
Genome-Wide Identification of Histone Modification Gene Families in the Model Legume Medicago truncatula and Their Expression Analysis in Nodules

 ,
,  ,
,  , and
, and 
Abstract
:1. Introduction
2. Results
2.1. M. truncatula Histone Methyltransferases (MtHMT)
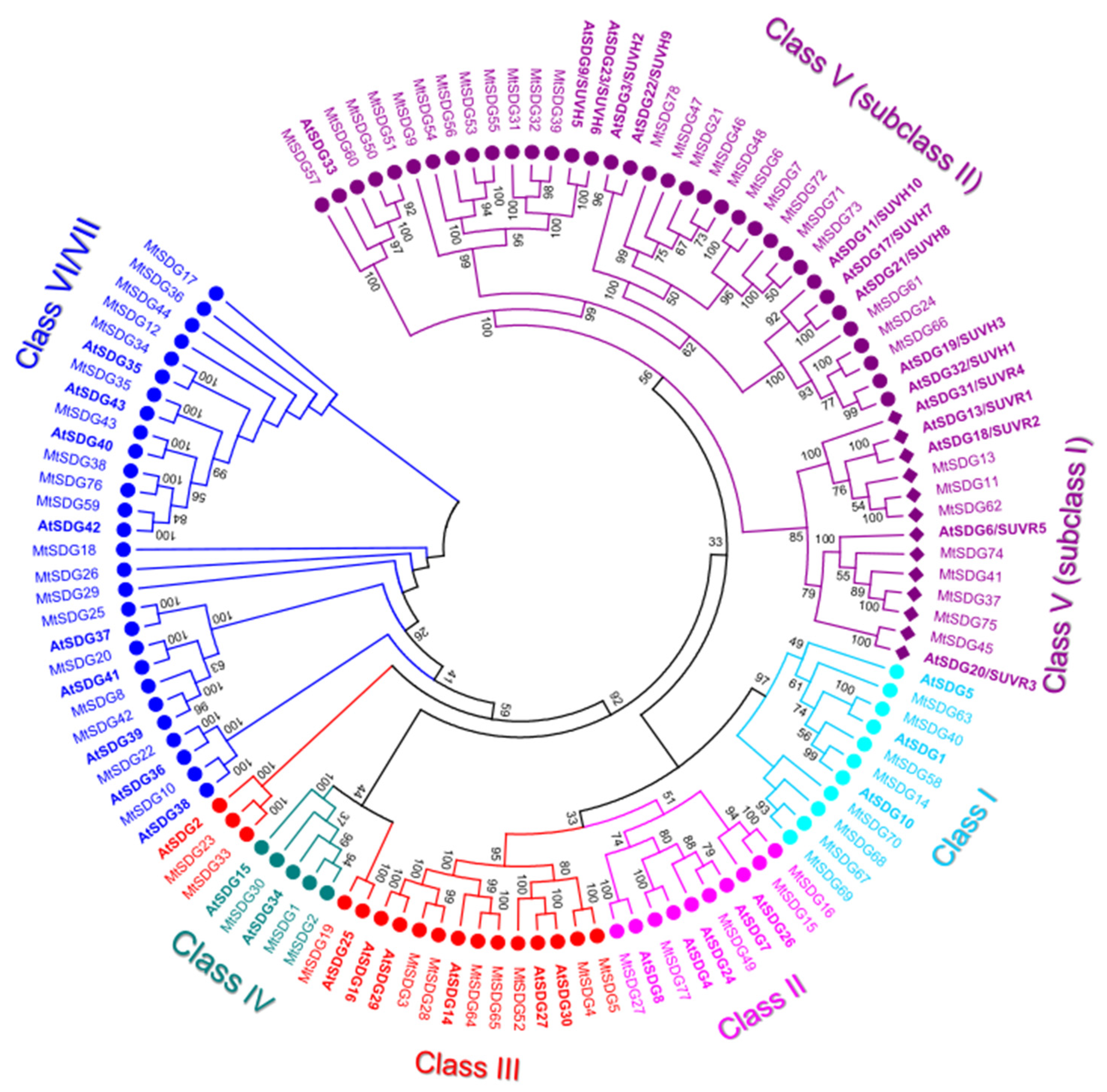
2.1.1. SDG Subfamily
- Class II, ASH1-like: M. truncatula Class II SDGs did not show number variation compared to Arabidopsis. All the proteins of the class have SET and post SET domains. MtSDG27, the largest protein of the class, has an additional N-terminal CW-type zinc finger domain as AtSDG8. MtSDG77 has two additional PHD zinc finger domains similarly to AtSDG4. MtSFG15 and MtSDG16, which derive from a tandem duplication on chromosome 1, has an additional AWS domain while MtSDG77 has two additional PHD zinc finger and a structure that is similar to AtSDG4. MtSDG49, instead, lacks the AWS domain.
- Class III, TRX-like: M. truncatula Class III SDGs showed a modest number increase when compared to Arabidopsis (Mt:At ratio equal to 1.43:1). MtSDG52 has the same domain composition of AtSDG27 and AtSDG30 including the FYR domain. MtSDG3, MtSDG27, MtSDG64, and MtSDG65 have the same domain composition of AtSDG14, AtSDG16, AtSDG29, and map within segmentally duplicated regions on chromosomes 1, 3, and 7 (Figure S1). MtSDG4 and 5 lack a PWWP domain. MtSDG19 and its orthologous gene AtSDG25 and MtSDG23, MtSDG33, and AtSDG2 identified two separate groups that were both closer to class IV than other class III SDGs. To keep coherence with the original Arabidopsis HMG classification, we included these MtSDG within class III although a classification into the separate class M was also proposed [36].
- Class IV: MtSDG1, 2, and 30 belong to class IV, although tandem duplicated genes MtSDG1 and MtSDG2 show remarkable differences for the intron exon structure and domain composition. Compared to MtSDG1, MtSDG2 has four additional exons at the 5′ and encodes for a larger protein with two additional N-terminal domains. Interestingly, as shown in Table S1, no similar proteins could be found in the other Medicago genomes that were analyzed.
- Class V, SU(VAR)3–9-like: M. truncatula class V is the largest SDG class with 33 loci. A total of eight are clustered with subclass I Arabidopsis proteins that contain Pre-SET, SET, and Post-SET domains. MtSDG11, 13, and 62 are clustered with AtSDG13, 18, and 31 but all lack the Post-SET domain and have the WIYLD domain at the N-terminal. MtSDG11 and 62 belong to segmentally duplicated regions on chromosomes 1 and 7 (Figure S1). MtSDG37, 41, and 75 have three C2H2 domains similarly to AtSDG6, MtSDG37 also has a zf-CCCH N-terminal domain. MtSDG41 and 75 belong to segmentally duplicated regions on chromosomes 5 and 8 (Figure S1). MtSDG74 is a tandem duplication of the 3′ portion of MtSDG75 harboring only Pre-SET and SET domains. MtSDG45 is more related to AtSDG20. In subclass II, MtSDG24, 61, and 66 are clustered with AtSDG19 and 32 and have the same domain composition except for the PostSET in MtSDG61. There are ten proteins that are clustered with AtSDG3 and 22 and have the same domain composition: SRA-PreSET-SET. Among them are several tandemly duplicated genes: MtSDG71–73 on chromosome 7, MtSDG6 and 7 on chromosome 1, and MtSDG46–48 on chromosome 6. A total of eight proteins are clustered with AtSDG9 and 23 that have the following domain composition: SRA-PreSET-SET-PostSET. MtSDG39 and the tandemly duplicated MtSDG31 and 32 have the expected domain composition. The four tandemly duplicated MtSDG53–56 lack both Pre-SET and Post-SET domains with the exception of MtSDG55 that retained the Post-SET domain. MtSDG9 lacks the Post-SET domain. A total of four proteins are clustered with AtSDG33: MtSDG50, 51, and 60 that have SRA-PreSET-SET-PostSET domains and MtSDG57 lacking SRA.
