 10, 632. [Google Scholar] [CrossRef] [Green Version]
- Bronzato Badial, A.; Sherman, D.; Stone, A.; Gopakumar, A.; Wilson, V.; Schneider, W.; King, J. Nanopore sequencing as a surveillance tool for plant pathogens in plant and insect tissues. Plant Dis. 2018, 102, 1648–1652. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Giordano, F.; Ning, Z. Oxford Nanopore MinION sequencing and genome assembly. Genom. Proteom. Bioinform. 2016, 14, 265–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, J.J.M.; Ip, Y.C.A.; Bauman, A.G.; Huang, D. MinION-in-ARMS: Nanopore sequencing to expedite barcoding of specimen-rich macrofaunal samples from autonomous reef monitoring structures. Front. Mar. Sci. 2020, 7, 448. [Google Scholar] [CrossRef]
- Van Dijk, E.L.; Jaszczyszyn, Y.; Naquin, D.; Thermes, C. The third revolution in sequencing technology. Trends Genet. 2018, 34, 666–681. [Google Scholar] [CrossRef] [PubMed]
- Liefting, L.W.; Waite, D.W.; Thompson, J.R. Application of Oxford Nanopore technology to plant virus detection. Viruses 2021, 13, 1424. [Google Scholar] [CrossRef]
- Redila, C.D.; Prakash, V.; Nouri, S. Metagenomics analysis of the wheat virome identifies novel plant and fungal-associated viral sequences. Viruses 2021, 13, 2457. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Jarošová, J.; Fousek, J.; Huan, C.; Kundu, J.K. Virome identification in wheat in the Czech Republic using small RNA deep sequencing. J. Integr. Agric. 2020, 19, 1825–1833. [Google Scholar] [CrossRef]
- Kondo, H.; Yoshida, N.; Fujita, M.; Maruyama, K.; Hyodo, K.; Hisano, H.; Tamada, T.; Andika, I.B.; Suzuki, N. Identification of a Novel Quinvirus in the Family Betaflexiviridae That Infects Winter Wheat. Front. Microbiol. 2021, 12, 715545. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Zhang, T.; He, M.; Sun, B.; Zhou, X.; Wu, J. Molecular characterization of a novel wheat-infecting virus of the family Betaflexiviridae. Arch. Virol. 2021, 166, 2875–2879. [Google Scholar] [CrossRef]
- Ranabhat, N.B.; Fellers, J.P.; Bruce, M.A.; Rupp, J.L.S. Brome mosaic virus detected in Kansas wheat co-infected with other common wheat viruses. Front. Plant Sci. 2023, 14, 633. [Google Scholar] [CrossRef]
- Hodge, B.; Paul, P.; Stewart, L.R. Occurrence and high-throughput sequencing of viruses in Ohio wheat. Plant Dis. 2020, 104, 1789–1800. [Google Scholar] [CrossRef]
- Fellers, J.P.; Webb, C.; Fellers, M.C.; Shoup Rupp, J.; De Wolf, E. Wheat virus identification within infected tissue using nanopore sequencing technology. Plant Dis. 2019, 103, 2199–2203. [Google Scholar] [CrossRef]
- Gallo, Y.; Marín, M.; Gutiérrez, P. Detection of RNA viruses in Solanum quitoense by high-throughput sequencing (HTS) using total and double stranded RNA inputs. Physiol. Mol. Plant Pathol. 2021, 113, 101570. [Google Scholar] [CrossRef]
- Petersen, L.M.; Martin, I.W.; Moschetti, W.E.; Kershaw, C.M.; Tsongalis, G.J. Third-generation sequencing in the clinical laboratory: Exploring the advantages and challenges of nanopore sequencing. J. Clin. Microbiol. 2019, 58, e01315–e01319. [Google Scholar] [CrossRef]
