
Sequence Capture of Mitochondrial Genome with PCR-Generated Baits Provides New Insights into the Biogeography of the Genus Abies Mill.

 and
and {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. MtDNA Tree
2.2. Possible Admixtute among the Fir Lineages
3. Discussion
3.1. Sequence Capture Data Shed New Light on the Evolution and History of the Colonization of Abies through Seed Dispersion
3.2. Solving the Problem of Sample Classification Errors
3.3. Pros and Cons of the Used Approach
4. Materials and Methods
4.1. Assembly of Abies Sibirica Genome and Mitochondrial Contigs Selection
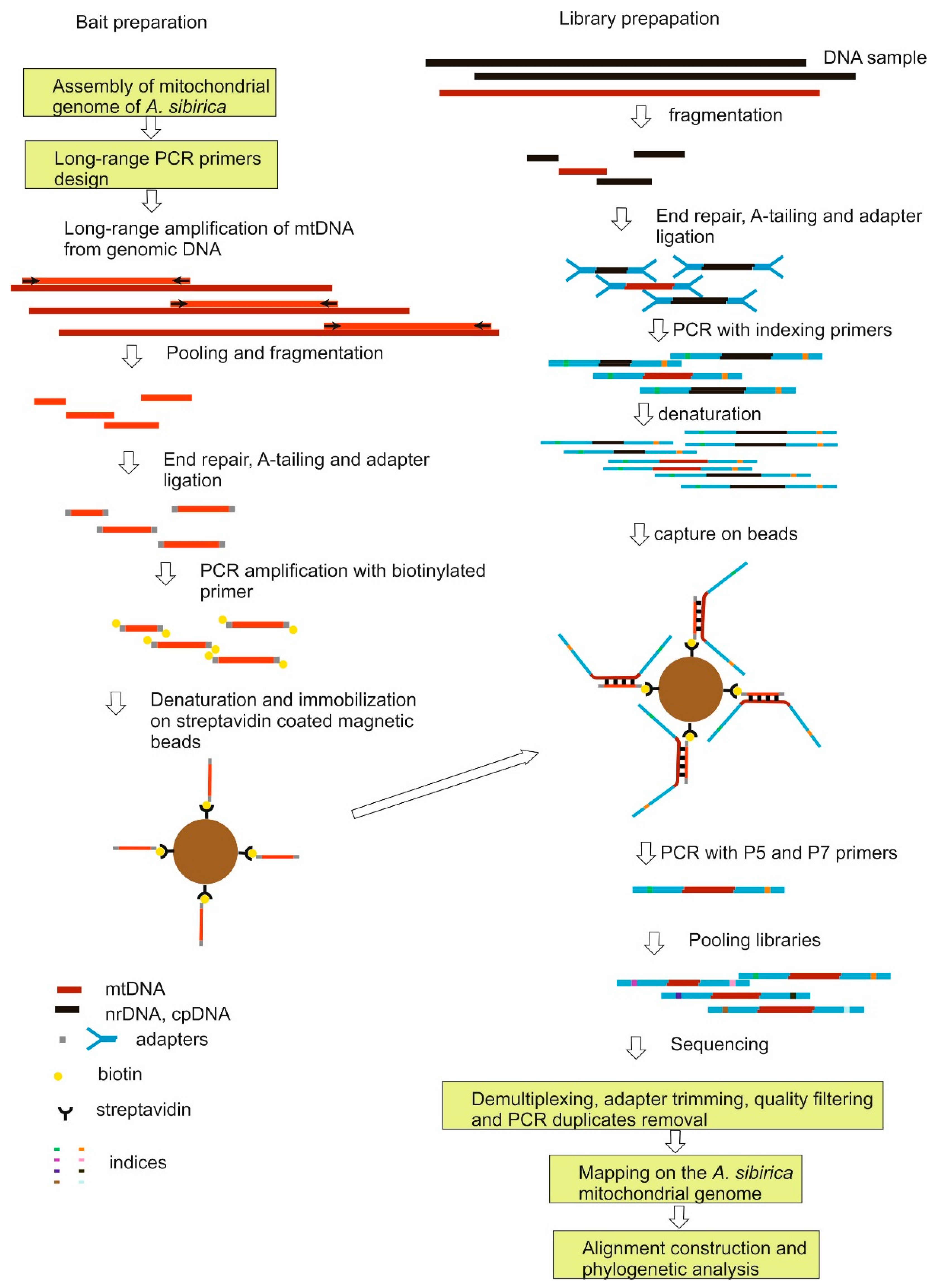
4.2. Preparing of Baits
4.3. Preparation of Libraries
4.4. Hybridization
4.5. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Petit, R.J.; Kremer, A.; Wagner, D.B. Finite Island Model for Organelle and Nuclear Genes in Plants. Heredity 1993, 71, 630–641. [Google Scholar] [CrossRef] [Green Version]
- Currat, M.; Ruedi, M.; Petit, R.J.; Excoffier, L. The hidden side of invasions: Massive introgression by local genes. Evolution 2008, 62, 1908–1920. [Google Scholar] [CrossRef] [PubMed]
- Bouille, M.; Senneville, S.; Bousquet, J. Discordant mtDNA and cpDNA phylogenies indicate geographic speciation and reticulation as driving factors for the diversification of the genus Picea. Tree Genet. Genomes 2011, 7, 469–484. [Google Scholar] [CrossRef]
- Tsutsui, K.; Suwa, A.; Sawada, K.; Kato, T.; Ohsawa, T.A.; Watano, Y. Incongruence among mitochondrial, chloroplast and nuclear gene trees in Pinus subgenus Strobus (Pinaceae). J. Plant Res. 2009, 122, 509–521. [Google Scholar] [CrossRef] [PubMed]
- Neale, D.B.; Sederoff, R.R. Paternal Inheritance of Chloroplast DNA and Maternal Inheritance of mitochondrial-DNA in loblolly-pine. Appl. Genet. 1989, 77, 212–216. [Google Scholar] [CrossRef] [PubMed]
- Jaramillo-Correa, J.P.; Bousquet, J.; Beaulieu, J.; Isabel, N.; Perron, M.; Bouille, M. Cross-species amplification of mitochondrial DNA sequence-tagged-site markers in conifers: The nature of polymorphism and variation within and among species in Picea. Appl. Genet. 2003, 106, 1353–1367. [Google Scholar] [CrossRef]
- Wolfe, K.H.; Li, W.H.; Sharp, P.M. Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast, and nuclear dnas. Proc. Natl. Acad. Sci. USA 1987, 84, 9054–9058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaramillo-Correa, J.P.; Bousquet, J. Mitochondrial genome recombination in the zone of contact between two hybridizing conifers. Genetics 2005, 171, 1951–1962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mamanova, L.; Coffey, A.J.; Scott, C.E.; Kozarewa, I.; Turner, E.H.; Kumar, A.; Howard, E.; Shendure, J.; Turner, D.J. Target-enrichment strategies for next-generation sequencing. Nat. Methods 2010, 7, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Semerikova, S.A.; Khrunyk, Y.Y.; Lascoux, M.; Semerikov, V.L. From America to Eurasia: A multigenomes history of the genus Abies. Mol. Phylogenet. Evol. 2018, 125, 14–28. [Google Scholar] [CrossRef]