- Class VI/VII: The 19 MtSDGs that harbor a truncated and/or interrupted SET domain, were classified as belonging to Class VI/VII. Of those, seven have an RBS domain. MtSDG29 has four tetratricopeptide (TPR) domains, MtSDG25 has SCOP d1elra_ domain, and MtSDG34 has SCOP d1ihga1 domain, both are related to TPR and thus are generally involved in protein–protein interactions. MtSDG36 has a SCOP d1dqaa1 domain that is the NAD-binding domain of HMG-CoA reductase in Homo sapiens.
2.1.2. Medicago Truncatula PRMT Subfamily
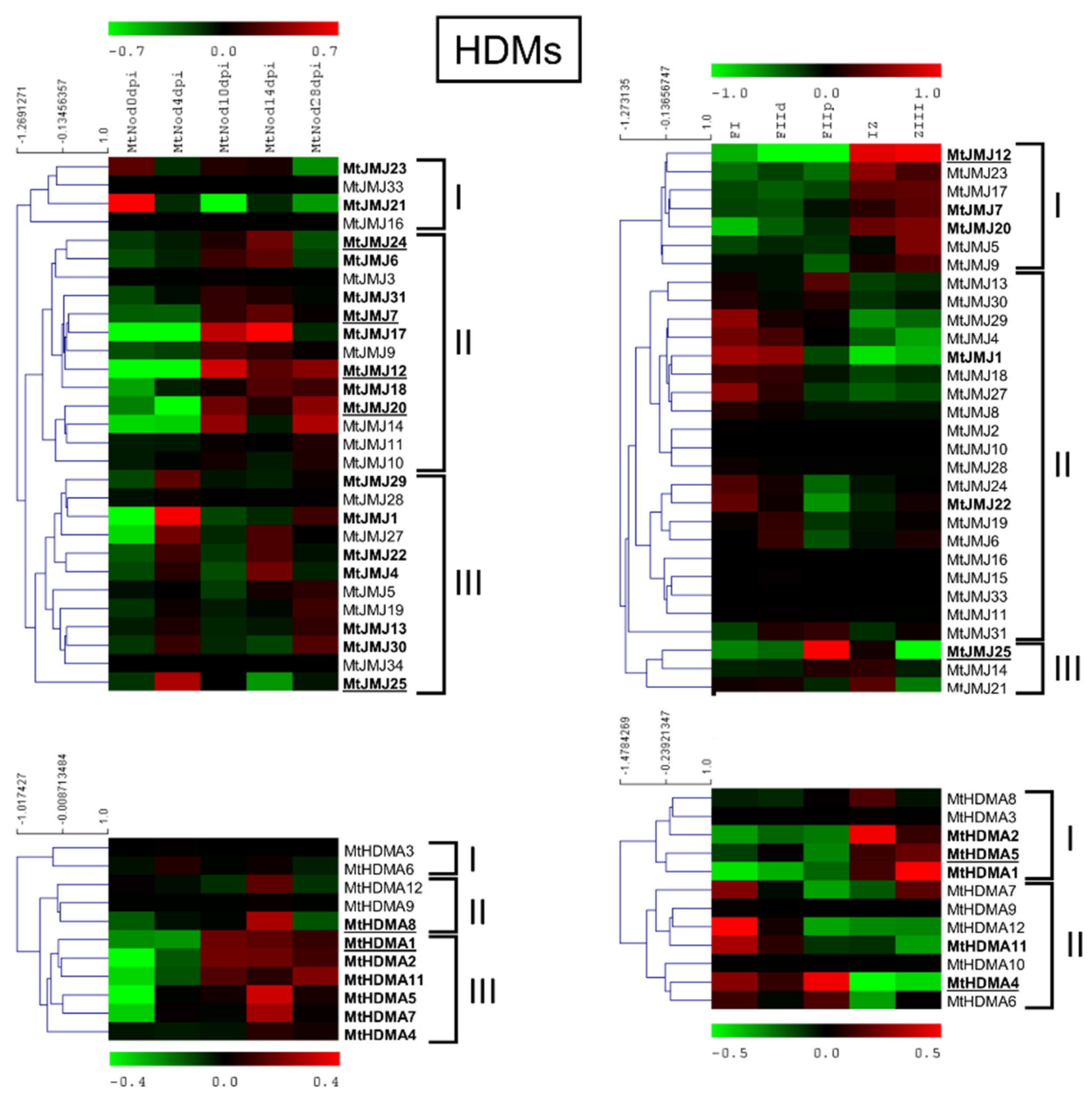
2.2. Medicago truncatula HDMs
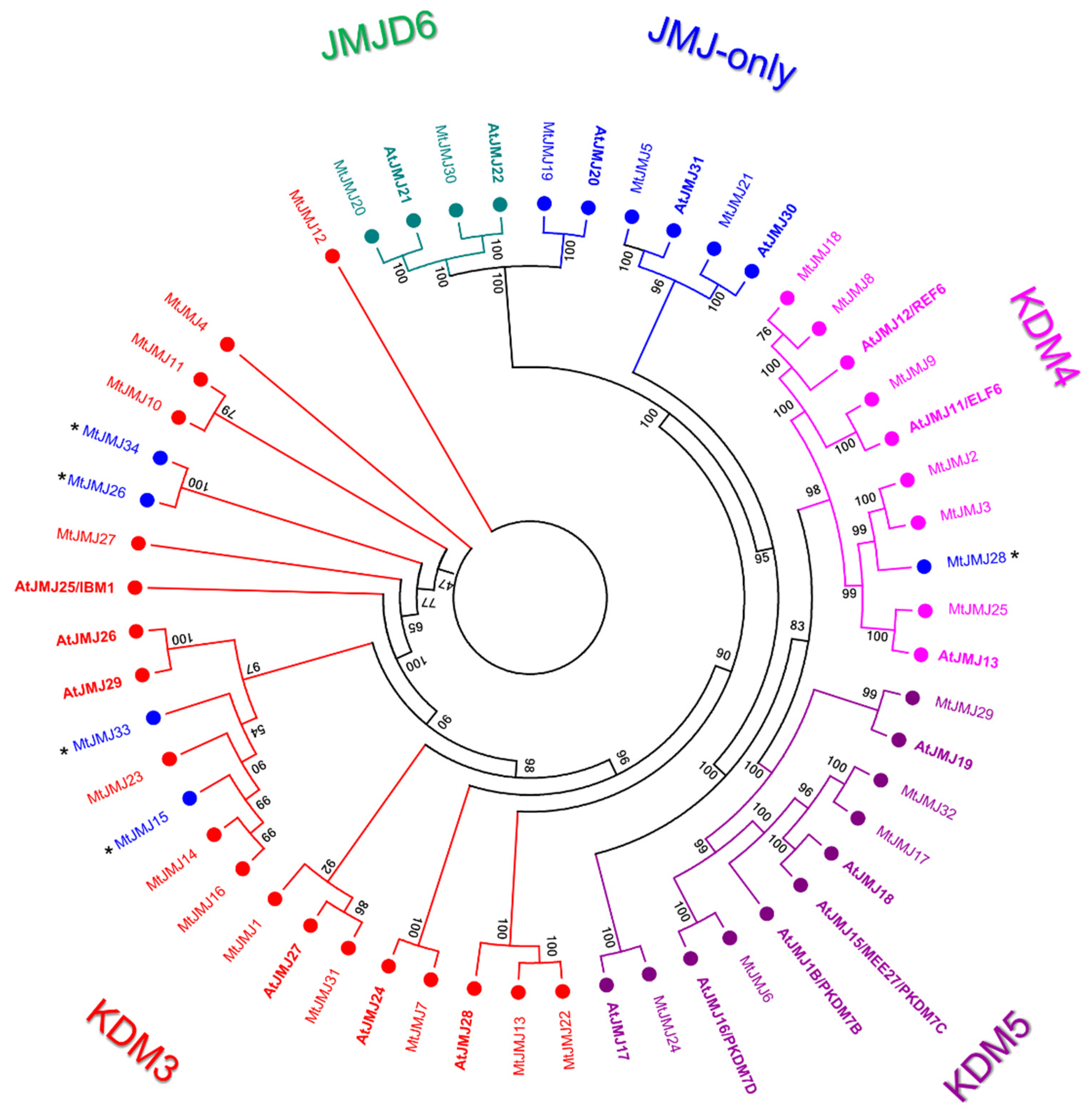
- JMJ-only: eight proteins possessed only the JMJC domain. Among these, three: MtJMJ5 and MtJMJ21 form a clade with AtJMJ30 and 31 while MtJMJ19 is clustered with AtJMJ20 and is related to JMJD6 Class. The other MtJMJ proteins with only the JMJC domain (15, 26, 28, 33, and 34) as above mentioned do not form a clade with the A. thaliana members.