- Park, C.; Oh, J.; Min, H.-G.; Lee, H.-K.; Lee, S.-H. First report of barley virus g in proso millet (Panicum miliaceum) in Korea. Plant Dis. 2017, 101, 393. [Google Scholar] [CrossRef]
- Oh, J.; Park, C.; Min, H.-G.; Lee, H.-K.; Yeom, Y.-A.; Yoon, Y.; Lee, S.-H. First report of barley virus g in foxtail millet (Setaria italica) in Korea. Plant Dis. 2017, 101, 1061. [Google Scholar] [CrossRef]
- Jo, Y.; Bae, J.-Y.; Kim, S.-M.; Choi, H.; Lee, B.C.; Cho, W.K. Barley RNA viromes in six different geographical regions in Korea. Sci. Rep. 2018, 8, 13237. [Google Scholar] [CrossRef] [Green Version]
- Zhao, F.; Lim, S.; Yoo, R.H.; Igori, D.; Kim, S.-M.; Kwak, D.Y.; Kim, S.L.; Lee, B.C.; Moon, J.S. The complete genomic sequence of a tentative new polerovirus identified in barley in South Korea. Arch. Virol. 2016, 161, 2047–2050. [Google Scholar] [CrossRef]
- Candresse, T.; Marais, A.; Sorrentino, R.; Faure, C.; Theil, S.; Cadot, V.; Rolland, M.; Villemot, J.; Rabenstein, F. Complete genomic sequence of barley (Hordeum vulgare) endornavirus (HvEV) determined by next-generation sequencing. Arch. Virol. 2016, 161, 741–743. [Google Scholar] [CrossRef]
- Fukuhara, T.; Gibbs, M. Family endornaviridae. In Virus Taxonomy: Ninth Report of the International Committee on Taxonomy of Viruses; Elsevier: Amsterdam, The Netherlands, 2012; pp. 519–521. [Google Scholar]
- Roossinck, M.J. Lifestyles of plant viruses. Philos. Trans. R. Soc. B Biol. Sci. 2010, 365, 1899–1905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walls, J., III; Rajotte, E.; Rosa, C. The past, present, and future of barley yellow dwarf management. Agriculture 2019, 9, 23. [Google Scholar] [CrossRef] [Green Version]
- Kim, N.-K.; Lee, H.-J.; Kim, S.-M.; Jeong, R.-D. Identification of viruses infecting oats in Korea by metatranscriptomics. Plants 2022, 11, 256. [Google Scholar] [CrossRef] [PubMed]
- ElSayed, A.I.; Komor, E.; Boulila, M.; Viswanathan, R.; Odero, D.C. Biology and management of sugarcane yellow leaf virus: An historical overview. Arch. Virol. 2015, 160, 2921–2934. [Google Scholar] [CrossRef] [PubMed]
- Bouallegue, M.; Mezghani-Khemakhem, M.; Makni, H.; Makni, M. First report of Sugarcane yellow leaf virus infecting barley in Tunisia. Plant Dis. 2014, 98, 1016. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Liu, Y.; Liu, W.; Cao, M.; Massart, S.; Wang, X. Identification, characterization and full-length sequence analysis of a novel polerovirus associated with wheat leaf yellowing disease. Front. Microbiol. 2017, 8, 1689. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-J.; Cho, I.-S.; Jeong, R.-D. Nanopore Metagenomics Sequencing for Rapid Diagnosis and Characterization of Lily Viruses. Plant Pathol. J. 2022, 38, 503–512. [Google Scholar] [CrossRef]
- Krueger, F. Trim Galore. A Wrapper Tool Around Cutadapt and FastQC to Consistently Apply Quality and Adapter Trimming to FastQ Files; Babraham Institute: Cambridge, UK, 2015; pp. 516–517. [Google Scholar]
- Schmieder, R.; Edwards, R. Fast identification and removal of sequence contamination from genomic and metagenomic datasets. PLoS ONE 2011, 6, e17288. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547. [Google Scholar] [CrossRef]
