

Metagenomic Study of Fungal Microbial Communities in Two PDO Somontano Vineyards (Huesca, Spain): Effects of Age, Plant Genotype, and Initial Phytosanitary Status on the Priming and Selection of their Associated Microorganisms

 ,
,  ,
,  ,
,  ,
,  ,
,
Abstract
:1. Introduction
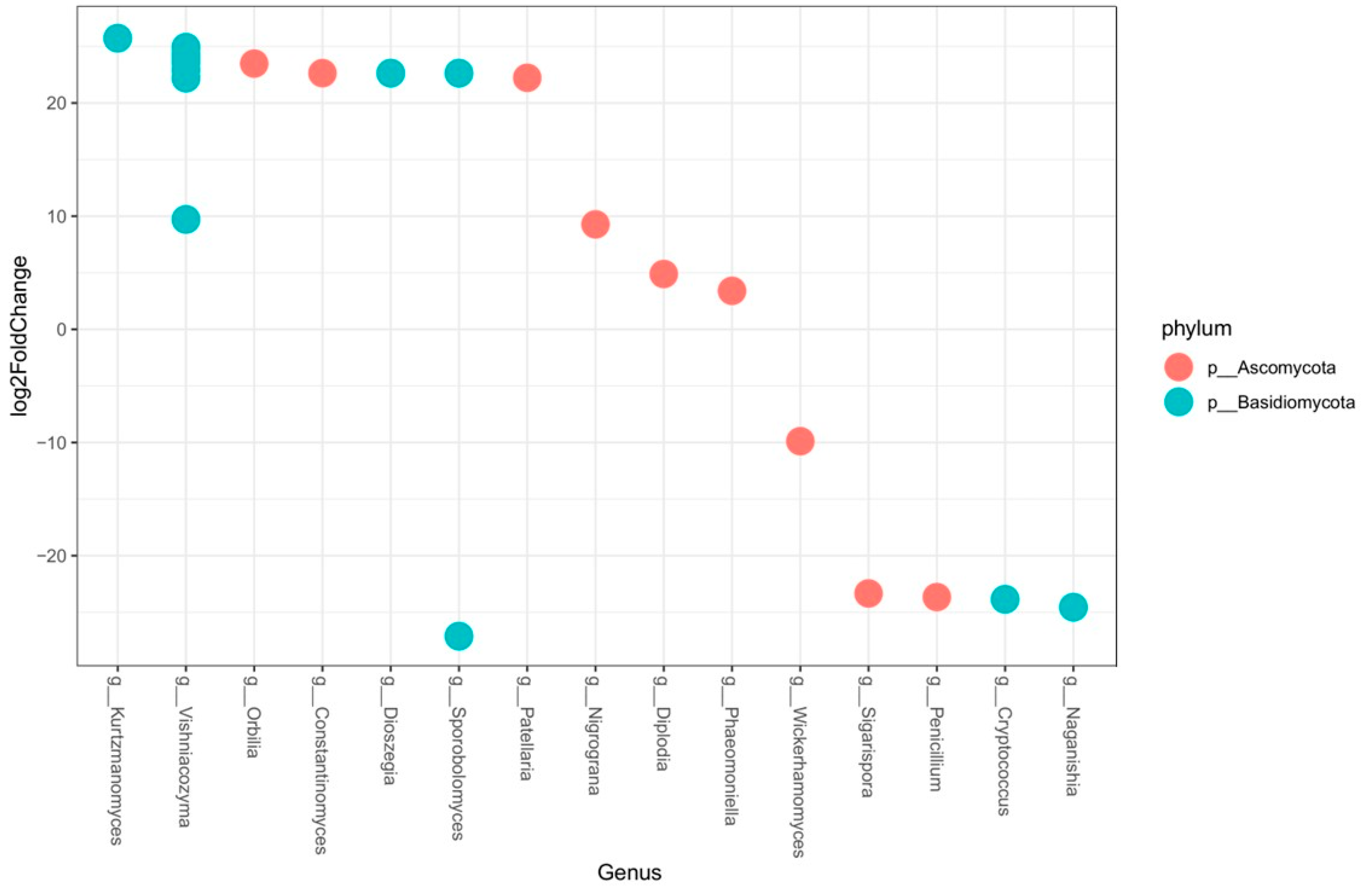
2. Results
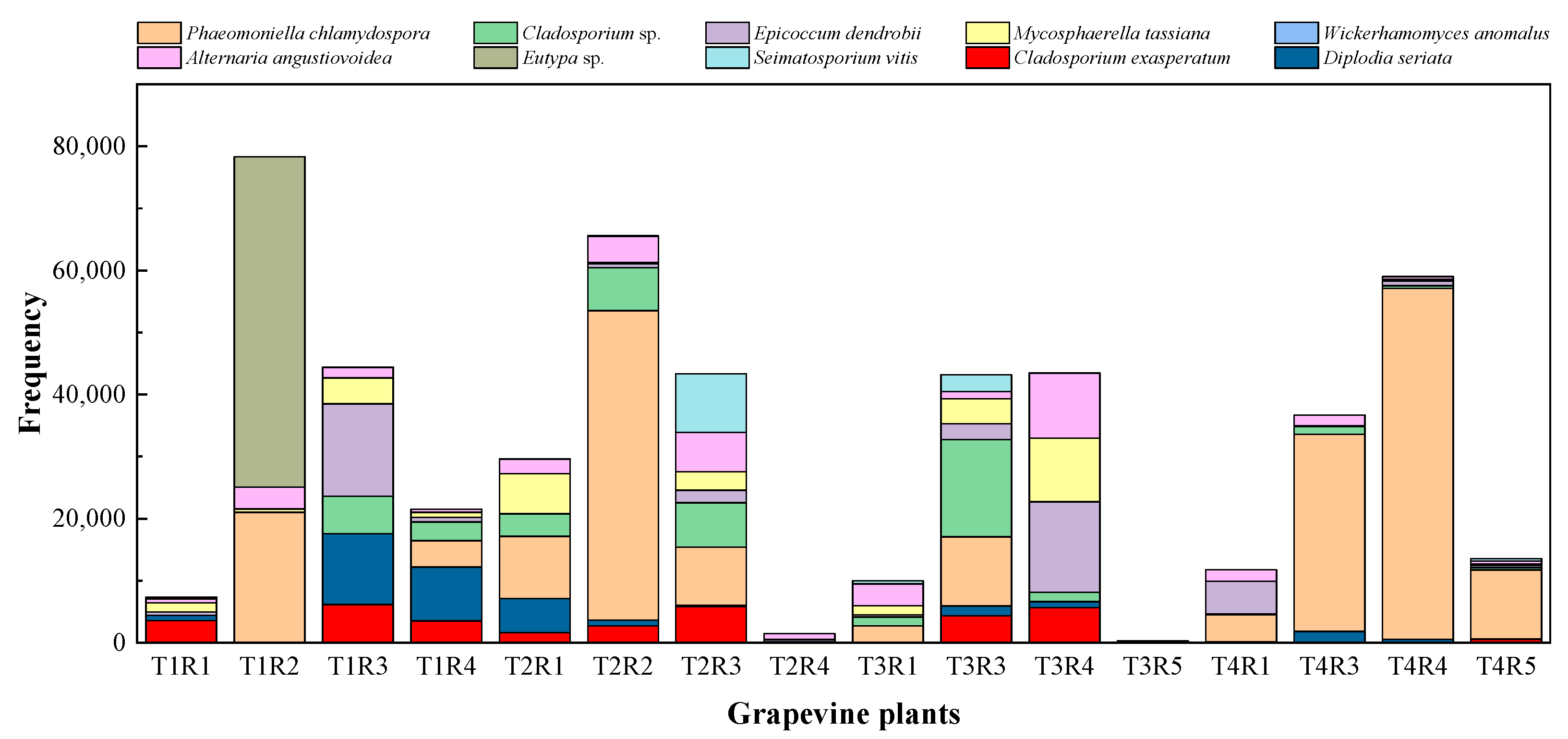
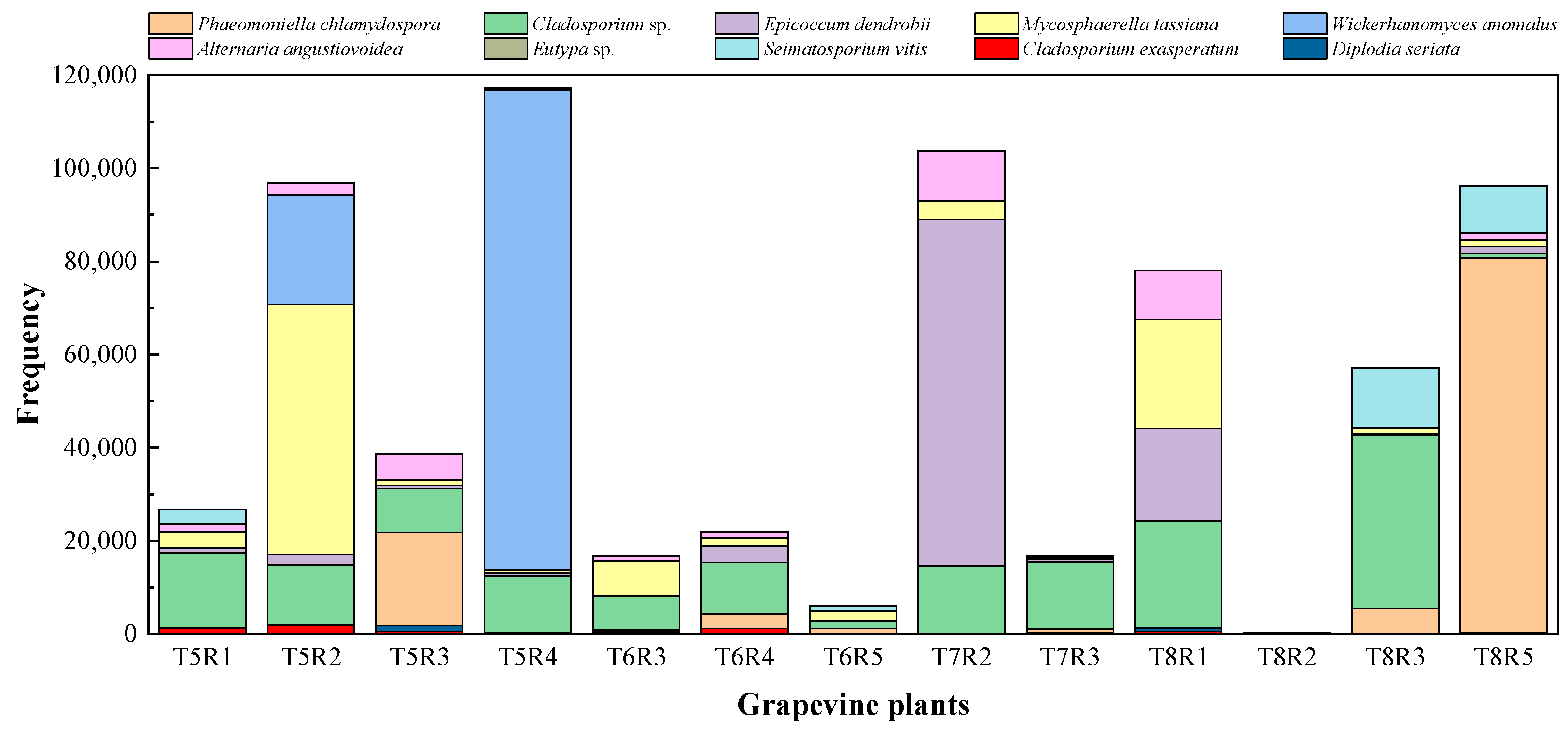
2.1. Isolation of Endophytic Fungi from Grapevine Plants
2.2. NGS Analyses of Fungal Communities
2.2.1. Alpha-Diversity of Fungal Microbiome
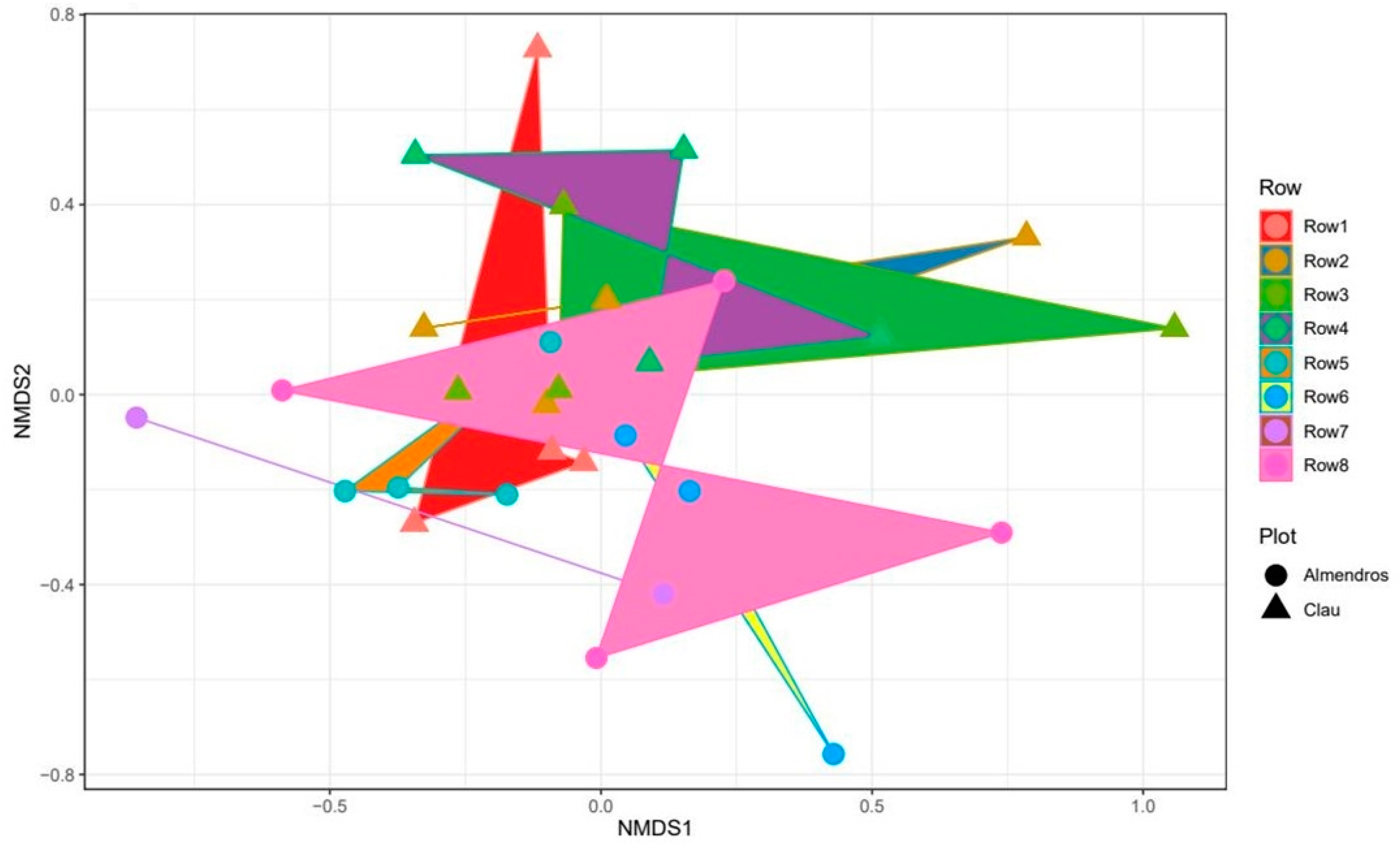
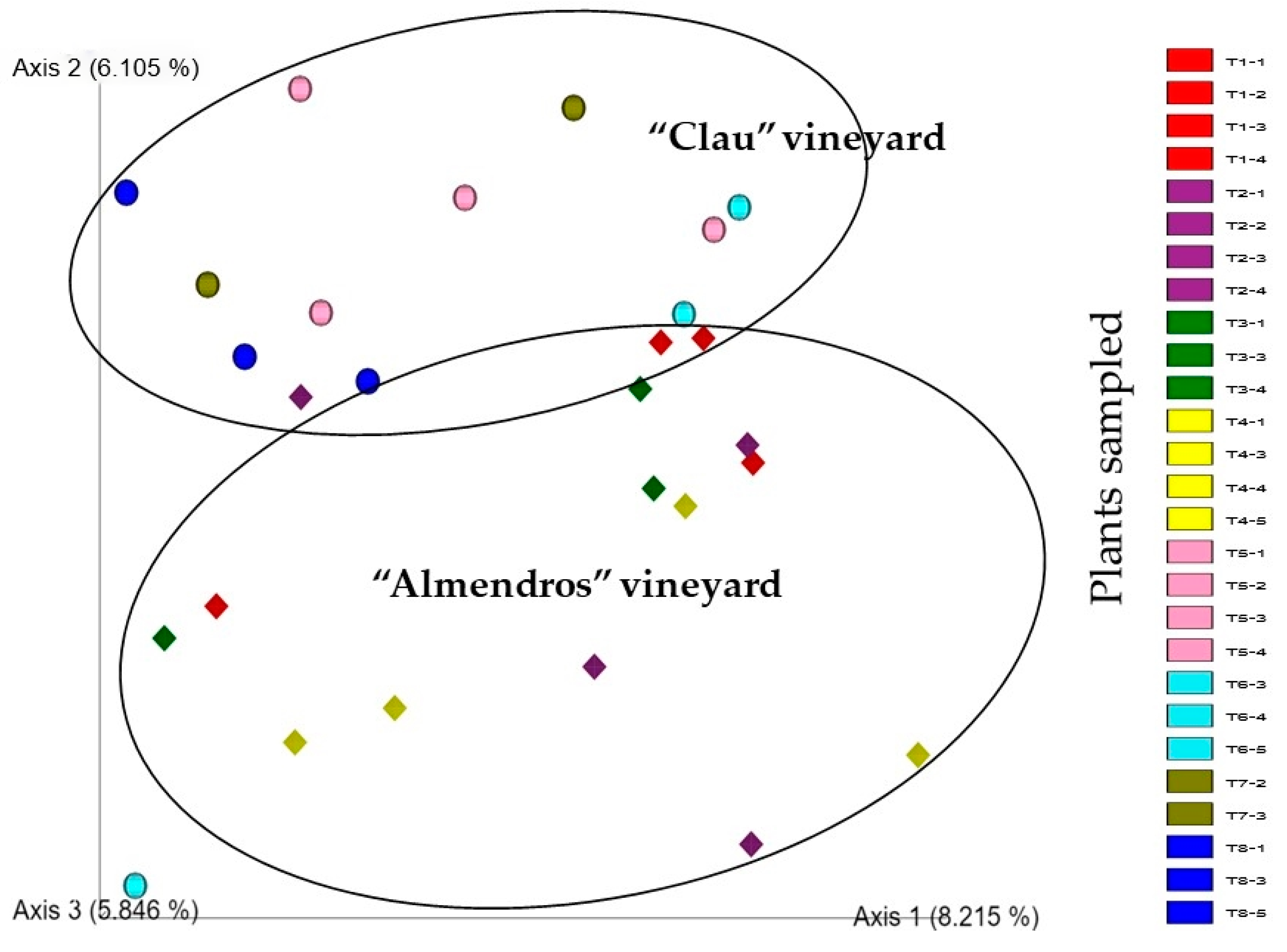
2.2.2. Comparison between Vineyards: Beta-Diversity
2.3. Correlation between NGS and Culture-Dependent Methods
3. Discussion
3.1. Grapevine Inner Wood Microbiome
3.2. Comparison of Microbial Communities: Beta-Diversity
3.3. Comparative Microbial Diversity According to Methodology: NGS vs. Culture-Dependent Techniques
4. Materials and Methods
4.1. Grapevine Plots
4.2. Wood Samples
4.3. Isolation of Grapevine Endophytic Fungi
4.4. DNA Extraction and Sequencing
4.5. Bioinformatics Procedure
4.6. Statistical Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gramaje, D.; Urbez-Torres, J.R.; Sosnowski, M.R. Managing grapevine trunk diseases with respect to etiology and epidemiology: Current strategies and future prospects. Plant Dis. 2018, 102, 12–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mondello, V.; Songy, A.; Battiston, E.; Pinto, C.; Coppin, C.; Trotel-Aziz, P.; Clement, C.; Mugnai, L.; Fontaine, F. Grapevine trunk diseases: A review of fifteen years of trials for their control with chemicals and biocontrol agents. Plant Dis. 2018, 102, 1189–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertsch, C.; Ramírez-Suero, M.; Magnin-Robert, M.; Larignon, P.; Chong, J.; Abou-Mansour, E.; Spagnolo, A.; Clément, C.; Fontaine, F. Grapevine trunk diseases: Complex and still poorly understood. Plant Pathol. 2013, 62, 243–265. [Google Scholar] [CrossRef] [Green Version]
