
Developmental Regulation of Transcription in Touriga Nacional Berries under Deficit Irrigation

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Results and Discussion
2.1. Bioinformatics Validation
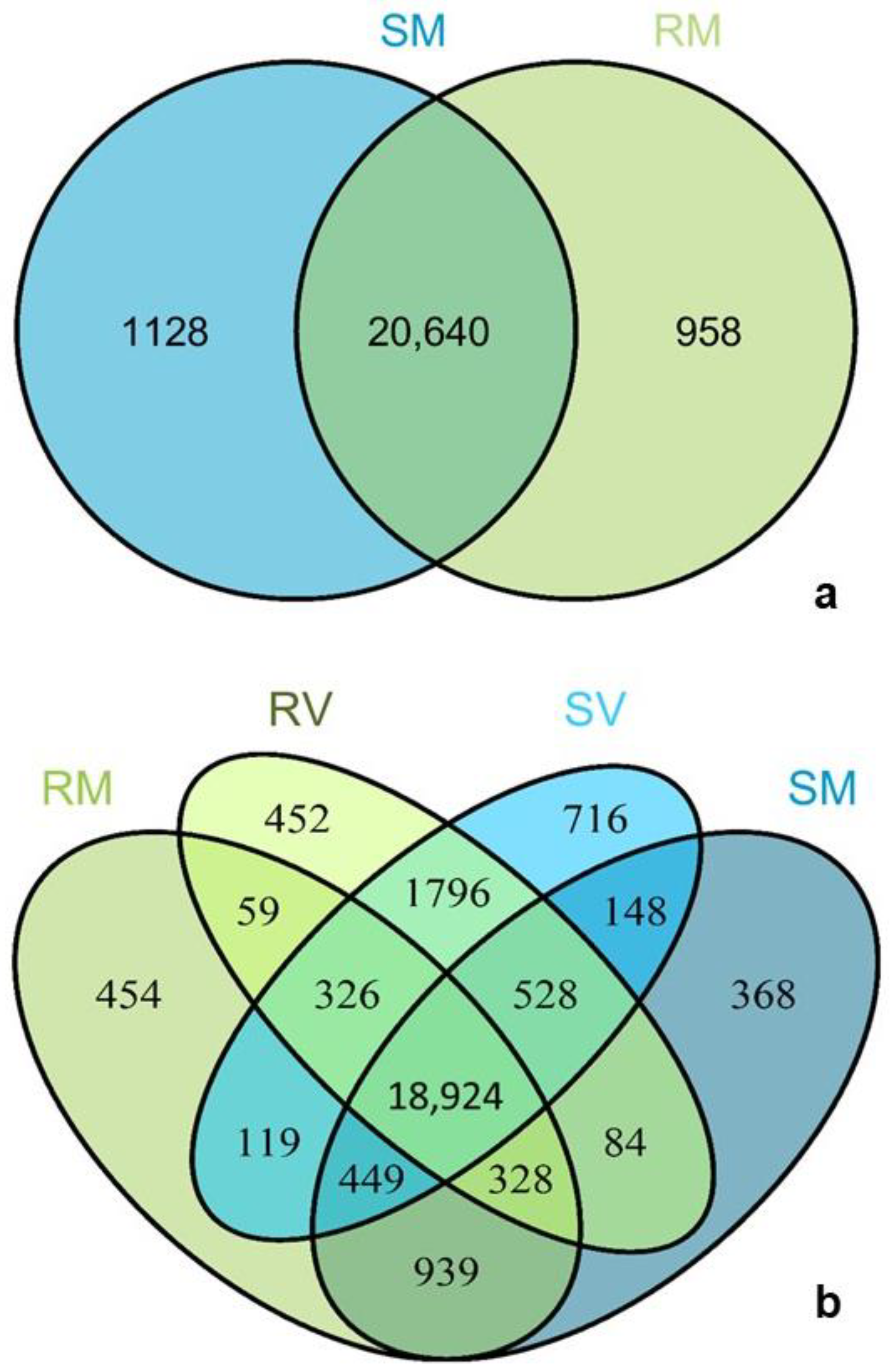
2.2. Revelations from the Whole Transcriptome
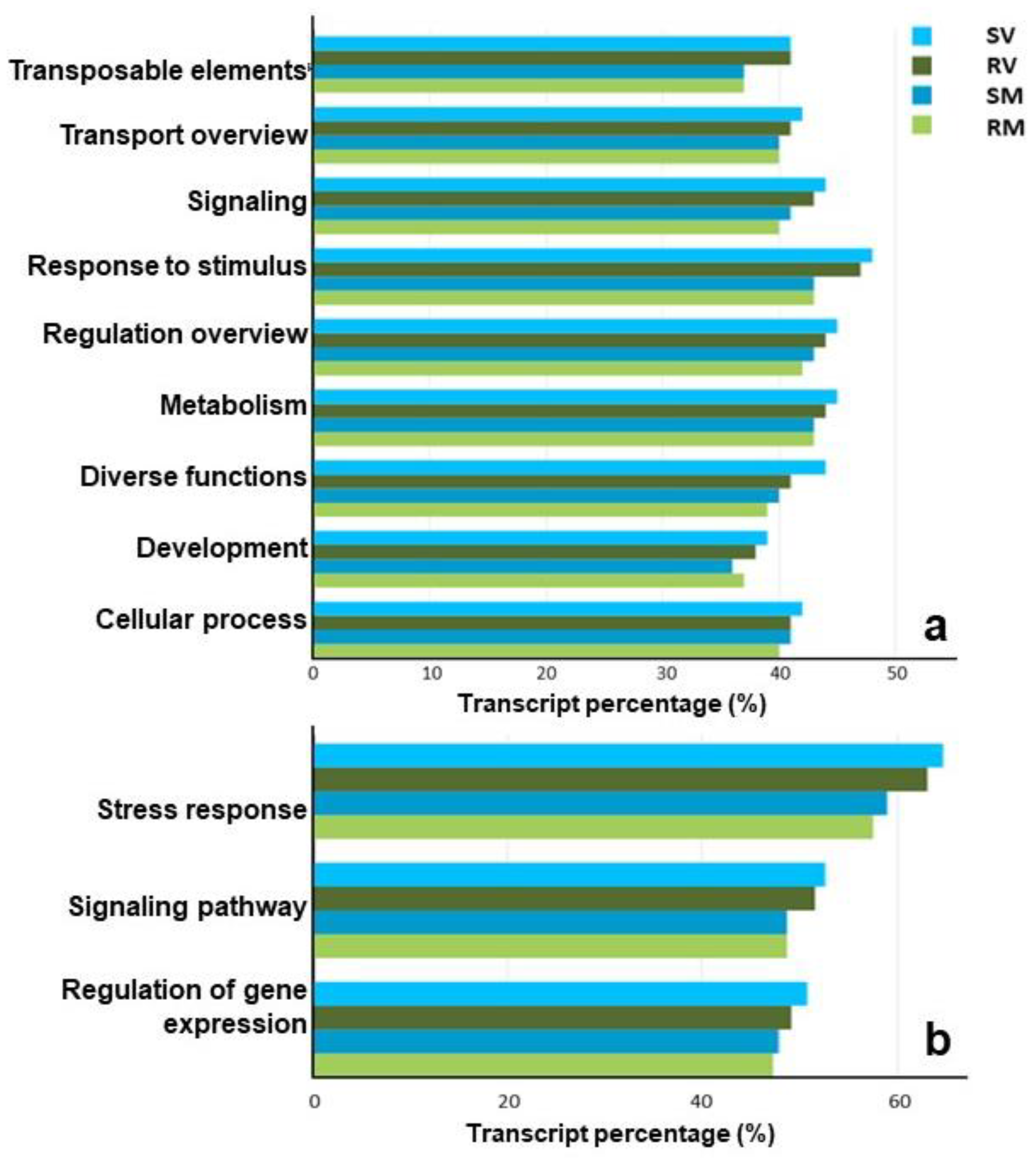
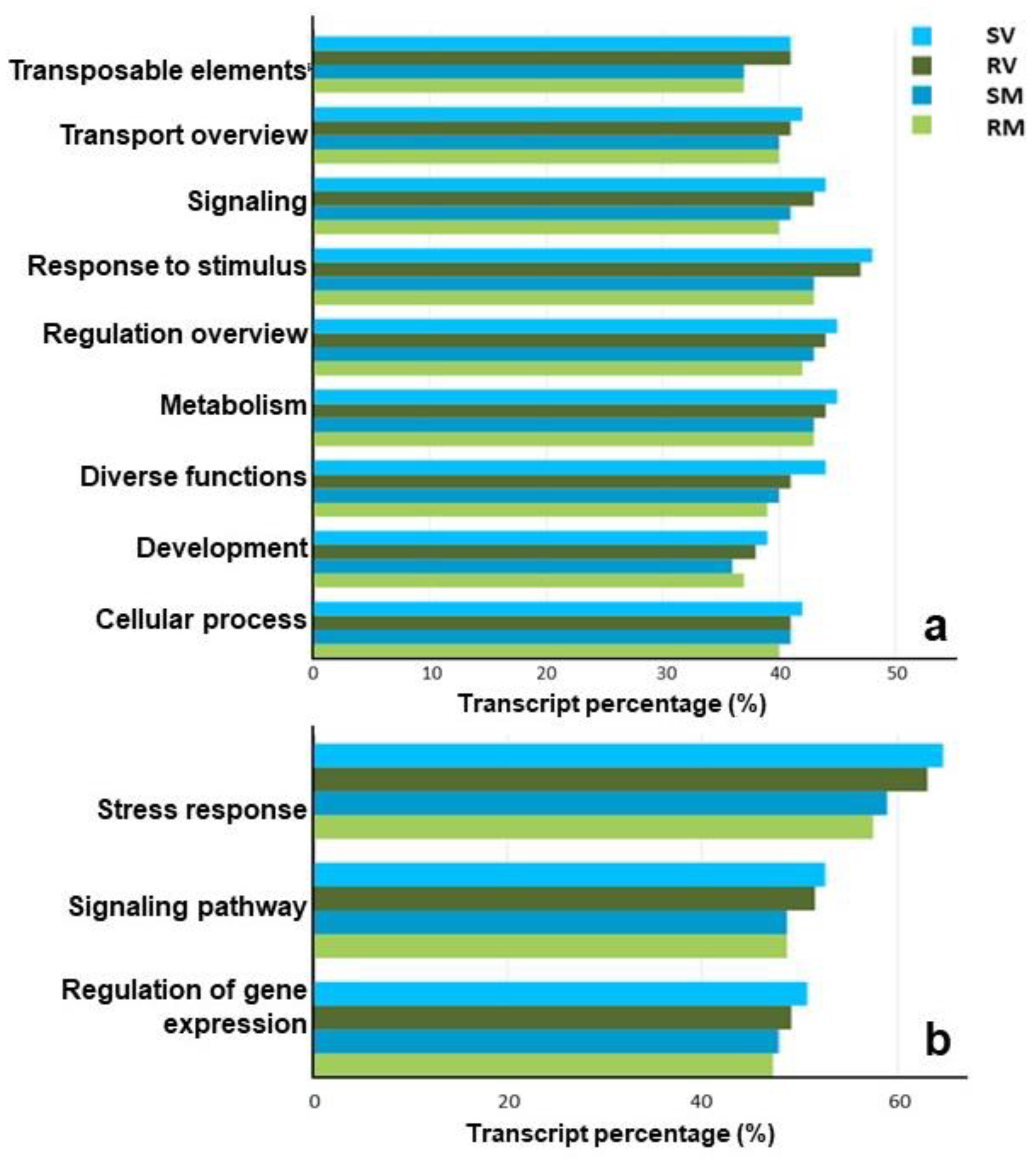
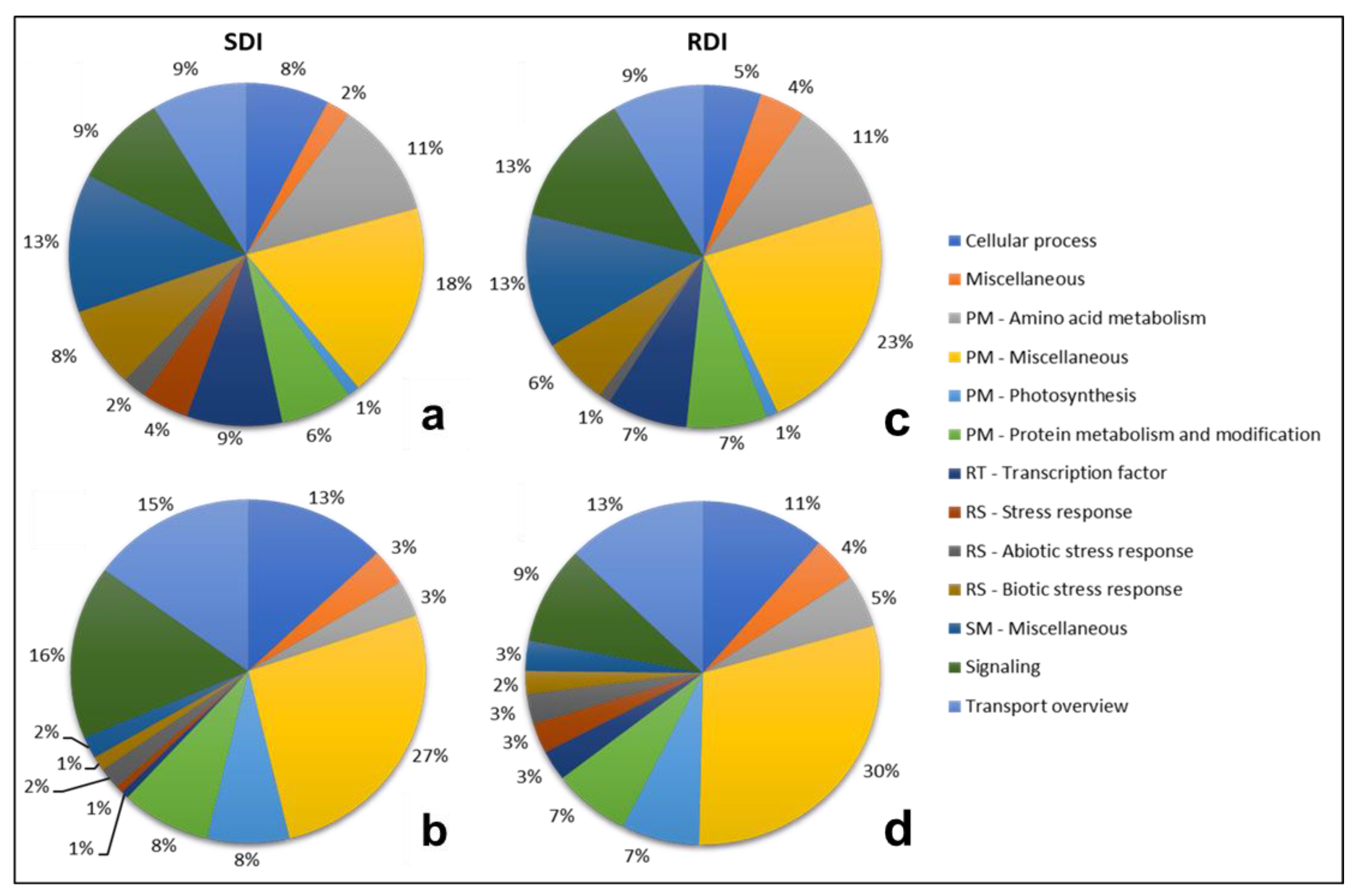
2.3. Functional Profiling of Transcripts
2.4. Correlation of Significantly Regulated DEGs with Leaf Water Potential
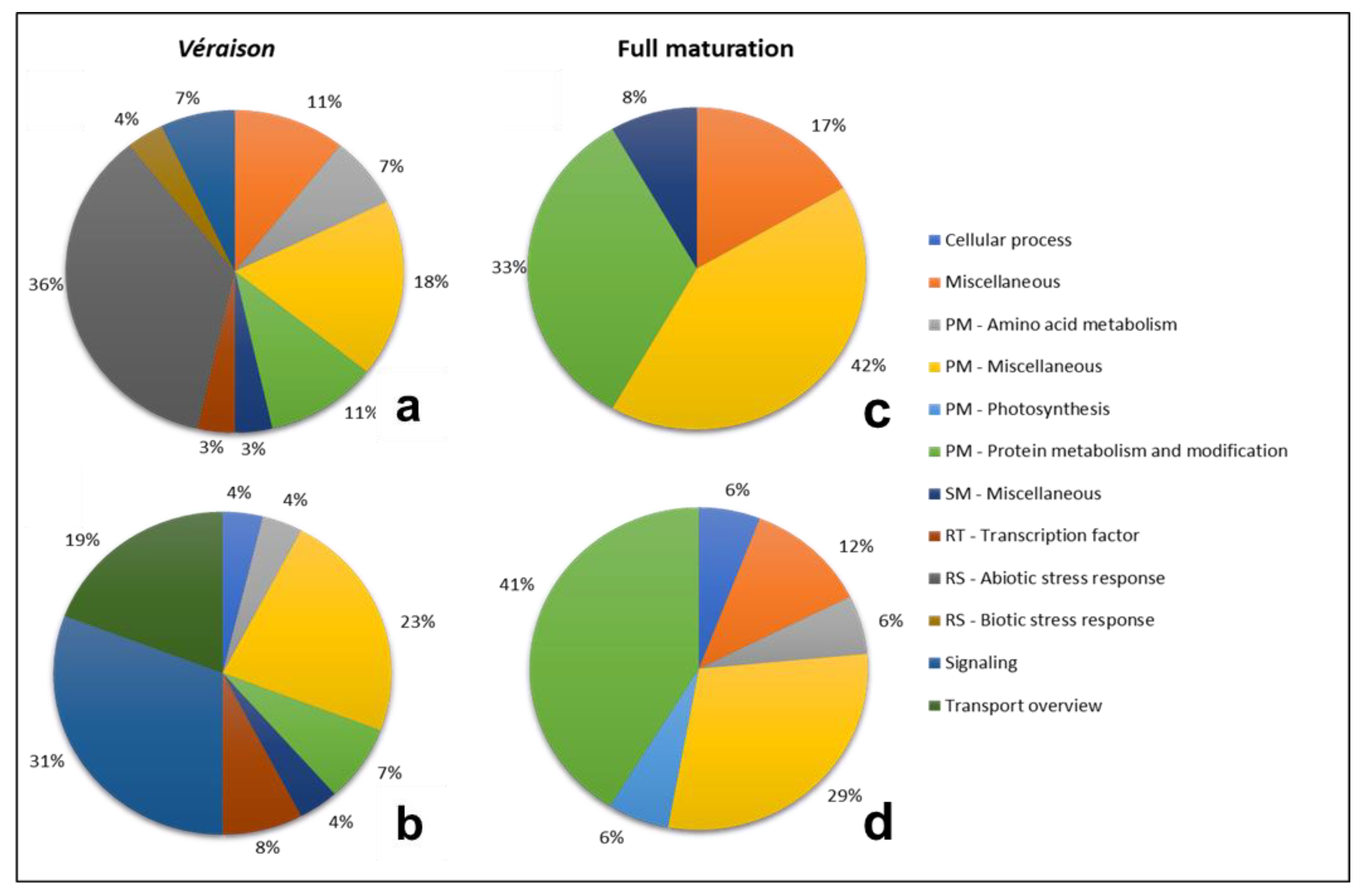
2.5. DEG Functional Profiling
2.5.1. Response to Water Stress
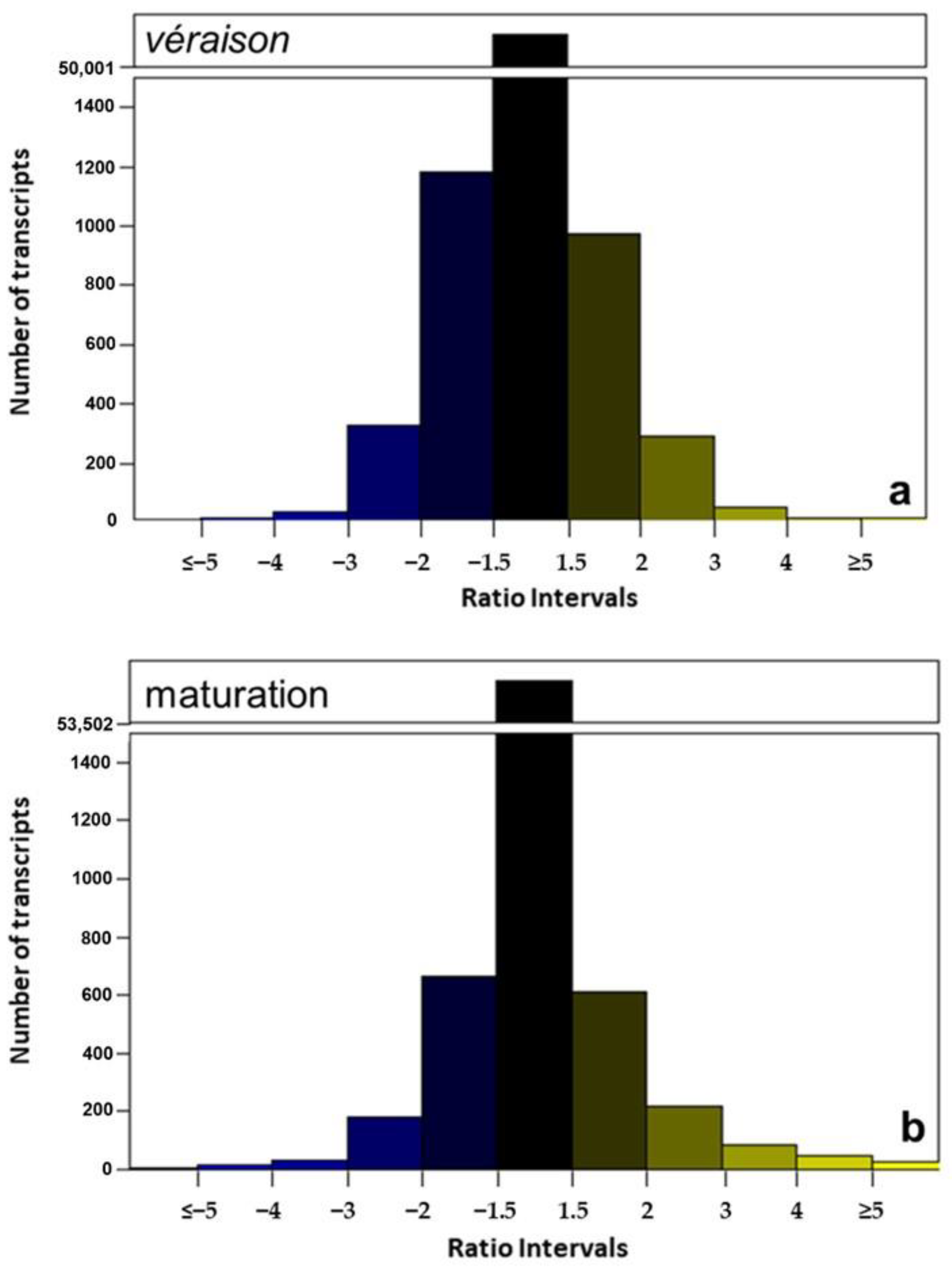
2.5.2. Differential Regulation between Véraison and Maturation
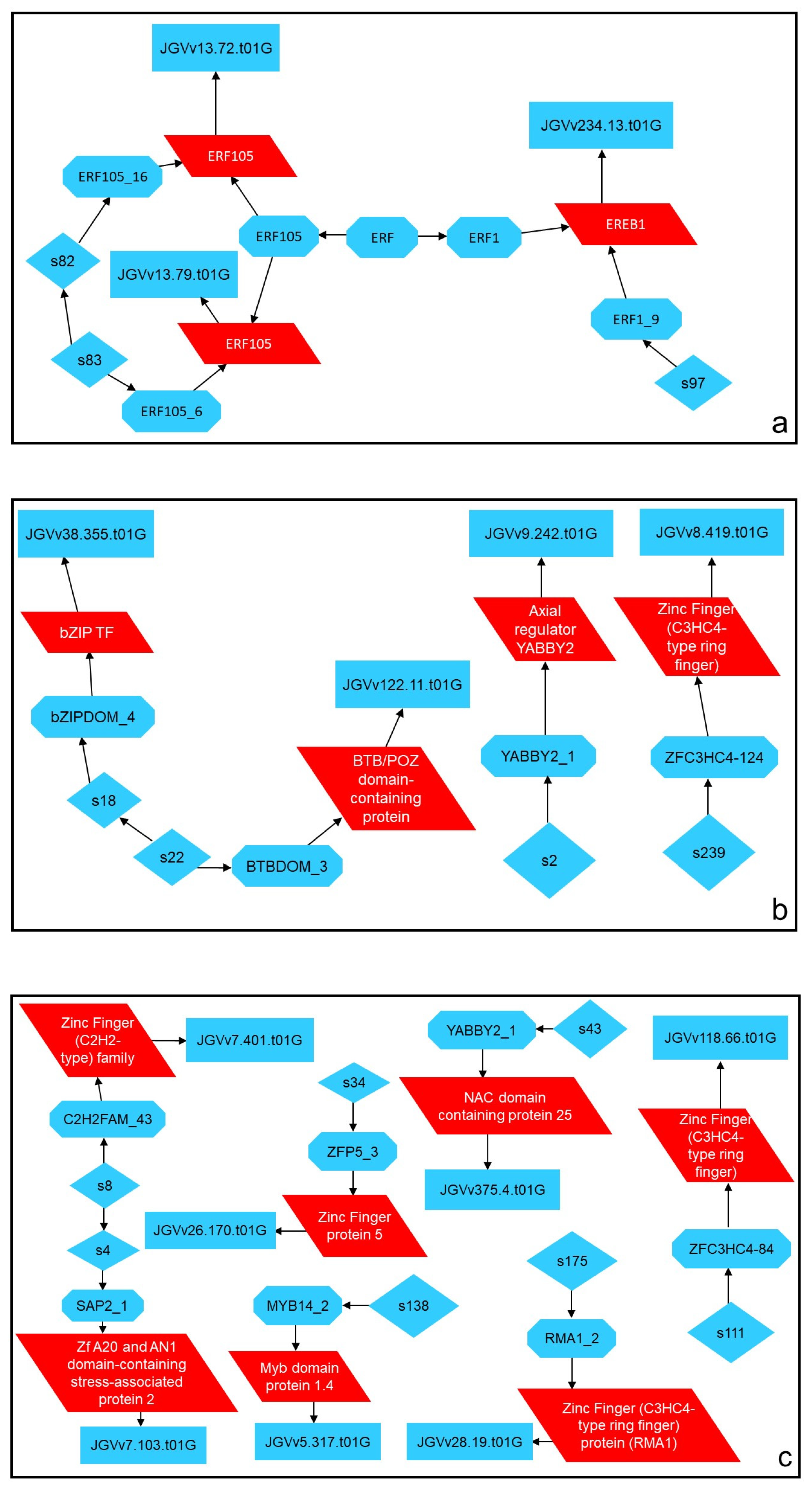
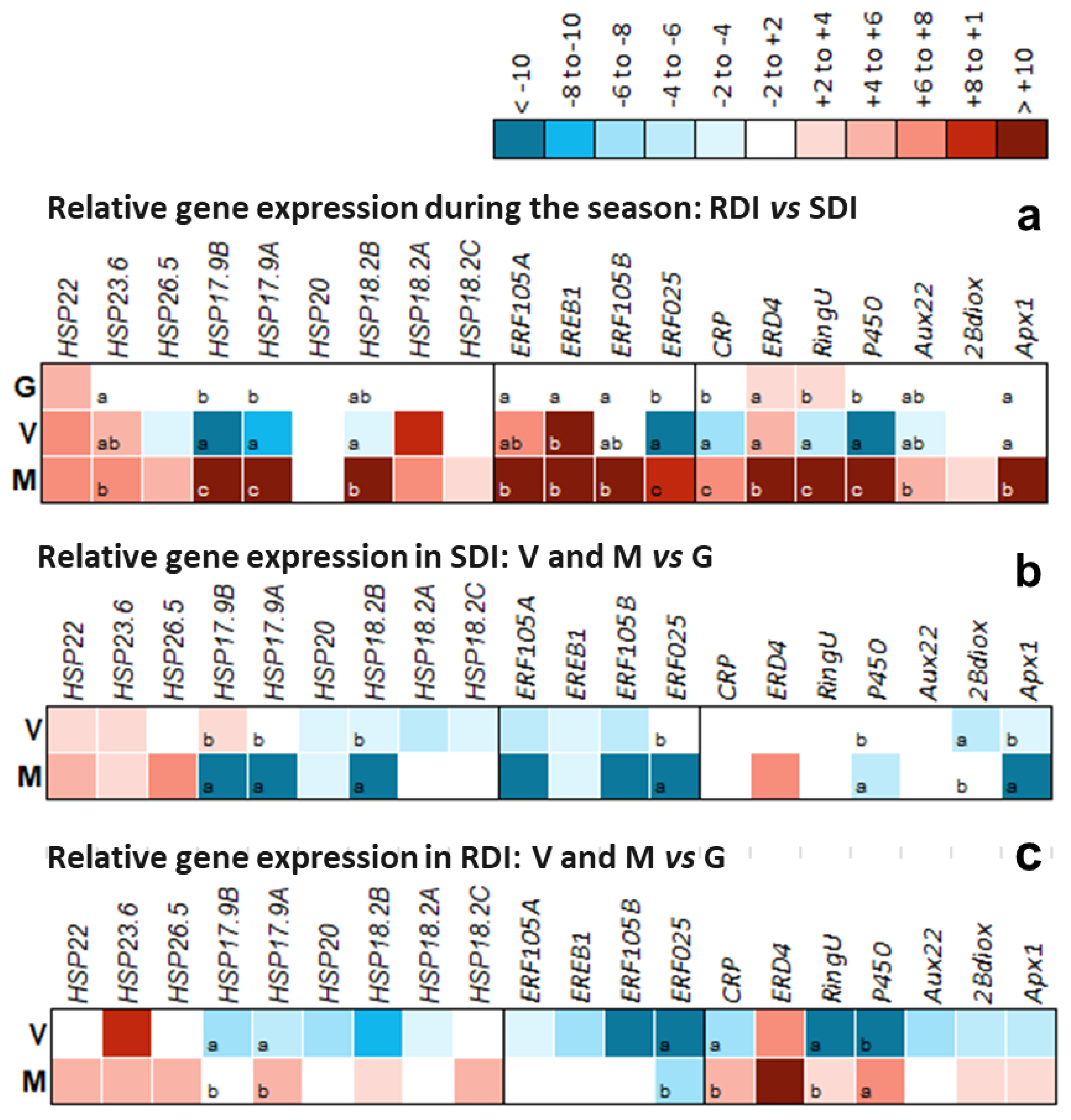
2.6. Close-Up on Relevant DEG Modulation and Its Significance
3. Materials and Methods
3.1. Field Conditions and Sampling
3.2. RNA Extraction, Quantification and Quality Evaluation
3.3. Transcriptome Sequencing and Mapping
3.4. Gene Expression and Validation
3.5. Bioinformatics Analysis
Bioinformatics Validation and Overview
3.6. Differentially Expressed Genes
3.7. Sample-Specific Transcripts
3.8. Correlation between RNA-Seq Results and Leaf Water Potential
3.9. Validation of Expression Profiles through Real Time Quantitative PCR (RT-qPCR)
3.10. Statistical Analysis of RT-qPCR Results
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- OIV, State of the Vitivinicultural Sector in 2020. 2021. Available online: https://www.oiv.int/public/medias/7909/oiv-state-of-the-world-vitivinicultural-sector-in-2020.pdf (accessed on 20 October 2021).
- Fraga, H. Climate Change: A New Challenge for the Winemaking Sector. Agronomy 2020, 10, 1465. [Google Scholar] [CrossRef]
- Carvalho, L.C.; Amâncio, S. Cutting the Gordian Knot of abiotic stress in grapevine: From the test tube to climate change adaptation. Physiol. Plant 2018, 165, 330–342. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, F.; Lionello, P. Climate change projections for the Mediterranean region. Glob. Planet Change 2008, 63, 90–104. [Google Scholar] [CrossRef]