- KDM3: KDM3 class has 13 M. truncatula members (1, 4, 7, 10 to 14, 16, 22, 23, 27, and 31), most of them are featured by a JMJC domain at the C-terminal with Ring finger domains (SM000184) ahead of it (Figure 4).
- KDM4: the six MtJMJs of KDM4 class have JMJC and JMJN domains. MtJMJ9 is clustered with AtJMJ11/ELF6 and it is characterized by four tandem repeats of ZnF_C2H2 domain (SM000355). MtJMJ3 and 25 contain a zf-C5HC2 domain (PF02928) at the C-terminal (Figure 4). MtJMJ8 is clustered with MtJMJ18 and AtJMJ12/REF6 but it is a shorter protein of 132 vs. 1543 amino acids that are encoded by a single orf that is probably derived from an aberrant duplication event (Table S2 and Figure 3).
- KDM5: MtJMJ6 is clustered with AtJMJ16/PKDM7D and has the same domain composition (JMJN-JMJC-zf-C5HC2-FYRN-FYRC) of MtJMJ17 and 32. In the same class, MtJMJ24 is clustered with AtJMJ17 and it is the only member with additional ARID, PHD, and PLU-1 domains.
2.3. Medicago truncatula HDMA Family
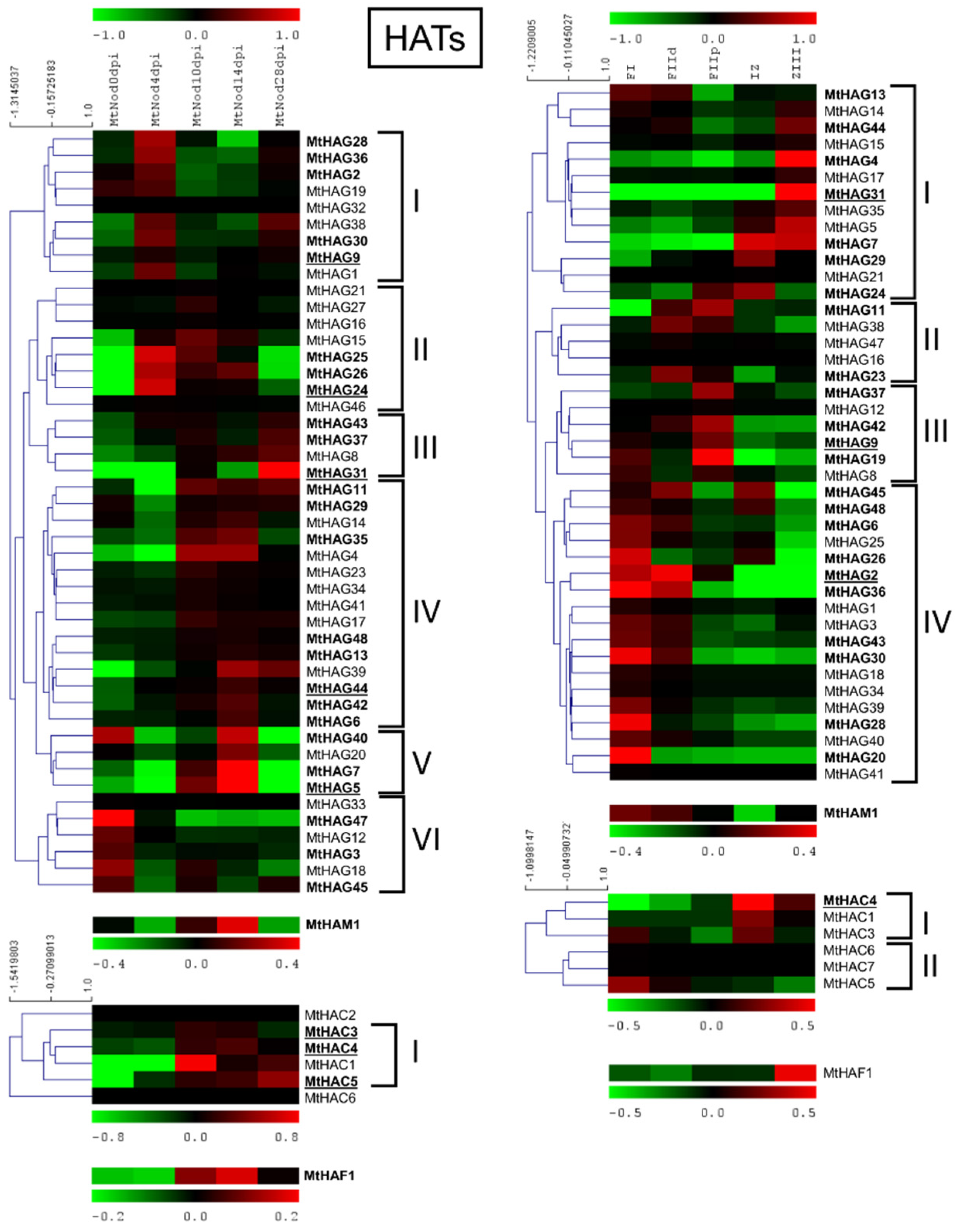
2.4. M. truncatula Histone Acetyltransferase Family (HAT)
- HAG: The genomic locations of MtHAGs show that many of them arose probably from tandem duplications: MtHAG4–5, 8–10, 15–17, 24–29, 30–31, 32–33, 34–35, and 39–41. MtHAG4–5 and 24–28 belong to segmentally duplicated regions on chromosomes 1 and 5. A further segmental duplication harbors MtHAG21 and 22 on chromosome 4 and MtHAG30–31 and 32–33 on chromosome 5 (Figure S1). MtHAG44 and MtHAG49 contain the BrD domain and are clustered with AtHAG1 in the GCN5 group (Figures S3 and S4). MtHAG36 is clustered with AtHAG2 in the HAT1 group and has the HAT1 domain. The ELP3 group includes AtHAG3 and MtHAG6 harboring the Elp3 domain. A total of 10 MtHAGs form a cluster with the ELP3 group but lack the Elp3 domain: MtHAG1, 2, 3, 13, 19, 34, 35, 37, 42, and 43. MtHAG34 has an Oleosin domain (PF01277) followed by a transmembrane region and is probably derived from a fusion between an oleosin and a tandem duplication of MtHAG35 (Figures S3 and S4). According to a M. truncatula gene expression study [37], MtHAG34 expression is higher in seeds between 16 and 36 dap as expected from Oleosins. A total of seven MtHAGs: 15, 16, 17, 21, 32, 33, and 46 form a cluster that is characterized by Jas-RING-PHD-AT1 domain composition with the exception of MtHAG21 lacking RING and MtHAG17 lacking Jas domains, this cluster is identified as PHD in Figure S3. There are two further clusters that are represented by MtHAG4, 5, 11, 24, 25, 26, 27, 28, 39, 40, 41, and 47 and by MtHAG8, 9, 10, 12, 18, 20, 22, 31, 48, and 50 that shows only the AT1 domain with the exception of MtHAG28 with a KIP1 domain and MtHAG10 with a C-terminal RRM domain. MtHAG7, 14, 23, 29, 30, 38, 45, and 51 all contained the AT1 domain but could not be included in defined clusters probably due to heavier rearrangements in their sequence/structure. Among them, MtHAG30 harbors AA_kinase and SCOP d1gs5a_ domains. Interestingly, the AAK domain was found also in two Solanum lycopersicum HAGs: SlNAGS1 and SlNAGS2 that were considered as probable not histone modifiers by the authors [38].