- Hrycan, J.; Hart, M.; Bowen, P.; Forge, T.; ÚRbez-Torres, J.R. Grapevine trunk disease fungi: Their roles as latent pathogens and stress factors that favour disease development and symptom expression. Phytopathol. Mediterr. 2020, 59, 395–424. [Google Scholar] [CrossRef]
- Fontaine, F.; Gramaje, D.; Armengol, J.; Smart, R.; Nagy, Z.A.; Borgo, M.; Rego, C.; Corio-Costet, M.-F. Grapevine Trunk Diseases. A Review; De la Fuente, M., Ed.; OIV Publications: Paris, France, 2016; p. 25. [Google Scholar]
- Bettenfeld, P.; Cadena i Canals, J.; Jacquens, L.; Fernandez, O.; Fontaine, F.; van Schaik, E.; Courty, P.-E.; Trouvelot, S. The microbiota of the grapevine holobiont: A key component of plant health. J. Adv. Res. 2022, 40, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Forbes, J.D.; Knox, N.C.; Ronholm, J.; Pagotto, F.; Reimer, A. Metagenomics: The Next Culture-Independent Game Changer. Front. Microbiol. 2017, 8, 1069. [Google Scholar] [CrossRef] [Green Version]
- Hinsu, A.; Dumadiya, A.; Joshi, A.; Kotadiya, R.; Andharia, K.; Koringa, P.; Kothari, R. To culture or not to culture: A snapshot of culture-dependent and culture-independent bacterial diversity from peanut rhizosphere. PeerJ 2021, 9, e12035. [Google Scholar] [CrossRef]
- Bekris, F.; Vasileiadis, S.; Papadopoulou, E.; Samaras, A.; Testempasis, S.; Gkizi, D.; Tavlaki, G.; Tzima, A.; Paplomatas, E.; Markakis, E.; et al. Grapevine wood microbiome analysis identifies key fungal pathogens and potential interactions with the bacterial community implicated in grapevine trunk disease appearance. Environ. Microbiome 2021, 16, 23. [Google Scholar] [CrossRef]
- Tiwari, M.; Pati, D.; Mohapatra, R.; Sahu, B.B.; Singh, P. The Impact of Microbes in Plant Immunity and Priming Induced Inheritance: A Sustainable Approach for Crop protection. Plant Stress 2022, 4, 100072. [Google Scholar] [CrossRef]
- Liu, D.; Howell, K. Community succession of the grapevine fungal microbiome in the annual growth cycle. Environ. Microbiol. 2021, 23, 1842–1857. [Google Scholar] [CrossRef]