- De Orduña, R.M. Climate change associated effects on grape and wine quality and production. Food Res. Int. 2010, 43, 1844–1855. [Google Scholar] [CrossRef]
- Olesen, J.E.; Trnka, M.; Kersebaum, K.C.; Skjelvåg, A.O.; Seguin, B.; Peltonen-Sainio, P.; Rossi, F.; Kozyra, J.; Micale, F. Impacts and adaptation of European crop production systems to climate change. Eur. J. Agron. 2011, 34, 96–112. [Google Scholar] [CrossRef]
- Dal Santo, S.; Zenoni, S.; Sandri, M.; De Lorenzis, G.; Magris, G.; De Paoli, E.; Di Gaspero, G.; Del Fabbro, C.; Morgante, M. Grapevine field experiments reveal the contribution of genotype, the influence of environment and the effect of their interaction (G×E) on the berry transcriptome. Plant J. 2018, 93, 1143–1159. [Google Scholar] [CrossRef]
- Carvalho, L.C.; Silva, M.; Coito, J.L.; Rocheta, M.P.; Amâncio, S. Design of a custom RT-qPCR array for assignment of abiotic stress tolerance in traditional Portuguese grapevine varieties. Front Plant Sci. 2017, 8, 1835. [Google Scholar] [CrossRef] [Green Version]
- Rocheta, M.; Coito, J.L.; Ramos, M.J.N.; Carvalho, L.C.; Becker, J.D.; Carbonell-Bejerano, P.; Amâncio, S. Transcriptomic comparison between two Vitis vinifera L. varieties (Trincadeira and Touriga Nacional) in abiotic stress conditions. BMC Plant Biol. 2016, 16, 224. [Google Scholar] [CrossRef] [Green Version]
- Girona, J.; Mata, M.; del Campo, J.; Arbonés, A.; Bartra, E.; Marsal, J. The use of midday leaf water potential for scheduling deficit irrigation in vineyards. Irrig. Sci. 2006, 24, 115–127. [Google Scholar] [CrossRef]
- Girona, J.; Marsal, J.; Mata, M.; Del Campo, J.; Basile, B. Phenological sensitivity of berry growth and composition of tempranillo grapevines (Vitis Vinifera L.) to water stress. Aust. J. Grape Wine Res. 2009, 15, 268–277. [Google Scholar] [CrossRef]
- Chaves, M.M.; Zarrouk, O.; Francisco, R.; Costa, J.M.; Santos, T.; Regalado, A.P.; Rodrigues, M.L.; Lopes, C.M. Grapevine under deficit irrigation: Hints from physiological and molecular data. Ann. Bot. 2010, 105, 661–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deytieux-Belleau, C.; Geny, L.; Roudet, J.; Maet, V.; Donèchoe, B.; Fermaud, M. Grape berry skin features related to ontogenic resistance to Botrytis cinerea. Eur. J. Plant Pathol. 2009, 125, 551–563. [Google Scholar] [CrossRef]