- HAC: AtHACs are characterized by the TAZ-PHD-KAT11-ZZ-TAZ domain composition with the exception of AtHAC2 lacking the N-terminal TAZ domain. Only MtHAC4 and MtHAC8 have all the characteristic domains. All the other MtHACs lack the ZZ domain (Figure S4). A total of five MtHACs (7–11) are tandemly duplicated on chromosome 6 between position 6,665,635 and 6,780,888 and present many rearrangements in sequence and structure. MtHAC2 and 3 are tandemly duplicated on chromosome 3 and have only PHD-KAT11-TAZ domains like MtHAC5, six that are on chromosome 5. MtHAC1 harbors only the KAT11 domain. MtHACs went through several duplication events in the M. truncatula genome that generated a series of probably not fully functional histone modifiers (Figures S1 and S4). A phylogenetic tree including MtHACs and AtHACs is displayed in Figure S5.
- HAM and HAF: Only one MtHAM and one MtHAF protein have been identified by our analysis. The domain composition is shown in Figure S4 and is similar to that of AtHAMs and AtHAFs.
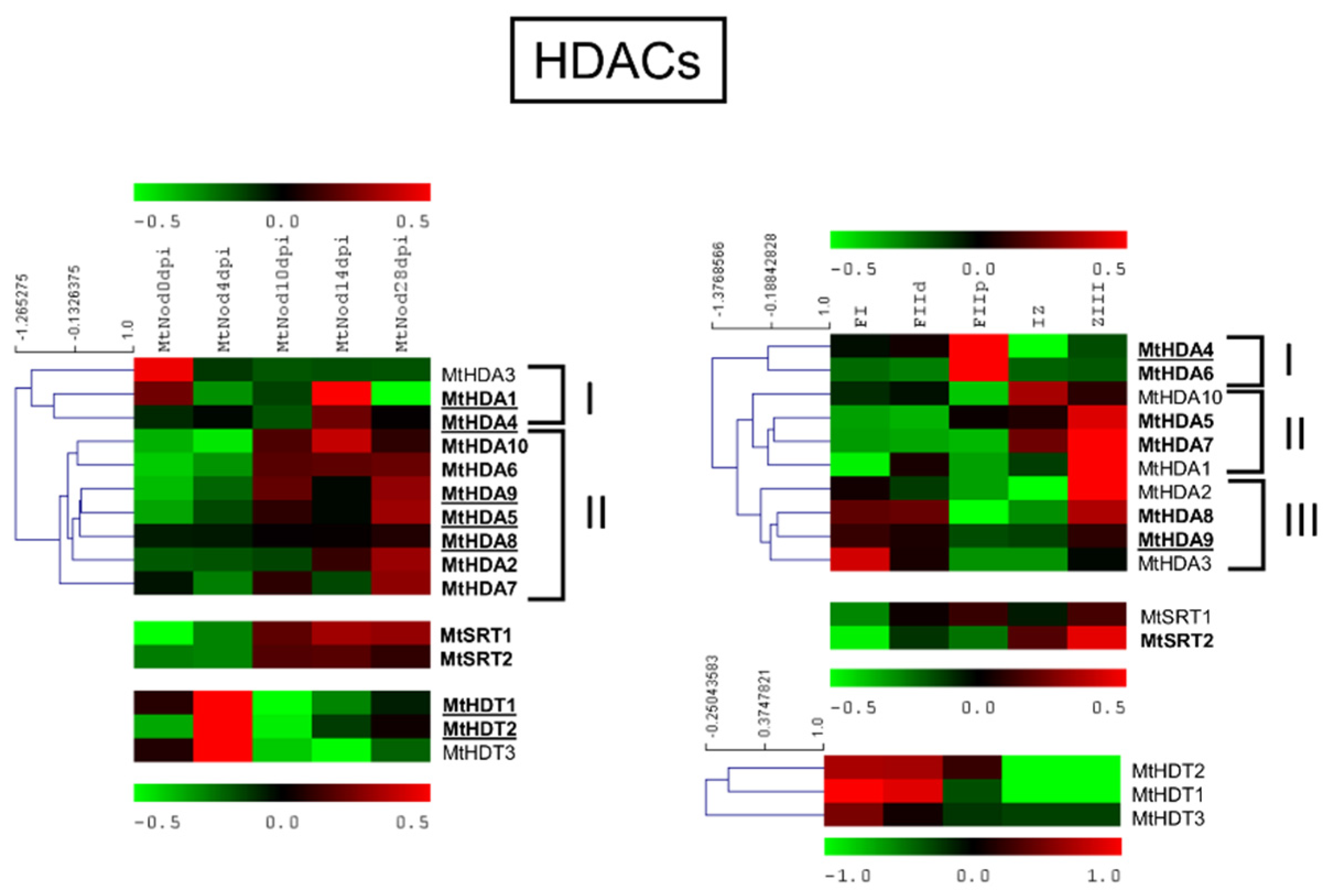
2.5. M. truncatula Histone Deacetylation Family (HDAC)
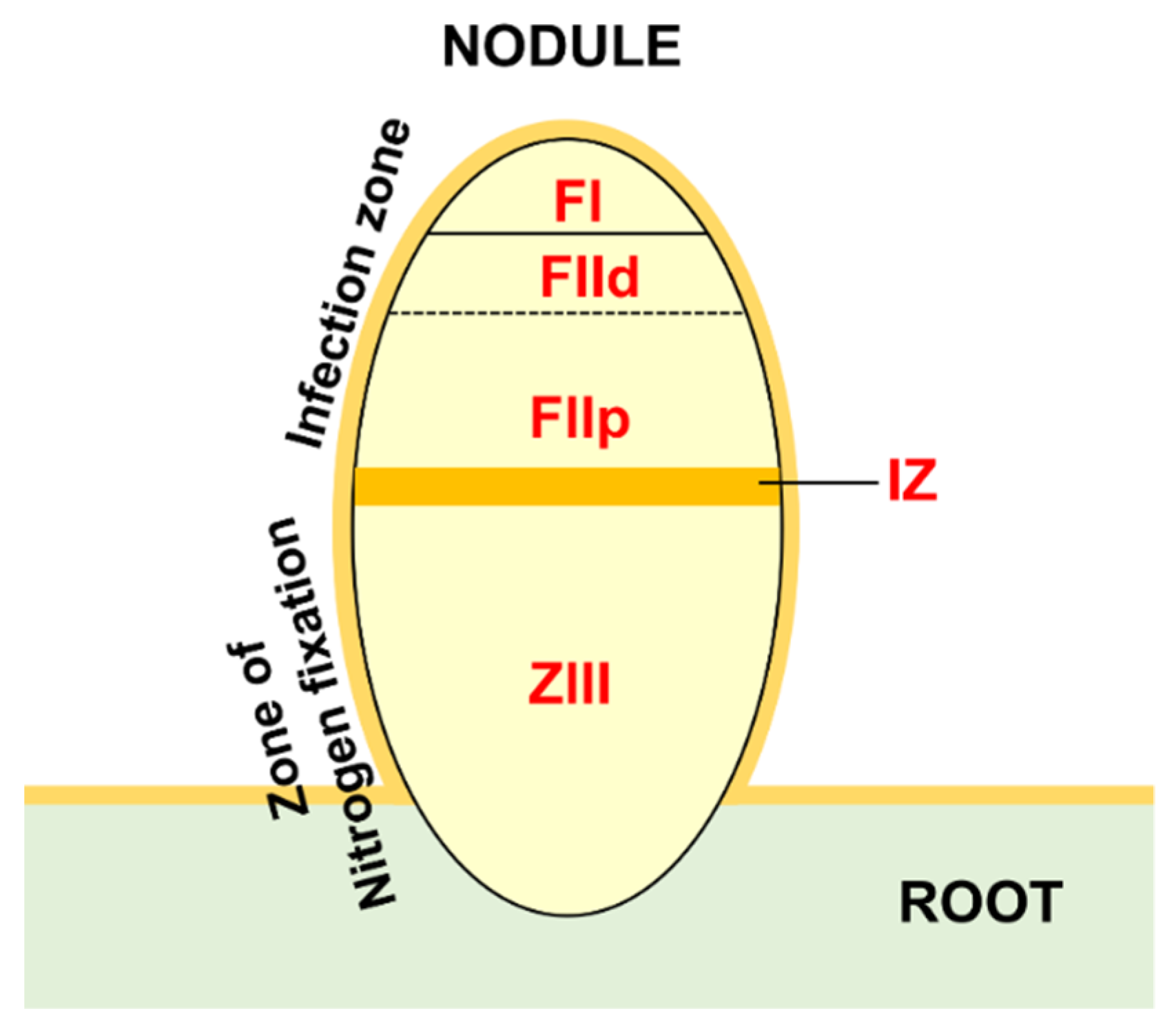
2.6. Gene Expression Analysis during Nodule Development and in Different Zones of Actively Fixing Nodules
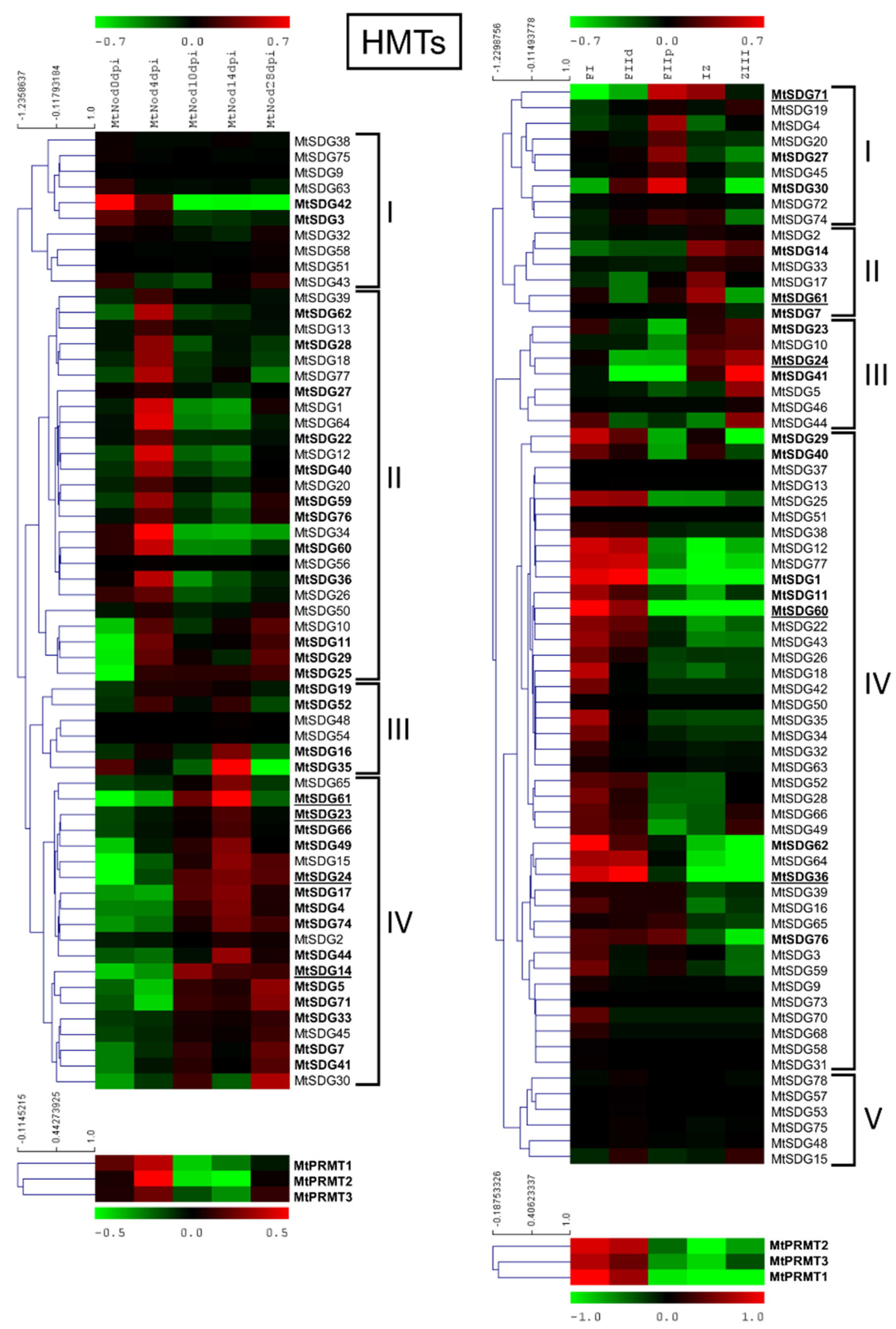
2.6.1. MtHMTs
2.6.2. MtHDMs
2.6.3. MtHATs
2.6.4. MtHDACs
3. Discussion
3.1. Histone Modifier Gene Families in Medicago truncatula
3.2. Expression Data Analysis Showed the Possible Involvement of HMGs in Different Aspects of Nodule Development and Function in Medicago truncatula
4. Materials and Methods
4.1. Genomic Sequences
4.2. In Silico Identification and Analysis of HMG Loci
4.3. Phylogenetic Analysis
4.4. Synteny Analysis of M. truncatula and Arabidopsis
4.5. Expression Analyses of M. truncatula HMGs in Nodules
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bressan, R.A.; Zhu, J.-K.; Van Oosten, M.J.; Maggio, A.; Bohnert, H.J.; Chinnusamy, V. Epigenetics Connects the Genome to Its Environment. Plant Breed. Rev. 2014, 38, 69–142. [Google Scholar]
- Vergara, Z.; Gutierrez, C. Emerging roles of chromatin in the maintenance of genome organization and function in plants. Genome Biol. 2017, 18, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin Modifications and Their Function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Lu, F.; Cui, X.; Cao, X. Histone Methylation in Higher Plants. Annu. Rev. Plant Biol. 2010, 61, 395–420. [Google Scholar] [CrossRef]
- Baumbusch, L.O.; Thorstensen, T.; Krauss, V.; Fischer, A.; Naumann, K.; Assalkhou, R.; Schulz, I.; Reuter, G.; Aalen, R.B. The Arabidopsis thaliana genome contains at least 29 active genes encoding SET domain proteins that can be assigned to four evolutionarily conserved classes. Nucleic Acids Res. 2001, 29, 4319–4333. [Google Scholar] [CrossRef]
- Zhao, Z.; Shen, W.-H. Plants contain a high number of proteins showing sequence similarity to the animal SUV39H family of histone methyltransferases. Ann. N. Y. Acad. Sci. 2004, 1030, 661–669. [Google Scholar] [CrossRef]
- Ahmad, A.; Dong, Y.; Cao, X. Characterization of the PRMT gene family in rice reveals conservation of arginine methylation. PLoS ONE 2011, 6, e22664. [Google Scholar] [CrossRef]
