
Compensatory Base Changes and Varying Phylogenetic Effects on Angiosperm ITS2 Genetic Distances


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. ITS2 Sequences and Their Genetic Distances among the Investigated Lineages
2.2. ITS2 Secondary Structure and the Structure-Based SSP Alignments
2.3. Comparison of the Best-Fitting DNA and RNA Models
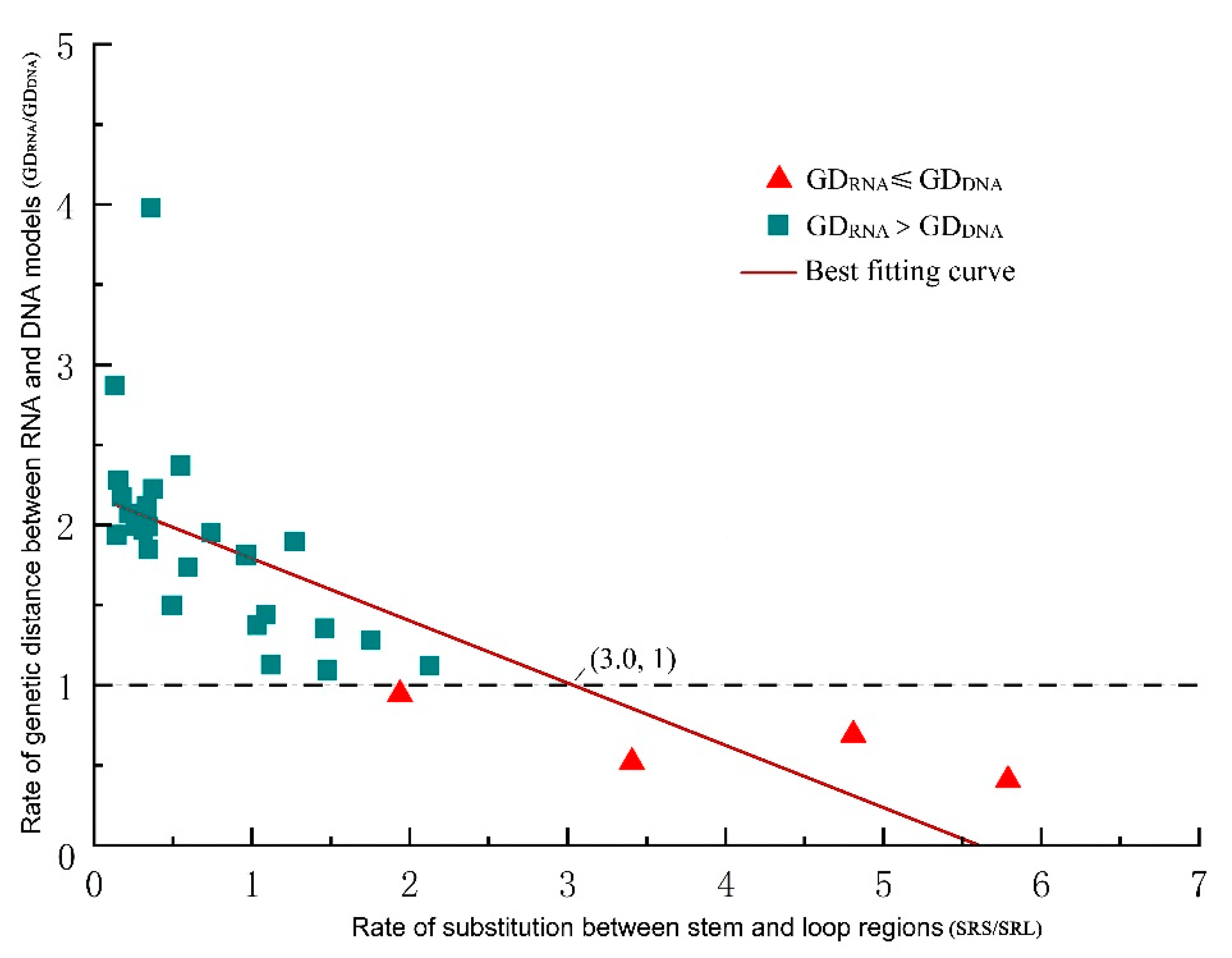
2.4. Comparison between GDRNA and GDDNA
3. Discussion
4. Materials and Methods
4.1. Lineage Sampling and Sister Species Pair Acquisition
4.2. ITS2 Sequence-Structure Alignment
4.3. Genetic Distance Acquisition Using DNA and RNA Substitution Models
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nei, M.; Kumar, S. Molecular Evolution and Phylogenetics; Oxford University Press: New York, NY, USA, 2000. [Google Scholar]
- Gu, X.; Li, W.-H. Estimation of evolutionary distances under stationary and nonstationary models of nucleotide substitution. Proc. Natl. Acad. Sci. USA 1998, 95, 5899–5905. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. Molecular Evolution: A Statistical Approach; Oxford University Press: New York, NY, USA, 2014. [Google Scholar]
- Xia, X.; Xie, Z.; Salemi, M.; Chen, L.; Wang, Y. An index of substitution saturation and its application. Mol. Phylogenet. Evol. 2002, 26, 1–7. [Google Scholar] [CrossRef]
- Philippe, H.; Brinkmann, H.; Lavrov, D.V.; Littlewood, D.T.J.; Manuel, M.; Wörheide, G.; Baurain, D. Resolving Difficult Phylogenetic Questions: Why More Sequences Are Not Enough. PLoS Biol. 2011, 9, e1000602. [Google Scholar] [CrossRef]
- Posada, D.; Crandall, K.A. Selecting the best-fit model of nucleotide substitution. Syst. Biol. 2001, 50, 580–601. [Google Scholar] [CrossRef] [PubMed]
- Rousset, F.; Pélandakis, M.; Solignac, M. Evolution of compensatory substitutions through G.U intermediate state in Drosophila rRNA. Proc. Natl. Acad. Sci. USA 1991, 88, 10032–10036. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M.; Chen, S.; Song, J.; Ankenbrand, M.; Müller, T. Compensatory base changes in ITS2 secondary structures correlate with the biological species concept despite intragenomic variability in ITS2 sequences—A proof of concept. PLoS ONE 2013, 8, e66726. [Google Scholar] [CrossRef]
- Li, M.; Zhao, H.; Zhao, F.; Jiang, L.; Peng, H.; Zhang, W.; Simmons, M.P. Alternative analyses of compensatory base changes in an ITS2 phylogeny of Corydalis (Papaveraceae). Ann. Bot. 2019, 124, 233–243. [Google Scholar] [CrossRef]