- Liu, H.; Brettell, L.E. Plant Defense by VOC-Induced Microbial Priming. Trends Plant Sci. 2019, 24, 187–189. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, E.; Zilber-Rosenberg, I.; Collier, R.J. Microbes Drive Evolution of Animals and Plants: The Hologenome Concept. mBio 2016, 7, e01395-15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gopal, M.; Gupta, A. Microbiome Selection Could Spur Next-Generation Plant Breeding Strategies. Front. Microbiol. 2016, 7, 1971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berg, G.; Rybakova, D.; Grube, M.; Köberl, M. The plant microbiome explored: Implications for experimental botany. J. Exp. Bot. 2016, 67, 995–1002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fournier, P.; Pellan, L.; Barroso-Bergadà, D.; Bohan, D.A.; Candresse, T.; Delmotte, F.; Dufour, M.-C.; Lauvergeat, V.; Le Marrec, C.; Marais, A.; et al. The functional microbiome of grapevine throughout plant evolutionary history and lifetime. Adv. Ecol. Res. 2022, 67, 27–99. [Google Scholar] [CrossRef]
- Morgan, H.H.; du Toit, M.; Setati, M.E. The Grapevine and Wine Microbiome: Insights from High-Throughput Amplicon Sequencing. Front. Microbiol. 2017, 8, 820. [Google Scholar] [CrossRef] [Green Version]
- Berlanas, C.; Berbegal, M.; Elena, G.; Laidani, M.; Cibriain, J.F.; Sagües, A.; Gramaje, D. The fungal and bacterial rhizosphere microbiome associated with grapevine rootstock genotypes in mature and young vineyards. Front. Microbiol. 2019, 10, 1142. [Google Scholar] [CrossRef] [Green Version]
- Hamaoka, K.; Aoki, Y.; Takahashi, S.; Enoki, S.; Yamamoto, K.; Tanaka, K.; Suzuki, S. Diversity of endophytic bacterial microbiota in grapevine shoot xylems varies depending on wine grape-growing region, cultivar, and shoot growth stage. Sci. Rep. 2022, 12, 15772. [Google Scholar] [CrossRef]
- Martínez-Diz, M.d.P.; Andrés-Sodupe, M.; Bujanda, R.; Díaz-Losada, E.; Eichmeier, A.; Gramaje, D. Soil-plant compartments affect fungal microbiome diversity and composition in grapevine. Fungal Ecol. 2019, 41, 234–244. [Google Scholar] [CrossRef]