- Carbonell-Bejerano, P.; Carvalho, L.C.; Dias, J.E.E.; Martinez-Zapater, J.M.; Amâncio, S. Exploiting Vitis genetic diversity to manage with stress. In Grapevine in a Changing Environment: A Molecular and Ecophysiological Perspective; Wiley-Blackwell: Hoboken, NJ, USA, 2015; pp. 347–380. [Google Scholar] [CrossRef]
- Savoi, S.; Wong, D.C.J.; Arapitsas, P.; Miculan, M.; Bucchetti, B.; Peterlunger, E.; Fait, A.; Mattivi, F.; Castellarin, S.D. Transcriptome and metabolite profiling reveals that prolonged drought modulates the phenylpropanoid and terpenoid pathway in white grapes (Vitis vinifera L.). BMC Plant Biol. 2016, 16, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castellarin, S.D.; Matthews, M.A.; Di Gaspero, G.; Gambetta, G.A. Water deficits accelerate ripening and induce changes in gene expression regulating flavonoid biosynthesis in grape berries. Planta 2007, 227, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Finotello, F.; Di Camillo, B. Measuring differential gene expression with RNA-seq: Challenges and strategies for data analysis. Brief. Funct. Genom. 2015, 14, 130–142. [Google Scholar] [CrossRef]
- Palumbo, M.C.; Zenoni, S.; Fasoli, M.; Massonnet, M.; Farina, L.; Castiglione, F.; Pezzotti, M.; Paci, P. Integrated Network Analysis Identifies Fight-Club Nodes as a Class of Hubs Encompassing Key Putative Switch Genes That Induce Major Transcriptome Reprogramming during Grapevine Development. Plant Cell 2014, 26, 4617–4635. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, L.C.; Coito, J.L.; Colaço, S.; Sangiogo, M.; Amâncio, S. Heat stress in grapevine: The pros and cons of acclimation. Plant Cell Environ. 2015, 38, 777–789. [Google Scholar] [CrossRef]
- Carvalho, L.C.; Coito, J.L.; Gonçalves, E.F.; Chaves, M.M.; Amâncio, S. Differential physiological response of the grapevine varieties Touriga Nacional and Trincadeira to combined heat, drought and light stresses. Plant Biol. 2016, 18, 101–111. [Google Scholar] [CrossRef]