- Tamura, K.; Nei, M.; Kumar, S. Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc. Natl. Acad. Sci. USA 2004, 101, 11030–11035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laney, A.G.; Acosta-Leal, R.; Rotenberg, D. Optimized yellow dwarf virus multiplex PCR assay reveals a common occurrence of Barley yellow dwarf virus-PAS in Kansas winter wheat. Plant Health Prog. 2018, 19, 37–43. [Google Scholar] [CrossRef] [Green Version]
- Malmstrom, C.M.; Shu, R. Multiplexed RT-PCR for streamlined detection and separation of barley and cereal yellow dwarf viruses. J. Virol. Methods 2004, 120, 69–78. [Google Scholar] [CrossRef]
- Viswanathan, R.; Karuppaiah, R.; Balamuralikrishnan, M. Detection of three major RNA viruses infecting sugarcane by multiplex reverse transcription-polymerase chain reaction (multiplex-RT-PCR). Australas. Plant Pathol. 2010, 39, 79–84. [Google Scholar] [CrossRef]
- Balaji, B.; Bucholtz, D.B.; Anderson, J.M. Barley yellow dwarf virus and Cereal yellow dwarf virus quantification by real-time polymerase chain reaction in resistant and susceptible plants. Phytopathology 2003, 93, 1386–1392. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HTS Platform | Library ID | Raw Reads | Trimmed Reads | SRA Accession Number | ||||||
| Total Reads | Mini Read Length | Max Read Length | Mean Read Length | Total Reads | Mini Read Length | Max Read Length | Mean Read Length | |||
| ONT | JN-ONT | 772,355 | 91 | 8222 | 412.8 | 761,277 | 1 | 8222 | 296.3 | SRR22371364 |
| JB-ONT | 490,030 | 55 | 12,301 | 340.1 | 485,491 | 1 | 12,301 | 272 | SRR22371365 | |
| GN-ONT | 1,177,844 | 82 | 78,921 | 325.1 | 1,160,201 | 1 | 78,921 | 221.8 | SRR22371366 | |
| HTS platform | Library ID | Total reads based (bp) | Total reads | Q30 (%) | Virus data | Virus annotated contigs | SRA Accession Number | |||
| Sum reads | Sum bases (bp) | |||||||||
| Illumina | JN-Ill | 5,478,870,712 | 36,283,912 | 95.52 | 19,138 | 2,550,819 | 21 | SRR16774430 | ||
| JB-Ill | 5,931,982,150 | 39,284,650 | 95.2 | 220,452 | 32,179,884 | 38 | SRR16774429 | |||
| GN-Ill | 5,327,334,360 | 35,280,360 | 95.04 | 120,742 | 17,615,293 | 46 | SRR16774428 | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, H.-J.; Kim, S.-M.; Jeong, R.-D. Analysis of Wheat Virome in Korea Using Illumina and Oxford Nanopore Sequencing Platforms. Plants 2023, 12, 2374. https://doi.org/10.3390/plants12122374
Lee H-J, Kim S-M, Jeong R-D. Analysis of Wheat Virome in Korea Using Illumina and Oxford Nanopore Sequencing Platforms. Plants. 2023; 12(12):2374. https://doi.org/10.3390/plants12122374
Chicago/Turabian StyleLee, Hyo-Jeong, Sang-Min Kim, and Rae-Dong Jeong. 2023. "Analysis of Wheat Virome in Korea Using Illumina and Oxford Nanopore Sequencing Platforms" Plants 12, no. 12: 2374. https://doi.org/10.3390/plants12122374
APA StyleLee, H.-J., Kim, S.-M., & Jeong, R.-D. (2023). Analysis of Wheat Virome in Korea Using Illumina and Oxford Nanopore Sequencing Platforms. Plants, 12(12), 2374. https://doi.org/10.3390/plants12122374