- Wei, Y.-J.; Wu, Y.; Yan, Y.-Z.; Zou, W.; Xue, J.; Ma, W.-R.; Wang, W.; Tian, G.; Wang, L.-Y. High-throughput sequencing of microbial community diversity in soil, grapes, leaves, grape juice and wine of grapevine from China. PLoS ONE 2018, 13, e0193097. [Google Scholar] [CrossRef] [Green Version]
- Niem, J.M.; Billones-Baaijens, R.; Stodart, B.; Savocchia, S. Diversity Profiling of Grapevine Microbial Endosphere and Antagonistic Potential of Endophytic Pseudomonas Against Grapevine Trunk Diseases. Front. Microbiol. 2020, 11, 477. [Google Scholar] [CrossRef] [PubMed]
- Cobos, R.; Ibañez, A.; Diez-Galán, A.; Calvo-Peña, C.; Ghoreshizadeh, S.; Coque, J.J.R. The Grapevine Microbiome to the Rescue: Implications for the Biocontrol of Trunk Diseases. Plants 2022, 11, 840. [Google Scholar] [CrossRef] [PubMed]
- Darriaut, R.; Lailheugue, V.; Masneuf-Pomarède, I.; Marguerit, E.; Martins, G.; Compant, S.; Ballestra, P.; Upton, S.; Ollat, N.; Lauvergeat, V. Grapevine rootstock and soil microbiome interactions: Keys for a resilient viticulture. Hortic. Res. 2022, 9, uhac019. [Google Scholar] [CrossRef] [PubMed]
- Pauvert, C.; Fort, T.; Calonnec, A.; Faivre d’Arcier, J.; Chancerel, E.; Massot, M.; Chiquet, J.; Robin, S.; Bohan, D.A.; Vallance, J.; et al. Microbial association networks give relevant insights into plant pathobiomes. bioRxiv 2020. [Google Scholar] [CrossRef]
- Del Frari, G.; Gobbi, A.; Aggerbeck, M.R.; Oliveira, H.; Hansen, L.H.; Ferreira, R.B. Characterization of the Wood Mycobiome of Vitis vinifera in a Vineyard Affected by Esca. Spatial Distribution of Fungal Communities and Their Putative Relation With Leaf Symptoms. Front. Plant Sci. 2019, 10, 910. [Google Scholar] [CrossRef] [Green Version]
- Cordero-Bueso, G.; Mangieri, N.; Maghradze, D.; Foschino, R.; Valdetara, F.; Cantoral, J.M.; Vigentini, I. Wild Grape-Associated Yeasts as Promising Biocontrol Agents against Vitis vinifera Fungal Pathogens. Front. Microbiol. 2017, 8, 2025. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Jiao, R.; Chen, S.; Ren, Y.; Zhang, L.; Zhang, D.; Chen, J.; Li, G. First Report of Fruit Rot of Grapes (Vitis vinifera) Caused by Cladosporium cladosporioides in Xinjiang, China. Plant Dis. 2022, 106, 315. [Google Scholar] [CrossRef]