- Aguirre-Planter, E.; Jaramillo-Correa, J.P.; Gomez-Acevedo, S.; Khasa, D.P.; Bousquet, J.; Eguiarte, L.E. Phylogeny, diversification rates and species boundaries of Mesoamerican firs (Abies, Pinaceae) in a genus-wide context. Mol. Phylogenet. Evol. 2012, 62, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Schwartz, S.; Wagner, L.; Miller, W. A greedy algorithm for aligning DNA sequences. J. Comput. Biol. 2000, 7, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Tsumura, Y.; Suyama, Y. Differentiation of mitochondrial DNA polymorphisms in populations of five Japanese Abies species. Evolution 1998, 52, 1031–1042. [Google Scholar] [CrossRef] [PubMed]
- Semerikov, V.L.; Semerikova, S.A.; Putintseva, Y.A. Mitochondrial DNA Confirms the American Origin of Modern Firs. Russ. J. Genet. 2021, 57, 1258–1262. [Google Scholar] [CrossRef]
- Liepelt, S.; Bialozyt, R.; Ziegenhagen, B. Wind-dispersed pollen mediates postglacial gene flow among refugia. Proc. Natl. Acad. Sci. USA 2002, 99, 14590–14594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balao, F.; Lorenzo, M.T.; Sanchez-Robles, J.M.; Paun, O.; Garcia-Castano, J.L.; Terrab, A. Early diversification and permeable species boundaries in the Mediterranean firs. Ann. Bot. 2020, 125, 495–507. [Google Scholar] [CrossRef] [PubMed]
- Schorn, H.E.; Wehr, W.C. Abies milleri, sp. nov., from the Middle Eocene Klondike Mountain Formation, Republic, Ferry County, Washington; Thomas Burke Memorial Washington State Museum: Seattle, WA, USA, 1986. [Google Scholar]
- Semerikov, V.L.; Semerikova, S.A.; Putintseva, Y.A.; Tarakanov, V.V.; Tikhonova, I.V.; Vidyakin, A.I.; Oreshkova, N.V.; Krutovsky, K.V. Colonization history of Scots pine in Eastern Europe and North Asia based on mitochondrial DNA variation. Tree Genet. Genomes 2018, 14, 8. [Google Scholar] [CrossRef] [Green Version]
- Semerikov, V.L.; Semerikova, S.A.; Putintseva, Y.A.; Oreshkova, N.V.; Krutovsky, K.V. Mitochondrial DNA in Siberian conifers indicates multiple postglacial colonization centers. Can. J. For. Res. 2019, 49, 875–883. [Google Scholar] [CrossRef] [Green Version]
- Donnelly, K.; Cottrell, J.; Ennos, R.A.; Vendramin, G.G.; A’Hara, S.; King, S.; Perry, A.; Wachowiak, W.; Cavers, S. Reconstructing the plant mitochondrial genome for marker discovery: A case study using Pinus. Mol. Ecol. Resour. 2017, 17, 943–954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semerikova, S.A.; Semerikov, V.L. Phylogeny of Firs (Genus Abies, Pinaceae) Based on Multilocus Nuclear Markers (AFLP). Russ. J. Genet. 2016, 52, 1164–1175. [Google Scholar] [CrossRef]
- Jackman, S.D.; Coombe, L.; Warren, R.L.; Kirk, H.; Trinh, E.; MacLeod, T.; Pleasance, S.; Pandoh, P.; Zhao, Y.J.; Coope, R.J.; et al. Complete Mitochondrial Genome of a Gymnosperm, Sitka Spruce (Picea sitchensis), Indicates a Complex Physical Structure. Genome Biol. Evol. 2020, 12, 1174–1179. [Google Scholar] [CrossRef] [PubMed]