- Hochberg, U.; Degu, A.; Toubiana, D.; Gendler, T.; Nikoloski, Z.; Rachmilevitch, S.; Fait, A. Metabolite profiling and network analysis reveal coordinated changes in grapevine water stress response. BMC Plant Biol. 2013, 13, 184–200. [Google Scholar] [CrossRef] [Green Version]
- Griesser, M.; Weingart, G.; Schoedl-Hummel, K.; Neumann, N.; Becker, M.; Varmuza, K.; Liebner, F.; Schuhmacher, R.; Forneck, A. Severe drought stress is affecting selected primary metabolites, polyphenols, and volatile metabolites in grapevine leaves (Vitis vinifera cv. Pinot noir). Plant Physiol. Biochem. 2015, 88, 17–26. [Google Scholar] [CrossRef]
- Santner, A.; Estelle, M. Recent advances and emerging trends in plant hormone signalling. Nature 2009, 459, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Argueso, C.T.; Ferreira, F.J.; Kieber, J.J. Environmental perception avenues: The interaction of cytokinin and environmental response pathways. Plant Cell Environ. 2009, 32, 1147–1160. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.W.; Li, C.H.; Cao, J.; Zhang, Y.C.; Zhang, S.Q.; Xia, Y.F.; Sun, D.Y.; Sun, Y. Altered Architecture and Enhanced Drought Tolerance in Rice via the Down-Regulation of Indole-3-Acetic Acid by TLD1/OsGH3.13 Activation. Plant Physiol. 2009, 151, 1889–1901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinozaki, K.; Yamaguchi-Shinozaki, K. Gene networks involved in drought stress response and tolerance. J. Exp. Bot. 2007, 58, 221–227. [Google Scholar] [CrossRef] [Green Version]
- Santana-Vieira, D.D.; Freschi, L.; Almeida, L.A.; Moraes, D.H.; Neves, D.M.; Santos, L.M.; Bertolde, F.Z.; Filho, W.S.S.; Filho, M.A.C.; Gesteira, A.S. Survival strategies of citrus rootstocks subjected to drought. Plant Cell Physiol. 2016, 6, 1–12. [Google Scholar] [CrossRef]
- Gimeno, J.; Gadea, J.; Forment, J.; Pérez-Valle, J.; Santiago, J.; Martínez-Godoy, M.A.; Yenush, L.; Bellés, J.M.; Brumós, J.; Colmenero-Flores, J.M.; et al. Shared and novel molecular responses of mandarin to drought. Plant Mol. Biol. 2009, 70, 403–420. [Google Scholar] [CrossRef]