- Lawrence, D.P.; Travadon, R.; Baumgartner, K. Novel Seimatosporium Species from Grapevine in Northern California and Their Interactions with Fungal Pathogens Involved in the Trunk-Disease Complex. Plant Dis. 2018, 102, 1081–1092. [Google Scholar] [CrossRef] [Green Version]
- Cloete, M.; Fischer, M.; Mostert, L.; Halleen, F. Hymenochaetales associated with esca-related wood rots on grapevine with a special emphasis on the status of esca in South African vineyards. Phytopathol. Mediterr. 2015, 54, 299–312. [Google Scholar] [CrossRef]
- Patanita, M.; Albuquerque, A.; Campos, M.D.; Materatski, P.; Varanda, C.M.R.; Ribeiro, J.A.; Félix, M.D.R. Metagenomic assessment unravels fungal microbiota associated to grapevine trunk diseases. Horticulturae 2022, 8, 288. [Google Scholar] [CrossRef]
- Knapp, D.G.; Lázár, A.; Molnár, A.; Vajna, B.; Karácsony, Z.; Váczy, K.Z.; Kovács, G.M. Above-ground parts of white grapevine Vitis vinifera cv. Furmint share core members of the fungal microbiome. Environ. Microbiol. Rep. 2021, 13, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Santoni, S.; Weber, A.; This, P.; Péros, J.-P. Understanding the phyllosphere microbiome assemblage in grape species (Vitaceae) with amplicon sequence data structures. Sci. Rep. 2019, 9, 14294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wicaksono, W.A.; Morauf, C.; Müller, H.; Abdelfattah, A.; Donat, C.; Berg, G. The mature phyllosphere microbiome of grapevine is associated with resistance against Plasmopara viticola. Front. Microbiol. 2023, 14, 1149307. [Google Scholar] [CrossRef] [PubMed]
- Gramaje, D.; Eichmeier, A.; Spetik, M.; Carbone, M.J.; Bujanda, R.; Vallance, J.; Rey, P. Exploring the temporal dynamics of the fungal microbiome in rootstocks, the lesser-known half of the grapevine crop. J. Fungi 2022, 8, 421. [Google Scholar] [CrossRef]