- Luo, M.; Hung, F.-Y.; Yang, S.; Liu, X.; Wu, K. Histone Lysine Demethylases and Their Functions in Plants. Plant Mol. Biol. Report. 2014, 32, 558–565. [Google Scholar] [CrossRef]
- Tsukada, Y.; Fang, J.; Erdjument-Bromage, H.; Warren, M.E.; Borchers, C.H.; Tempst, P.; Zhang, Y. Histone demethylation by a family of JmjC domain-containing proteins. Nature 2006, 439, 811–816. [Google Scholar] [CrossRef]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A.; Shi, Y. Histone Demethylation Mediated by the Nuclear Amine Oxidase Homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsukada, Y. Hydroxylation mediates chromatin demethylation. J. Biochem. 2012, 151, 229–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klose, R.J.; Zhang, Y. Regulation of histone methylation by demethylimination and demethylation. Nat. Rev. Mol. Cell Biol. 2007, 8, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Asensi-Fabado, M.-A.; Amtmann, A.; Perrella, G. Plant responses to abiotic stress: The chromatin context of transcriptional regulation. Biochim. Biophys. Acta-Gene Regul. Mech. 2017, 1860, 106–122. [Google Scholar] [CrossRef] [Green Version]
- Pandey, R.; Müller, A.; Napoli, C.A.; Selinger, D.A.; Pikaard, C.S.; Richards, E.J.; Bender, J.; Mount, D.W.; Jorgensen, R.A. Analysis of histone acetyltransferase and histone deacetylase families of Arabidopsis thaliana suggests functional diversification of chromatin modification among multicellular eukaryotes. Nucleic Acids Res. 2002, 30, 5036–5055. [Google Scholar] [CrossRef] [Green Version]
- Servet, C.; Conde e Silva, N.; Zhou, D.-X. Histone Acetyltransferase AtGCN5/HAG1 Is a Versatile Regulator of Developmental and Inducible Gene Expression in Arabidopsis. Mol. Plant 2010, 3, 670–677. [Google Scholar] [CrossRef]
- Ding, Y.; Mou, Z. Elongator and its epigenetic role in plant development and responses to abiotic and biotic stresses. Front. Plant Sci. 2015, 6, 296. [Google Scholar] [CrossRef] [Green Version]
- Roca Paixão, J.F.; Gillet, F.-X.; Ribeiro, T.P.; Bournaud, C.; Lourenço-Tessutti, I.T.; Noriega, D.D.; de Melo, B.P.; de Almeida-Engler, J.; Grossi-de-Sa, M.F. Improved drought stress tolerance in Arabidopsis by CRISPR/dCas9 fusion with a Histone AcetylTransferase. Sci. Rep. 2019, 9, 8080. [Google Scholar] [CrossRef] [Green Version]
- Latrasse, D.; Benhamed, M.; Henry, Y.; Domenichini, S.; Kim, W.; Zhou, D.-X.; Delarue, M. The MYST histone acetyltransferases are essential for gametophyte development in Arabidopsis. BMC Plant Biol. 2008, 8, 121. [Google Scholar] [CrossRef] [Green Version]
- Ueda, M.; Matsui, A.; Tanaka, M.; Nakamura, T.; Abe, T.; Sako, K.; Sasaki, T.; Kim, J.-M.; Ito, A.; Nishino, N.; et al. The Distinct Roles of Class I and II RPD3-Like Histone Deacetylases in Salinity Stress Response. Plant Physiol. 2017, 175, 1760–1773. [Google Scholar] [CrossRef] [Green Version]
- de Rooij, P.G.H.; Perrella, G.; Kaiserli, E.; van Zanten, M. The diverse and unanticipated roles of histone deacetylase 9 in coordinating plant development and environmental acclimation. J. Exp. Bot. 2020, 71, 6211–6225. [Google Scholar] [CrossRef] [PubMed]