- Huang, X.S.; Liu, J.H.; Chen, X.J. Overexpression of PtrABF gene; a bZIP transcription factor isolated from Poncirus trifoliate, enhances dehydration and drought tolerance in tobacco via scavenging ROS and modulating expression of stress-responsive genes. BMC Plant Biol. 2010, 10, 230–248. [Google Scholar] [CrossRef] [Green Version]
- Muthamilarasan, M.; Bonthala, V.S.; Mishra, A.K.; Khandelwal, R.; Khan, Y.; Roy, R.; Manoj, P. C2H2 type of zinc finger transcription factors in foxtail millet define response to abiotic stresses. Funct. Integr. Genom. 2014, 14, 531–543. [Google Scholar] [CrossRef]
- Jung, Y.Y.; Lee, I.H.; Nou, I.S.; Lee, K.D.; Rashotte, A.M.; Kang, K.K. BrRZFP1 a Brassica rapa C3HC4-type RING zinc finger protein involved in cold, salt and dehydration stress. Plant Biol. 2013, 15, 274–283. [Google Scholar] [CrossRef]
- Carvalho, L.C.; Vilela, B.J.; Mullineaux, P.M.; Amâncio, S. Comparative Transcriptomic Profiling of Vitis vinifera Under High Light Using a Custom-Made Array and the Affymetrix GeneChip. Mol. Plant 2011, 4, 1038–1051. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Vinocur, B.; Altman, A. Plant responses to drought, salinity and extreme temperatures: Towards genetic engineering for stress tolerance. Planta 2003, 218, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Rocheta, M.; Becker, J.D.; Coito, J.L.; Carvalho, L.C.; Amâncio, S. Heat and water stress induce unique transcriptional signatures of heat-shock proteins and transcription factors in grapevine. Funct. Integr. Genom. 2014, 14, 135–148. [Google Scholar] [CrossRef] [Green Version]
- Barua, D.; Downs, C.; Geckathorn, S. Variation in chloroplast small heat-shock protein function is a major determinant of variation in thermotolerance of photosynthetic electron transport among ecotypes of Chenopodium album. Func. Plant Biol. 2003, 30, 1071–1079. [Google Scholar] [CrossRef] [PubMed]
- Punja, Z.K.; Zhang, Y.Y. Plant chitinases and their roles in resistance to fungal diseases. J. Nematol. 1993, 25, 526–540. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Zheng, Y.; Xin, H.; Fang, L.; Li, S. Comprehensive analysis of NAC domain transcription factor gene family in Vitis vinifera. Plant Cell Rep. 2013, 32, 61–75. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, Y.; Jia, L.; Yang, G.; Xu, X.; Zhai, H.; He, H.; Li, J.; Dai, X.; Qin, N.; et al. Involvement of an ABI-like protein and a Ca2+-ATPase in drought tolerance as revealed by transcript profiling of a sweet potato somatic hybrid and its parents Ipomoea batatas (L.) Lam. and I. triloba L. PLoS ONE 2018, 13, e0193193. [Google Scholar] [CrossRef]