- Carbone, M.J.; Alaniz, S.; Mondino, P.; Gelabert, M.; Eichmeier, A.; Tekielska, D.; Bujanda, R.; Gramaje, D. Drought influences fungal community dynamics in the grapevine rhizosphere and root microbiome. J. Fungi 2021, 7, 686. [Google Scholar] [CrossRef]
- Claverie, M.; Notaro, M.; Fontaine, F.; Wery, J. Current knowledge on grapevine trunk diseases with complex etiology: A systemic approach. Phytopathol. Mediterr. 2020, 59, 29–53. [Google Scholar] [CrossRef]
- Kenfaoui, J.; Radouane, N.; Mennani, M.; Tahiri, A.; El Ghadraoui, L.; Belabess, Z.; Fontaine, F.; El Hamss, H.; Amiri, S.; Lahlali, R.; et al. A panoramic view on grapevine trunk diseases threats: Case of Eutypa dieback, Botryosphaeria dieback, and esca disease. J. Fungi 2022, 8, 595. [Google Scholar] [CrossRef]
- Azevedo-Nogueira, F.; Rego, C.; Gonçalves, H.M.R.; Fortes, A.M.; Gramaje, D.; Martins-Lopes, P. The road to molecular identification and detection of fungal grapevine trunk diseases. Front. Plant Sci. 2022, 13, 960289. [Google Scholar] [CrossRef]
- Morales-Cruz, A.; Figueroa-Balderas, R.; García, J.F.; Tran, E.; Rolshausen, P.E.; Baumgartner, K.; Cantu, D. Profiling grapevine trunk pathogens in planta: A case for community-targeted DNA metabarcoding. BMC Microbiol. 2018, 18, 214. [Google Scholar] [CrossRef] [Green Version]
- Lade, S.B.; Štraus, D.; Oliva, J. Variation in fungal community in grapevine (Vitis vinifera) nursery stock depends on nursery, variety and rootstock. J. Fungi 2022, 8, 47. [Google Scholar] [CrossRef]
- Bruez, E.; Vallance, J.; Gautier, A.; Laval, V.; Compant, S.; Maurer, W.; Sessitsch, A.; Lebrun, M.H.; Rey, P. Major changes in grapevine wood microbiota are associated with the onset of esca, a devastating trunk disease. Environ. Microbiol. 2020, 22, 5189–5206. [Google Scholar] [CrossRef] [PubMed]