- Wheeler, W.C.; Honeycutt, R.L. Paired sequence difference in ribosomal RNAs: Evolutionary and phylogenetic implications. Mol. Biol. Evol. 1988, 5, 90–96. [Google Scholar] [CrossRef]
- Dixon, M.T.; Hillis, D.M. Ribosomal RNA secondary structure: Compensatory mutations and implications for phylogenetic analysis. Mol. Biol. Evol. 1993, 10, 256–267. [Google Scholar] [CrossRef][Green Version]
- Galtier, N. Sampling properties of the bootstrap support in molecular phylogeny: Influence of nonindependence among sites. Syst. Biol. 2004, 53, 38–46. [Google Scholar] [CrossRef]
- Tillier, E.R.M.; Collins, A.R. High Apparent Rate of Simultaneous Compensatory Base-Pair Substitutions in Ribosomal RNA. Genetics 1998, 148, 1993–2002. [Google Scholar] [CrossRef] [PubMed]
- Savill, N.J.; Hoyle, D.C.; Higgs, P.G. RNA Sequence Evolution with Secondary Structure Constraints: Comparison of Substitution Rate Models Using Maximum-Likelihood Methods. Genetics 2001, 157, 399–411. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.E.; Whelan, S. Assessing the State of Substitution Models Describing Noncoding RNA Evolution. Genome Biol. Evol. 2014, 6, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Jow, H.; Hudelot, C.; Rattray, M.; Higgs, P.G. Bayesian Phylogenetics Using an RNA Substitution Model Applied to Early Mammalian Evolution. Mol. Biol. Evol. 2002, 19, 1591–1601. [Google Scholar] [CrossRef] [PubMed]
- Telford, M.J.; Wise, M.J.; Gowri-Shankar, V. Consideration of RNA Secondary Structure Significantly Improves Likelihood-Based Estimates of Phylogeny: Examples from the Bilateria. Mol. Biol. Evol. 2005, 22, 1129–1136. [Google Scholar] [CrossRef]
- Letsch, O.H.; Kjer, K.M. Potential pitfalls of modelling ribosomal RNA data in phylogenetic tree reconstruction: Evidence from case studies in the Metazoa. BMC Evol. Biol. 2011, 11, 146. [Google Scholar] [CrossRef]
- Schultz, J.; Maisel, S.; Gerlach, D.; Müller, T.; Wolf, M. A common core of secondary structure of the internal transcribed spacer 2 (ITS2) throughout the Eukaryota. RNA 2005, 11, 361–364. [Google Scholar] [CrossRef]
- Coleman, A.W. Nuclear rRNA transcript processing versus internal transcribed spacer secondary structure. Trends Genet. 2015, 31, 157–163. [Google Scholar] [CrossRef]
- Zhang, W.; Tian, W.; Gao, Z.; Wang, G.; Zhao, H. Phylogenetic Utility of rRNA ITS2 Sequence-Structure under Functional Constraint. Int. J. Mol. Sci. 2020, 21, 6395. [Google Scholar] [CrossRef]