- Ueda, M.; Seki, M. Histone Modifications Form Epigenetic Regulatory Networks to Regulate Abiotic Stress Response1 [OPEN]. Plant Physiol. 2020, 182, 15–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thudi, M.; Palakurthi, R.; Schnable, J.C.; Chitikineni, A.; Dreisigacker, S.; Mace, E.; Srivastava, R.K.; Satyavathi, C.T.; Odeny, D.; Tiwari, V.K.; et al. Genomic resources in plant breeding for sustainable agriculture. J. Plant Physiol. 2021, 257, 153351. [Google Scholar] [CrossRef] [PubMed]
- Nagymihály, M.; Veluchamy, A.; Györgypál, Z.; Ariel, F.; Jégu, T.; Benhamed, M.; Szücs, A.; Kereszt, A.; Mergaert, P.; Kondorosi, É. Ploidy-Dependent changes in the epigenome of symbiotic cells correlate with specific patterns of gene expression. Proc. Natl. Acad. Sci. USA 2017, 114, 4543–4548. [Google Scholar] [CrossRef] [Green Version]
- Pecrix, Y.; Staton, S.E.; Sallet, E.; Lelandais-Brière, C.; Moreau, S.; Carrère, S.; Blein, T.; Jardinaud, M.F.; Latrasse, D.; Zouine, M.; et al. Whole-Genome landscape of Medicago truncatula symbiotic genes. Nat. Plants 2018, 4, 1017–1025. [Google Scholar] [CrossRef]
- Mergaert, P.; Kereszt, A.; Kondorosi, E. Gene Expression in Nitrogen-Fixing Symbiotic Nodule Cells in Medicago truncatula and Other Nodulating Plants. Plant Cell 2020, 32, 42–68. [Google Scholar] [CrossRef]
- Roux, B.; Rodde, N.; Jardinaud, M.F.; Timmers, T.; Sauviac, L.; Cottret, L.; Carrère, S.; Sallet, E.; Courcelle, E.; Moreau, S.; et al. An integrated analysis of plant and bacterial gene expression in symbiotic root nodules using laser-capture microdissection coupled to RNA sequencing. Plant J. 2014, 77, 817–837. [Google Scholar] [CrossRef]
- Li, H.; Schilderink, S.; Cao, Q.; Kulikova, O.; Bisseling, T. Plant-Specific histone deacetylases are essential for early and late stages of Medicago nodule development. Plant Physiol. 2021, 186, 1–15. [Google Scholar] [CrossRef]
- Boycheva, I.; Vassileva, V.; Revalska, M.; Zehirov, G.; Iantcheva, A. Different functions of the histone acetyltransferase HAC1 gene traced in the model species Medicago truncatula, Lotus japonicus and Arabidopsis thaliana. Protoplasma 2017, 254, 697–711. [Google Scholar] [CrossRef]
- Shen, Y.; Wu, X.; Liu, D.; Song, S.; Liu, D.; Wang, H. Cold-Dependent alternative splicing of a Jumonji C domain-containing gene MtJMJC5 in Medicago truncatula. Biochem. Biophys. Res. Commun. 2016, 474, 271–276. [Google Scholar] [CrossRef]
- Stanton-Geddes, J.; Paape, T.; Epstein, B.; Briskine, R.; Yoder, J.; Mudge, J.; Bharti, A.K.; Farmer, A.D.; Zhou, P.; Denny, R.; et al. Candidate Genes and Genetic Architecture of Symbiotic and Agronomic Traits Revealed by Whole-Genome, Sequence-Based Association Genetics in Medicago truncatula. PLoS ONE 2013, 8, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Bang, T.C.; Lundquist, P.K.; Dai, X.; Boschiero, C.; Zhuang, Z.; Pant, P.; Torres-Jerez, I.; Roy, S.; Nogales, J.; Veerappan, V.; et al. Genome-Wide identification of medicago peptides involved in macronutrient responses and nodulation. Plant Physiol. 2017, 175, 1669–1689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- HAPMAP2. Available online: https://medicagohapmap2.org/ (accessed on 10 November 2021).