- Putintseva, Y.A.; Bondar, E.I.; Simonov, E.P.; Sharov, V.V.; Oreshkova, N.V.; Kuzmin, D.A.; Konstantinov, Y.M.; Shmakov, V.N.; Belkov, V.I.; Sadovsky, M.G.; et al. Siberian larch (Larix sibirica Ledeb.) mitochondrial genome assembled using both short and long nucleotide sequence reads is currently the largest known mitogenome. BMC Genom. 2020, 21, 654. [Google Scholar] [CrossRef] [PubMed]
- Kan, S.L.; Shen, T.T.; Gong, P.; Ran, J.H.; Wang, X.Q. The complete mitochondrial genome of Taxus cuspidata (Taxaceae): Eight protein-coding genes have transferred to the nuclear genome. BMC Evol. Biol. 2020, 20, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, A.R.; Eldfjell, Y.; Schiffthaler, B.; Delhomme, N.; Asp, T.; Hebelstrup, K.H.; Keech, O.; Oberg, L.; Moller, I.M.; Arvestad, L.; et al. The Mitogenome of Norway Spruce and a Reappraisal of Mitochondrial Recombination in Plants. Genome Biol. Evol. 2020, 12, 3586–3598. [Google Scholar] [CrossRef] [PubMed]
- Nystedt, B.; Street, N.R.; Wetterbom, A.; Zuccolo, A.; Lin, Y.C.; Scofield, D.G.; Vezzi, F.; Delhomme, N.; Giacomello, S.; Alexeyenko, A.; et al. The Norway spruce genome sequence and conifer genome evolution. Nature 2013, 497, 579–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, J.Y.; Dong, S.S.; Wang, M.Q.; Song, T.Q.; Zhou, G.C.; Li, Z.; Van de Peer, Y.; Shao, Z.Q.; Wang, W.; Chen, M.; et al. Mitochondrial genes from 18 angiosperms fill sampling gaps for phylogenomic inferences of the early diversification of flowering plants. J. Syst. Evol. 2021. [Google Scholar] [CrossRef]
- Bell, D.; Lin, Q.S.; Gerelle, W.K.; Joya, S.; Chang, Y.; Taylor, Z.N.; Rothfels, C.J.; Larsson, A.; Villarreal, J.C.; Li, F.W.; et al. Organellomic data sets confirm a cryptic consensus on (unrooted) land-plant relationships and provide new insights into bryophyte molecular evolution. Am. J. Bot. 2020, 107, 91–115. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Cox, C.J.; Wang, W.; Goffinet, B. Mitochondrial Phylogenomics of Early Land Plants: Mitigating the Effects of Saturation, Compositional Heterogeneity, and Codon-Usage Bias. Syst. Biol. 2014, 63, 862–878. [Google Scholar] [CrossRef] [Green Version]
- Palmer, J.D.; Herbon, L.A. Plant mitochondrial-DNA evolves rapidly in structure, but slowly in sequence. J. Mol. Evol. 1988, 28, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Sloan, D.B. One ring to rule them all? Genome sequencing provides new insights into the ‘master circle’ model of plant mitochondrial DNA structure. New Phytol. 2013, 200, 978–985. [Google Scholar] [CrossRef]