- Pastori, G.M.; Foyer, C.H. Common components, networks, and pathways of cross-tolerance to stress: The central role of ‘redox’ and abscisic acid-mediated controls. Plant Physiol. 2002, 129, 460–468. [Google Scholar] [CrossRef] [Green Version]
- Cheng, M.C.; Liao, P.M.; Kuo, W.W.; Lin, T.P. The Arabidopsis Ethylene Response Factor1 regulates abiotic stress-responsive gene expression by binding to different cis-acting elements in response to different stress signals. Plant Physiol. 2013, 162, 1566–1582. [Google Scholar] [CrossRef] [Green Version]
- Müller, M.; Munné-Bosch, S. Ethylene Response Factors: A Key Regulatory Hub in Hormone and Stress Signaling. Plant Physiol. 2015, 169, 32–41. [Google Scholar] [CrossRef] [Green Version]
- Dubois, M.; Skirycz, A.; Claeys, H.; Maleux, K.; Dhondt, S.; De Bodt, S.; Bossche, R.V.; De Milde, L.; Yoshizumi, T.; Matsui, M.; et al. Ethylene Response Factor6 acts as a central regulator of leaf growth under water limiting conditions in Arabidopsis. Plant Physiol. 2013, 162, 319–332. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.; Xu, J.; He, Y.; Yang, K.Y.; Mordorski, B.; Liu, Y.; Zhang, S. Phosphorylation of an ERF transcription factor by Arabidopsis MPK3/ MPK6 regulates plant defense gene induction and fungal resistance. Plant Cell 2013, 25, 1126–1142. [Google Scholar] [CrossRef] [Green Version]
- Sewelam, N.; Kazan, K.; Thomas-Hall, S.R.; Kidd, B.N.; Manners, J.M.; Schenk, P.M. Ethylene response factor 6 is a regulator of reactive oxygen species signaling in Arabidopsis. PLoS ONE 2013, 8, e7028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davletova, S.; Rizhsky, L.; Liang, H.; Shengqiang, Z.; Oliver, D.J.; Coutu, J.; Shulaev, V.; Schlauch, K.; Mittler, R. Cytosolic Ascorbate Peroxidase 1 is a Central Component of the Reactive Oxygen Gene Network of Arabidopsis. Plant Cell 2005, 17, 268–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genomics and Transcriptomics Platform. Available online: https://cpt.univr.it/en/genomics-and-transcriptomics-platform/ (accessed on 1 February 2017).