- Springer, N.M.; Napoli, C.A.; Selinger, D.A.; Pandey, R.; Cone, K.C.; Chandler, V.L.; Kaeppler, H.F.; Kaeppler, S.M. Comparative analysis of SET domain proteins in maize and Arabidopsis reveals multiple duplications preceding the divergence of monocots and dicots. Plant Physiol. 2003, 132, 907–925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, D.W.-K.; Wang, T.; Chandrasekharan, M.B.; Aramayo, R.; Kertbundit, S.; Hall, T.C. Plant SET Domain-containing Proteins: Structure, Function and Regulation. Biochim. Biophys. Acta 2007, 1769, 316. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.H.; Qiu, H.L.; Huang, Y.; Zhang, L.; Si, J.P. Genome-Wide identification and expression profiling of SET DOMAIN GROUP family in Dendrobium catenatum. BMC Plant Biol. 2020, 20, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Benedito, V.A.; Torres-Jerez, I.; Murray, J.D.; Andriankaja, A.; Allen, S.; Kakar, K.; Wandrey, M.; Verdier, J.; Zuber, H.; Ott, T.; et al. A gene expression atlas of the model legume Medicago truncatula. Plant J. 2008, 55, 504–513. [Google Scholar] [CrossRef]
- Aiese Cigliano, R.; Sanseverino, W.; Cremona, G.; Ercolano, M.R.; Conicella, C.; Consiglio, F.M. Genome-Wide analysis of histone modifiers in tomato: Gaining an insight into their developmental roles. BMC Genom. 2013, 14, 57. [Google Scholar] [CrossRef] [Green Version]
- Perrella, G.; Kaiserli, E. Light behind the curtain: Photoregulation of nuclear architecture and chromatin dynamics in plants. New Phytol. 2016, 212, 908–919. [Google Scholar] [CrossRef] [Green Version]
- Zhai, H.; Zhang, X.; You, Y.; Lin, L.; Zhou, W.; Li, C. SEUSS integrates transcriptional and epigenetic control of root stem cell organizer specification. EMBO J. 2020, 39, 1–14. [Google Scholar] [CrossRef]
- Zhang, X.; Ménard, R.; Li, Y.; Coruzzi, G.M.; Heitz, T.; Shen, W.H.; Berr, A. Arabidopsis SDG8 Potentiates the Sustainable Transcriptional Induction of the Pathogenesis-Related Genes PR1 and PR2 During Plant Defense Response. Front. Plant Sci. 2020, 11, 277. [Google Scholar] [CrossRef] [Green Version]
- Xu, F.; Kuo, T.; Rosli, Y.; Liu, M.S.; Wu, L.; Chen, L.F.O.; Fletcher, J.C.; Sung, Z.R.; Pu, L. Trithorax Group Proteins Act Together with a Polycomb Group Protein to Maintain Chromatin Integrity for Epigenetic Silencing during Seed Germination in Arabidopsis. Mol. Plant 2018, 11, 659–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gan, E.S.; Xu, Y.; Wong, J.Y.; Geraldine Goh, J.; Sun, B.; Wee, W.Y.; Huang, J.; Ito, T. Jumonji demethylases moderate precocious flowering at elevated temperature via regulation of FLC in Arabidopsis. Nat. Commun. 2014, 5, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, J.N.; Ryu, J.Y.; Jeong, Y.M.; Park, J.; Song, J.J.; Amasino, R.M.; Noh, B.; Noh, Y.S. Control of Seed Germination by Light-Induced Histone Arginine Demethylation Activity. Dev. Cell 2012, 22, 736–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antunez-Sanchez, J.; Naish, M.; Ramirez-Prado, J.S.; Ohno, S.; Huang, Y.; Dawson, A.; Opassathian, K.; Manza-Mianza, D.; Ariel, F.; Raynaud, C.; et al. A new role for histone demethylases in the maintenance of plant genome integrity. Elife 2020, 9, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.; Cui, X.; Zhang, S.; Jenuwein, T.; Cao, X. Arabidopsis REF6 is a histone H3 lysine 27 demethylase. Nat. Genet. 2011, 43, 715–719. [Google Scholar] [CrossRef] [PubMed]
- True, J.R.; Carroll, S.B. Gene Co-Option in Physiological and Morphological Evolution. Annu. Rev. Cell Dev. Biol. 2002, 18, 53–80. [Google Scholar] [CrossRef] [PubMed]
- Winkler, G.S.; Kristjuhan, A.; Erdjument-Bromage, H.; Tempst, P.; Svejstrup, J.Q. Elongator is a histone H3 and H4 acetyltransferase important for normal histone acetylation levels in vivo. Proc. Natl. Acad. Sci. USA 2002, 99, 3517–3522. [Google Scholar] [CrossRef] [Green Version]
- Peng, M.; Ying, P.; Liu, X.; Li, C.; Xia, R.; Li, J.; Zhao, M. Genome-Wide Identification of Histone Modifiers and Their Expression Patterns during Fruit Abscission in Litchi. Front. Plant Sci. 2017, 8, 639. [Google Scholar] [CrossRef]
- Zhang, Y.; Xue, Y.; Shi, J.; Ahn, J.; Mi, W.; Ali, M.; Wang, X.; Klein, B.J.; Wen, H.; Li, W.; et al. The ZZ domain of p300 mediates specificity of the adjacent HAT domain for histone H3. Nat. Struct. Mol. Biol. 2019, 25, 841–849. [Google Scholar] [CrossRef]
- Wälde, S.; Thakar, K.; Hutten, S.; Spillner, C.; Nath, A.; Rothbauer, U.; Wiemann, S.; Kehlenbach, R.H. The Nucleoporin Nup358/RanBP2 Promotes Nuclear Import in a Cargo- and Transport Receptor-Specific Manner. Traffic 2012, 13, 218–233. [Google Scholar] [CrossRef]
- Santi, C.; Bogusz, D.; Franche, C. Biological nitrogen fixation in non-legume plants. Ann. Bot. 2013, 111, 743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.-H.; Huang, Y.; Jiang, C.; Si, J.-P. Chromatin-Based Regulation of Plant Root Development. Front. Plant Sci. 2018, 9, 1509. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.N.; Kim, J.H.; Jeong, C.Y.; Hong, S.-W.; Lee, H. Inhibition of histone deacetylation alters Arabidopsis root growth in response to auxin via PIN1 degradation. Plant Cell Rep. 2013, 32, 1625–1636. [Google Scholar] [CrossRef] [PubMed]
- Manzano, C.; Ramirez-Parra, E.; Casimiro, I.; Otero, S.; Desvoyes, B.; de Rybel, B.; Beeckman, T.; Casero, P.; Gutierrez, C.; del Pozo, J.C. Auxin and Epigenetic Regulation of SKP2B, an F-Box That Represses Lateral Root Formation. Plant Physiol. 2012, 160, 749–762. [Google Scholar] [CrossRef] [Green Version]
- Yao, X.; Feng, H.; Yu, Y.; Dong, A.; Shen, W.-H. SDG2-Mediated H3K4 Methylation Is Required for Proper Arabidopsis Root Growth and Development. PLoS ONE 2013, 8, e56537. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.; Park, O.-S.; Seo, P.J. JMJ30-mediated demethylation of H3K9me3 drives tissue identity changes to promote callus formation in Arabidopsis. Plant J. 2018, 95, 961–975. [Google Scholar] [CrossRef] [Green Version]