- Bogarin, D.; Perez-Escobar, O.A.; Groenenberg, D.; Holland, S.D.; Karremans, A.P.; Lemmon, E.M.; Lemmon, A.R.; Pupulin, F.; Smets, E.; Gravendeel, B. Anchored hybrid enrichment generated nuclear, plastid and mitochondrial markers resolve the Lepanthes horrida (Orchidaceae: Pleurothallidinae) species complex. Mol. Phylogenet. Evol. 2018, 129, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Schulte, L.; Bernhardt, N.; Stoof-Leichsenring, K.; Zimmermann, H.H.; Pestryakova, L.A.; Epp, L.S.; Herzschuh, U. Hybridization capture of larch (Larix Mill.) chloroplast genomes from sedimentary ancient DNA reveals past changes of Siberian forest. Mol. Ecol. Resour. 2021, 21, 801–815. [Google Scholar] [CrossRef] [PubMed]
- Mariac, C.; Scarcelli, N.; Pouzadou, J.; Barnaud, A.; Billot, C.; Faye, A.; Kougbeadjo, A.; Maillol, V.; Martin, G.; Sabot, F.; et al. Cost-effective enrichment hybridization capture of chloroplast genomes at deep multiplexing levels for population genetics and phylogeography studies. Mol. Ecol. Resour. 2014, 14, 1103–1113. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Deng, S.H.; Liang, D.; Zhang, P. Sequence capture across large phylogenetic scales by using pooled PCR-generated baits: A case study of Lepidoptera. Mol. Ecol. Resour. 2019, 19, 1037–1051. [Google Scholar] [CrossRef] [PubMed]
- Farjon, A.; Rushforth, K. A classification of Abies Miller (Pinaceae). Notes R. Bot. Gard. Edinburg 1989, 46, 59–79. [Google Scholar]
- Maricic, T.; Whitten, M.; Paabo, S. Multiplexed DNA Sequence Capture of Mitochondrial Genomes Using PCR Products. PLoS ONE 2010, 5, e14004. [Google Scholar] [CrossRef] [PubMed]
- Rohland, N.; Reich, D. Cost-effective, high-throughput DNA sequencing libraries for multiplexed target capture. Genome Res. 2012, 22, 939–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohri, D.; Khoshoo, T.N. Genome size in gymnosperms. Plant Syst. Evol. 1986, 153, 119–132. [Google Scholar] [CrossRef]
- FastQC. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 12 March 2022).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Laslett, D.; Canback, B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004, 32, 11–16. [Google Scholar] [CrossRef]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef] [PubMed]
- Lagesen, K.; Hallin, P.; Rodland, E.A.; Staerfeldt, H.H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef] [PubMed]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3-new capabilities and interfaces. Nucleic Acids Res. 2012, 40, 811–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for windows 95/98/ NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Swofford, D. PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods). Version 4.0b10; Smithsonian Institution: Washington, DC, USA, 2002. [Google Scholar]
- Smith, S.A.; O’Meara, B.C. treePL: Divergence time estimation using penalized likelihood for large phylogenies. Bioinformatics 2012, 28, 2689–2690. [Google Scholar] [CrossRef] [PubMed]
- Rothwell, G.W.; Mapes, G.; Stockey, R.A.; Hilton, J. The seed cone Eathiestrobus gen. nov.: Fossil evidence for a jurassic origin of pinaceae. Am. J. Bot. 2012, 99, 708–720. [Google Scholar] [CrossRef]