- Falgueras, J.; Lara, A.J.; Fernandez-Pozo, N.; Canton, F.R.; Perez-Trabado, G.; Claros, M.G. SeqTrim: A high-throughput pipeline for preprocessing any type of sequence reads. BMC Bioinform. 2010, 11, 38–50. [Google Scholar] [CrossRef] [Green Version]
- SeqtrimNEXT. Available online: https://github.com/dariogf/SeqtrimNext (accessed on 15 September 2016).
- CRIBI. Available online:https://genomes.cribi.unipd.it/grape/ (accessed on 21 November 2016).
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Kelley, J.L.; Passow, C.N.; Plath, M.; Rodriguez, L.A.; Yee, M.-C.; Tobler, M. Genomic resources for a model in adaptation and speciation research: Characterization of the Poecilia mexicana transcriptome. BMC Genom. 2012, 13, 652. [Google Scholar] [CrossRef] [Green Version]
- RStudio Team. RStudio: Integrated Development for R. RStudio; PBC: Boston, MA, USA, 2020; Available online: http://www.rstudio.com/ (accessed on 1 February 2017).
- Chang, Z.; Wang, Z.; Li, G. The impacts of read length and transcriptome complexity for de novo assembly: A simulation study. PLoS ONE 2014, 9, e94825. [Google Scholar] [CrossRef]
- Ramos, M.J.N.; Coito, J.L.; Fino, J.; Cunha, J.; Silva, H.; de Almeida, P.G.; Costa, M.M.R.; Amâncio, S.; Paulo, O.S.; Rocheta, M. Deep analysis of wild Vitis flower transcriptome reveals unexplored genome regions associated with sex specification. Plant Mol. Biol. 2017, 93, 151–170. [Google Scholar] [CrossRef] [Green Version]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; ISBN 978-3-319-24277-4. [Google Scholar]
- Plotrix Package—Rdocumentation. Available online: https://www.rdocumentation.org/packages/plotrix/versions/3.7 (accessed on 1 February 2017).
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nuc. Acid Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [Green Version]
- TAIR, the Arabidopsis Thaliana Genome Database. Available online: https://www.arabidopsis.org (accessed on 1 February 2017).