- Paolinelli, M.; Escoriaza, G.; Cesari, C.; Garcia-Lampasona, S.; Hernandez-Martinez, R. Metatranscriptomic approach for microbiome characterization and host gene expression evaluation for “Hoja de malvón” disease in Vitis vinifera cv. Malbec (preprint). Res. Sq. 2020, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Del Frari, G.; Oliveira, H.; Ferreira, R.B. White rot fungi (Hymenochaetales) and esca of grapevine: Insights from recent microbiome studies. J. Fungi 2021, 7, 770. [Google Scholar] [CrossRef] [PubMed]
- Vacher, C.; Francioni, C.; Michel, M.; Fort, T.; Faivre d’Arcier, J.; Chancerel, E.; Delmotte, F.; Delmas, C.E.L. Fungal metabarcoding data for two grapevine varieties (Regent and Vitis vinifera ‘Cabernet-Sauvignon’) inoculated with powdery mildew (Erysiphe necator) under drought conditions. Phytobiomes J. 2022, 6, 358–367. [Google Scholar] [CrossRef]
- Awad, M.; Giannopoulos, G.; Mylona, P.V.; Polidoros, A.N. Genotype may influence bacterial diversity in bark and bud of Vitis vinifera cultivars grown under the same environment. Appl. Sci. 2020, 10, 8405. [Google Scholar] [CrossRef]
- Dissanayake, A.J.; Purahong, W.; Wubet, T.; Hyde, K.D.; Zhang, W.; Xu, H.; Zhang, G.; Fu, C.; Liu, M.; Xing, Q.; et al. Direct comparison of culture-dependent and culture-independent molecular approaches reveal the diversity of fungal endophytic communities in stems of grapevine (Vitis vinifera). Fungal Divers. 2018, 90, 85–107. [Google Scholar] [CrossRef]
- Jayawardena, R.S.; Purahong, W.; Zhang, W.; Wubet, T.; Li, X.; Liu, M.; Zhao, W.; Hyde, K.D.; Liu, J.; Yan, J. Biodiversity of fungi on Vitis vinifera L. revealed by traditional and high-resolution culture-independent approaches. Fungal Divers. 2018, 90, 1–84. [Google Scholar] [CrossRef] [Green Version]
- Johnson, M.; Zaretskaya, I.; Raytselis, Y.; Merezhuk, Y.; McGinnis, S.; Madden, T.L. NCBI BLAST: A better web interface. Nucleic Acids Res. 2008, 36, W5–W9. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Abarenkov, K.; Zirk, A.; Piirmann, T.; Pöhönen, R.; Ivanov, F.; Nilsson, R.H.; Kõljalg, U. UNITE General FASTA Release for Fungi 2; Version 10.05.2021; UNITE Community. 2021. Available online: https://doi.plutof.ut.ee/doi/10.15156/BIO/1280089 (accessed on 5 June 2023).