- Qin, Y.; Li, M.; Cao, Y.; Gao, Y.; Zhang, W. Molecular thresholds of ITS2 and their implications for molecular evolution and species identification in seed plants. Sci. Rep. 2017, 7, 17316. [Google Scholar] [CrossRef]
- Álvarez, I.J.F.W.; Wendel, J.F. Ribosomal ITS sequences and plant phylogenetic inference. Mol. Phylogenet. Evol. 2003, 29, 417–434. [Google Scholar] [CrossRef]
- Chen, S.; Yao, H.; Han, J.; Liu, C.; Song, J.; Shi, L.; Zhu, Y.; Ma, X.; Gao, T.; Pang, X.; et al. Validation of the ITS2 Region as a Novel DNA Barcode for Identifying Medicinal Plant Species. PLoS ONE 2010, 5, e8613. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Cao, Y.; Zhang, W.; Simmons, M.P. Adenine·cytosine substitutions are an alternative pathway of compensatory mutation in angiosperm ITS2. RNA 2019, 26, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Ankenbrand, M.; Keller, A.; Wolf, M.; Schultz, J.; Förster, F. ITS2 Database V: Twice as Much: Table 1. Mol. Biol. Evol. 2015, 32, 3030–3032. [Google Scholar] [CrossRef]
- Seibel, P.N.; Müller, T.; Dandekar, T.; Schultz, J.; Wolf, M. 4SALE–a tool for synchronous RNA sequence and secondary structure alignment and editing. BMC Bioinform. 2006, 7, 498. [Google Scholar] [CrossRef]
- Wolf, M.; Koetschan, C.; Müller, T. ITS2, 18S, 16S or any other RNA—Simply aligning sequences and their individual secondary structures simultaneously by an automatic approach. Gene 2014, 546, 145–149. [Google Scholar] [CrossRef]
- PHASE: A Softwzare Package for Phylogenetics and Sequence Evolution. Available online: http://www.bioinf.man.ac.uk/resources/phase/ (accessed on 1 March 2022).



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, R.; Tong, S.; Luan, T.; Zheng, H.; Zhang, W. Compensatory Base Changes and Varying Phylogenetic Effects on Angiosperm ITS2 Genetic Distances. Plants 2022, 11, 929. https://doi.org/10.3390/plants11070929
Cao R, Tong S, Luan T, Zheng H, Zhang W. Compensatory Base Changes and Varying Phylogenetic Effects on Angiosperm ITS2 Genetic Distances. Plants. 2022; 11(7):929. https://doi.org/10.3390/plants11070929
Chicago/Turabian StyleCao, Ruixin, Shuyan Tong, Tianjing Luan, Hanyun Zheng, and Wei Zhang. 2022. "Compensatory Base Changes and Varying Phylogenetic Effects on Angiosperm ITS2 Genetic Distances" Plants 11, no. 7: 929. https://doi.org/10.3390/plants11070929
APA StyleCao, R., Tong, S., Luan, T., Zheng, H., & Zhang, W. (2022). Compensatory Base Changes and Varying Phylogenetic Effects on Angiosperm ITS2 Genetic Distances. Plants, 11(7), 929. https://doi.org/10.3390/plants11070929