- Chen, H.; Boutros, P.C. VennDiagram: A package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinform. 2011, 12, 35. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔC(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Coito, J.L.; Rocheta, M.; Carvalho, L.C.; Amâncio, S. Microarray-based uncovering reference genes for quantitative real time PCR in grapevine under abiotic stress. BMC Res. Notes 2012, 5, 220–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]

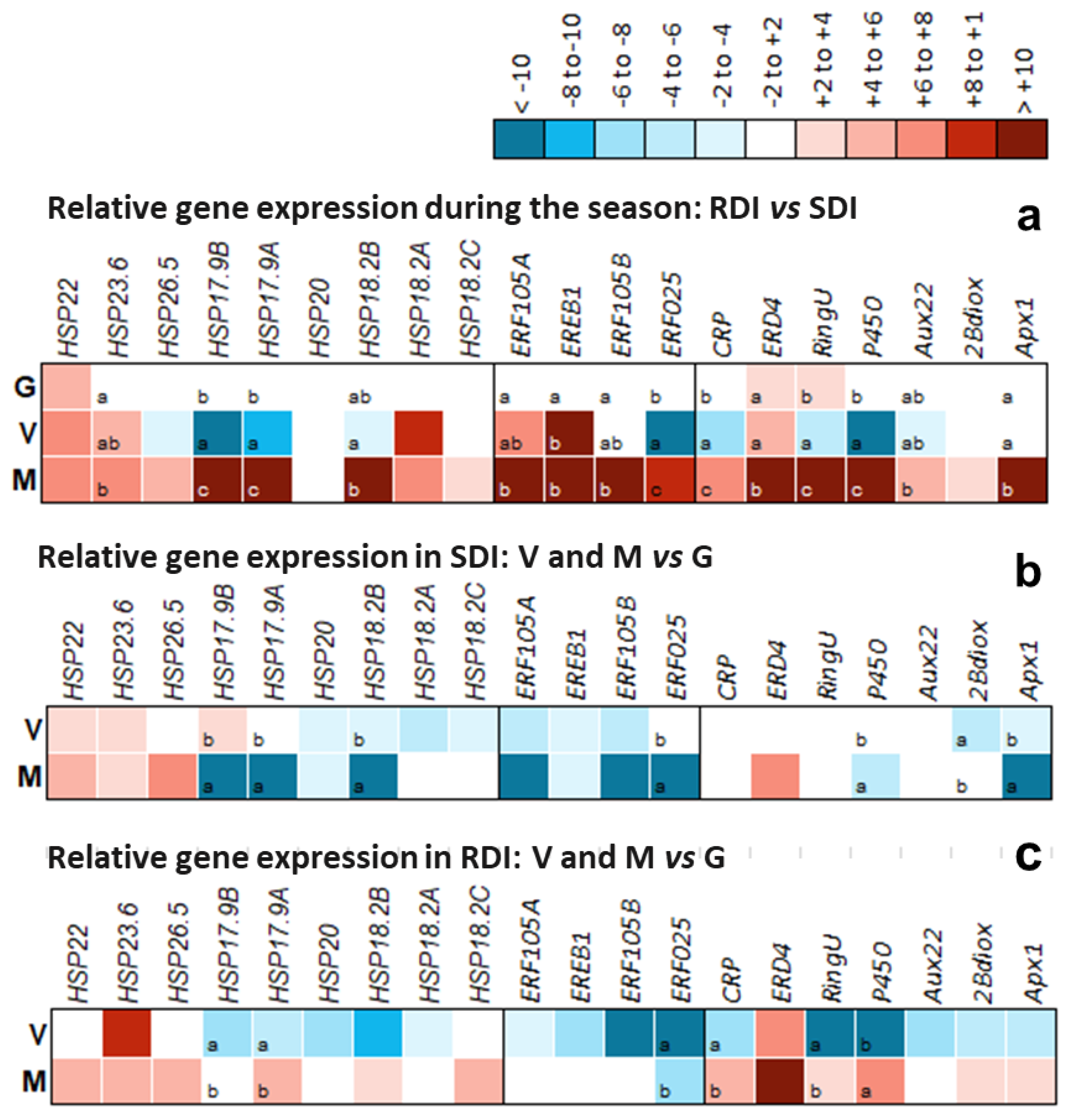

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Irrigation Strategy | Phenological Stage | R |
|---|---|---|
| SDI | Véraison | 0.984 |
| Maturation | 0.989 | |
| RDI | Véraison | 0.989 |
| Maturation | 0.974 |
| % | SV | RV | SM | RM |
|---|---|---|---|---|
| Unknown | 47 | 46 | 45 | 45 |
| Unclear | 43 | 42 | 41 | 40 |
| Unclassified | 45 | 44 | 42 | 41 |
| No Name | 27 | 27 | 26 | 26 |
| No Hit | 26 | 25 | 22 | 23 |
| Signaling | RT—Transcription Factors | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pathway | ABA | Auxin | Brassinosteroid | Cytokinin | Ethylene | Gibberellic Acid | Jasmonate Salicylate | bZIP | C2C2-YABBY | C2H2 | Homeobox Domain | MYB | NAC | TRAF | Zinc Finger AN1 | Zinc Finger C3HC4 | ||
| RDI | ↑ | 6 | - | 2 | - | - | 1 | 1 | 1 | - | - | 2 | - | 1 | 1 | - | 1 | 2 |
| ↓ | 4 | - | 7 | - | - | 2 | - | - | 1 | 1 | - | - | - | - | 1 | - | 1 | |
| SDI | ↑ | 3 | - | 1 | 1 | - | 1 | - | 1 | - | - | 3 | - | - | 2 | - | - | 3 |
| ↓ | 12 | 2 | 5 | - | 1 | 3 | 1 | - | - | - | - | 1 | - | - | - | - | - | |
| Signaling | RT—Transcription Factors | |||||||
|---|---|---|---|---|---|---|---|---|
| Pathway | Auxin | Ethylene | Jasmonate Salicylate | bZIP | WRKY | Zinc Finger C3HC4 | ||
| V | ↑ | - | 1 | - | 1 | 1 | - | - |
| ↓ | 2 | 2 | 3 | - | - | 1 | 1 | |
| M | ↑ | - | - | - | - | - | - | - |
| ↓ | - | - | - | - | - | - | - | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carvalho, L.C.; Ramos, M.J.N.; Faísca-Silva, D.; van der Kellen, D.; Fernandes, J.C.; Egipto, R.; Lopes, C.M.; Amâncio, S. Developmental Regulation of Transcription in Touriga Nacional Berries under Deficit Irrigation. Plants 2022, 11, 827. https://doi.org/10.3390/plants11060827
Carvalho LC, Ramos MJN, Faísca-Silva D, van der Kellen D, Fernandes JC, Egipto R, Lopes CM, Amâncio S. Developmental Regulation of Transcription in Touriga Nacional Berries under Deficit Irrigation. Plants. 2022; 11(6):827. https://doi.org/10.3390/plants11060827
Chicago/Turabian StyleCarvalho, Luísa C., Miguel J. N. Ramos, David Faísca-Silva, David van der Kellen, João C. Fernandes, Ricardo Egipto, Carlos M. Lopes, and Sara Amâncio. 2022. "Developmental Regulation of Transcription in Touriga Nacional Berries under Deficit Irrigation" Plants 11, no. 6: 827. https://doi.org/10.3390/plants11060827
APA StyleCarvalho, L. C., Ramos, M. J. N., Faísca-Silva, D., van der Kellen, D., Fernandes, J. C., Egipto, R., Lopes, C. M., & Amâncio, S. (2022). Developmental Regulation of Transcription in Touriga Nacional Berries under Deficit Irrigation. Plants, 11(6), 827. https://doi.org/10.3390/plants11060827