- Watson, M.; McMurdie, P.J.; Holmes, S. phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, N.H.; Song, Z.; Bates, S.T.; Branco, S.; Tedersoo, L.; Menke, J.; Schilling, J.S.; Kennedy, P.G. FUNGuild: An open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecol. 2016, 20, 241–248. [Google Scholar] [CrossRef]
- Bioinformatics & Evolutionary Genomics. Calculate and Draw Custom Venn Diagrams. Available online: http://bioinformatics.psb.ugent.be/webtools/Venn/ (accessed on 2 May 2023).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Taxonomy | Frequency | Fungal Guild |
|---|---|---|
| Cladosporium allicinum | 4601.6 | Pathotroph |
| Epicoccum nigrum (V) | 5101 | Pathotroph–Saprotroph–Symbiotroph |
| Stemphyliuym majusculum | 148.1 | Pathotroph–Saprotroph |
| Cystofilobasidium macerans (V) | 138 | Saprotroph |
| Seimatosporium vitis (V) | 1414.3 | Pathotroph |
| Phaeomoniella chlamydospora (V) | 12,741.1 | Pathotroph |
| Filobasidium stepposum (V) | 1028.8 | Saprotroph |
| Knufia perforans | 308.9 | Pathotroph–Saprotroph |
| Aureobasidium pullulans (V) | 683 | Pathotroph–Saprotroph–Symbiotroph |
| Filobasidium magnum (V) | 69.2 | Saprotroph |
| Rhinocladiella sp. (V) | 175 | Pathotroph |
| Cladosporium grevilleae (V) | 358.1 | Saprotroph |
| Cyphellophora sp. | 174.9 | Pathotroph–Saprotroph |
| Cladosporium cladosporioides (V) | 8257.4 | Saprotroph |
| Angustimassarina acerina (V) | 954.7 | Saprotroph |
| Myrmecridium banksiae | 65.3 | Saprotroph |
| Vishniacozyma victoriae (V) | 122.4 | Saprotroph |
| Aspergillus undulatus | 643.2 | Pathotroph–Saprotroph |
| Populocrescentia forlicesenensis | 1198.2 | Pathotroph–Saprotroph |
| Ord. Malasseziales | 23.2 | Pathotroph–Saprotroph |
| Acericola italica | 410.6 | Pathotroph–Saprotroph |
| Sporobolomyces roseus | 128.8 | Pathotroph–Saprotroph |
| Neoscytalidium sp. (V) | 43.8 | Pathotroph–Saprotroph |
| Alternaria infectoria | 359.2 | Pathotroph–Saprotroph–Symbiotroph |
| Neosetophoma lunariae (V) | 289.2 | Pathotroph |
| Vishniacozyma carnescens (V) | 751.5 | Saprotroph |
| Cladosporium exasperatum | 1392.3 | Saprotroph |
| Alternaria alternata (V) | 2608.3 | Pathotroph–Saprotroph–Symbiotroph |
| Cyphellophora oxyspora | 14.4 | Pathotroph–Saprotroph |
| Knufia mediterranea | 221.7 | Pathotroph–Saprotroph |
| Plot Name | “Clau” | “Almendros” |
|---|---|---|
| Var./Rootstock | “Cabernet Sauvignon” clone 170/SO4 | “Sauvignon Blanc”/376 and R140 |
| Year Established | 2000 | 2015 |
| Management/ Plantation frame | Cover crop, trellis formation system, and double cordon/3 × 1 m | Cover crop, trellis formation system, and double cordon/3 × 1.2 m |
| Soil Type | Loam texture; calcisol (accumulation of calcium carbonate at a certain depth, basic pH) | Loam texture; calcisol |
| Height (m.a.s.l) | 375 | 401 |
| Temperature, Rainfall, and Climate | 14.23 °C, 486 mm, and continental Mediterranean climate | 13.00 °C, 486 mm, and continental Mediterranean climate |
| Average Yield (last 3 years) | 6000 kg/ha | 9000 kg/ha |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Langa-Lomba, N.; Grimplet, J.; Sánchez-Hernández, E.; Martín-Ramos, P.; Casanova-Gascón, J.; Julián-Lagunas, C.; González-García, V. Metagenomic Study of Fungal Microbial Communities in Two PDO Somontano Vineyards (Huesca, Spain): Effects of Age, Plant Genotype, and Initial Phytosanitary Status on the Priming and Selection of their Associated Microorganisms. Plants 2023, 12, 2251. https://doi.org/10.3390/plants12122251
Langa-Lomba N, Grimplet J, Sánchez-Hernández E, Martín-Ramos P, Casanova-Gascón J, Julián-Lagunas C, González-García V. Metagenomic Study of Fungal Microbial Communities in Two PDO Somontano Vineyards (Huesca, Spain): Effects of Age, Plant Genotype, and Initial Phytosanitary Status on the Priming and Selection of their Associated Microorganisms. Plants. 2023; 12(12):2251. https://doi.org/10.3390/plants12122251
Chicago/Turabian StyleLanga-Lomba, Natalia, Jerome Grimplet, Eva Sánchez-Hernández, Pablo Martín-Ramos, José Casanova-Gascón, Carmen Julián-Lagunas, and Vicente González-García. 2023. "Metagenomic Study of Fungal Microbial Communities in Two PDO Somontano Vineyards (Huesca, Spain): Effects of Age, Plant Genotype, and Initial Phytosanitary Status on the Priming and Selection of their Associated Microorganisms" Plants 12, no. 12: 2251. https://doi.org/10.3390/plants12122251
APA StyleLanga-Lomba, N., Grimplet, J., Sánchez-Hernández, E., Martín-Ramos, P., Casanova-Gascón, J., Julián-Lagunas, C., & González-García, V. (2023). Metagenomic Study of Fungal Microbial Communities in Two PDO Somontano Vineyards (Huesca, Spain): Effects of Age, Plant Genotype, and Initial Phytosanitary Status on the Priming and Selection of their Associated Microorganisms. Plants, 12(12), 2251. https://doi.org/10.3390/plants12122251