- Gu, X.; Xu, T.; He, Y. A Histone H3 Lysine-27 Methyltransferase Complex Represses Lateral Root Formation in Arabidopsis thaliana. Mol. Plant 2014, 7, 977–988. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Gao, J.; Gao, S.; Li, Z.; Kuai, B.; Ren, G. REF6 promotes lateral root formation through de-repression of PIN1/3/7 genes. J. Integr. Plant Biol. 2019, 61, 383–387. [Google Scholar] [CrossRef]
- Singh, S.; Singh, A.; Roy, S.; Sarkar, A.K. SWP1 negatively regulates lateral root initiation and elongation in Arabidopsis. Plant Signal. Behav. 2012, 7, 1522–1525. [Google Scholar] [CrossRef] [Green Version]
- Naumann, K.; Fischer, A.; Hofmann, I.; Krauss, V.; Phalke, S.; Irmler, K.; Hause, G.; Aurich, A.-C.; Dorn, R.; Jenuwein, T.; et al. Pivotal role of AtSUVH2 in heterochromatic histone methylation and gene silencing in Arabidopsis. EMBO J. 2005, 24, 1418–1429. [Google Scholar] [CrossRef]
- Jackson, J.P.; Lindroth, A.M.; Cao, X.; Jacobsen, S.E. Control of CpNpG DNA methylation by the KRYPTONITE histone H3 methyltransferase. Nature 2002, 416, 556–560. [Google Scholar] [CrossRef]
- Berr, A.; McCallum, E.J.; Ménard, R.; Meyer, D.; Fuchs, J.; Dong, A.; Shen, W.H. Arabidopsis SET DOMAIN GROUP2 Is Required for H3K4 Trimethylation and Is Crucial for Both Sporophyte and Gametophyte Development. Plant Cell 2010, 22, 3232–3248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, H.; Krishnakumar, V.; Bidwell, S.; Rosen, B.; Chan, A.; Zhou, S.; Gentzbittel, L.; Childs, K.L.; Yandell, M.; Gundlach, H.; et al. An improved genome release (version Mt4.0) for the model legume Medicago truncatula. BMC Genom. 2014, 15, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, N.D.; Debellé, F.; Oldroyd, G.E.D.; Geurts, R.; Cannon, S.B.; Udvardi, M.K.; Benedito, V.A.; Mayer, K.F.X.; Gouzy, J.; Schoof, H.; et al. The Medicago genome provides insight into the evolution of rhizobial symbioses. Nature 2011, 480, 520–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phytozome. Available online: https://phytozome-next.jgi.doe.gov/ (accessed on 12 November 2021).
- Shen, C.; Du, H.; Chen, Z.; Lu, H.; Zhu, F.; Chen, H.; Meng, X.; Liu, Q.; Liu, P.; Zheng, L.; et al. The Chromosome-Level Genome Sequence of the Autotetraploid Alfalfa and Resequencing of Core Germplasms Provide Genomic Resources for Alfalfa Research. Mol. Plant 2020, 13, 1250–1261. [Google Scholar] [CrossRef]
- Camiolo, S.; Porceddu, A. Gff2sequence, a new user friendly tool for the generation of genomic sequences. BioData Min. 2013, 6, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Letunic, I.; Bork, P. 20 years of the SMART protein domain annotation resource. Nucleic Acids Res. 2018, 46, D493–D496. [Google Scholar] [CrossRef]
- Letunic, I.; Khedkar, S.; Bork, P. SMART: Recent updates, new developments and status in 2020. Nucleic Acids Res. 2021, 49, D458–D460. [Google Scholar] [CrossRef]
- Hu, B.; Jin, J.; Guo, A.Y.; Zhang, H.; Luo, J.; Gao, G. GSDS 2.0: An upgraded gene feature visualization server. Bioinformatics 2015, 31, 1296–1297. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Tang, H.; Debarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.H.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [Green Version]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [Green Version]
- Dai, X.; Zhuang, Z.; Boschiero, C.; Dong, Y.; Zhao, P.X. LegumeIP V3: From models to crops-An integrative gene discovery platform for translational genomics in legumes. Nucleic Acids Res. 2021, 49, D1472–D1479. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saeed, A.I.; Sharov, V.; White, J.; Li, J.; Liang, W.; Bhagabati, N.; Braisted, J.; Klapa, M.; Currier, T.; Thiagarajan, M.; et al. TM4: A free, open-source system for microarray data management and analysis. Biotechniques 2003, 34, 374–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| M. truncatula | M. sativa | |||||
|---|---|---|---|---|---|---|
| Type | Family | PFAM | A17 v.4.01 | R108 | CADL | Zhongmu No.1 |
| HMTs | SDGs | PF00856 | 78 | 67 | 130 | 68 |
| PRMTs | PF05185 | 3 | 3 | 6 | 4 | |
| HDMs | JMJs | PF02373 | 34 | 29 | 54 | 27 |
| HDMAs | PF04433 | 12 | 12 | 21 | 12 | |
| HATs | HAGs | PF00583 | 51 | 53 | 94 | 51 |
| HAMs | PF01853 | 1 | 1 | 2 | 1 | |
| HACs | PF08214 | 11 | 8 | 22 | 14 | |
| HAFs | PF09247 | 1 | 1 | 2 | 1 | |
| HDACs | HDAs | PF00850 | 10 | 11 | 19 | 12 |
| SRTs | PF02146 | 2 | 2 | 4 | 2 | |
| HDTs | None | 3 | 8 | 16 | 2 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lopez, L.; Perrella, G.; Calderini, O.; Porceddu, A.; Panara, F. Genome-Wide Identification of Histone Modification Gene Families in the Model Legume Medicago truncatula and Their Expression Analysis in Nodules. Plants 2022, 11, 322. https://doi.org/10.3390/plants11030322
Lopez L, Perrella G, Calderini O, Porceddu A, Panara F. Genome-Wide Identification of Histone Modification Gene Families in the Model Legume Medicago truncatula and Their Expression Analysis in Nodules. Plants. 2022; 11(3):322. https://doi.org/10.3390/plants11030322
Chicago/Turabian StyleLopez, Loredana, Giorgio Perrella, Ornella Calderini, Andrea Porceddu, and Francesco Panara. 2022. "Genome-Wide Identification of Histone Modification Gene Families in the Model Legume Medicago truncatula and Their Expression Analysis in Nodules" Plants 11, no. 3: 322. https://doi.org/10.3390/plants11030322
APA StyleLopez, L., Perrella, G., Calderini, O., Porceddu, A., & Panara, F. (2022). Genome-Wide Identification of Histone Modification Gene Families in the Model Legume Medicago truncatula and Their Expression Analysis in Nodules. Plants, 11(3), 322. https://doi.org/10.3390/plants11030322