- Rothwell, G.W.; Mapes, G.; Mapes, R.H. Late Paleozoic conifers of North America: Structure, diversity and occurrences. Rev. Palaeobot. Palynol. 1997, 95, 95–113. [Google Scholar] [CrossRef]
- Erwin, D.M.; Schorn, H.E. Revision of the conifers from the Eocene Thunder Mountain flora, Idaho, USA. Rev. Palaeobot. Palynol. 2005, 137, 125–145. [Google Scholar] [CrossRef]
- Kremp, G.O.W.; Traverse, A.; Ames, H.T. Catalog of Fossil Spores and Pollen: Permian-Triassic Transition and Mesozoic Spores and Pollen; Pennsylvania State University: University Park, PA, USA, 1967. [Google Scholar]
- Liu, T.; Liu, T.S. A Monograph of the Genus Abies; Department of Forestry, College of Agriculture, National Taiwan University: Taipei, Taiwan, 1971. [Google Scholar]
- Xiang, X.G.; Cao, M.; Zhou, Z.K. Fossil history and modern distribution of the genus Abies (Pinaceae). Front. For. China 2007, 2, 355–365. [Google Scholar] [CrossRef]
- Axelrod, D.I. Evolution of the Santa Lucia Fir (Abies bracteata) Ecosystem. Ann. Mo. Bot. Gard. 1976, 63, 24–41. [Google Scholar] [CrossRef]
- Falcon-Lang, H.J.; Mages, V.; Collinson, M. The oldest Pinus and its preservation by fire. Geology 2016, 44, 303–306. [Google Scholar] [CrossRef] [Green Version]
- Maurin, K.J. An empirical guide for producing a dated phylogeny with treePL in a maximum likelihood framework. arXiv 2008, arXiv:2008.07054 2020. [Google Scholar]
- Bouckaert, R.; Heled, J.; Kuhnert, D.; Vaughan, T.; Wu, C.H.; Xie, D.; Suchard, M.A.; Rambaut, A.; Drummond, A.J. BEAST 2: A Software Platform for Bayesian Evolutionary Analysis. PLoS Comput. Biol. 2014, 10, e1003537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durand, E.Y.; Patterson, N.; Reich, D.; Slatkin, M. Testing for Ancient Admixture between Closely Related Populations. Mol. Biol. Evol. 2011, 28, 2239–2252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Semerikov, V.L.; Semerikova, S.A.; Khrunyk, Y.Y.; Putintseva, Y.A. Sequence Capture of Mitochondrial Genome with PCR-Generated Baits Provides New Insights into the Biogeography of the Genus Abies Mill. Plants 2022, 11, 762. https://doi.org/10.3390/plants11060762
Semerikov VL, Semerikova SA, Khrunyk YY, Putintseva YA. Sequence Capture of Mitochondrial Genome with PCR-Generated Baits Provides New Insights into the Biogeography of the Genus Abies Mill. Plants. 2022; 11(6):762. https://doi.org/10.3390/plants11060762
Chicago/Turabian StyleSemerikov, Vladimir L., Svetlana A. Semerikova, Yuliya Y. Khrunyk, and Yuliya A. Putintseva. 2022. "Sequence Capture of Mitochondrial Genome with PCR-Generated Baits Provides New Insights into the Biogeography of the Genus Abies Mill." Plants 11, no. 6: 762. https://doi.org/10.3390/plants11060762
APA StyleSemerikov, V. L., Semerikova, S. A., Khrunyk, Y. Y., & Putintseva, Y. A. (2022). Sequence Capture of Mitochondrial Genome with PCR-Generated Baits Provides New Insights into the Biogeography of the Genus Abies Mill. Plants, 11(6), 762. https://doi.org/10.3390/plants11060762