
Identification of Genomic Loci and Candidate Genes Related to Seed Tocopherol Content in Soybean

 ,
,  ,
,  ,
,  and
and 
Abstract
:1. Introduction
2. Results
2.1. Pooling of the Accessions with Extreme Tocopherol Content
2.2. Whole-Genome Resequencing
2.3. Genomic Variation (SNP/InDel) Detection and Annotation
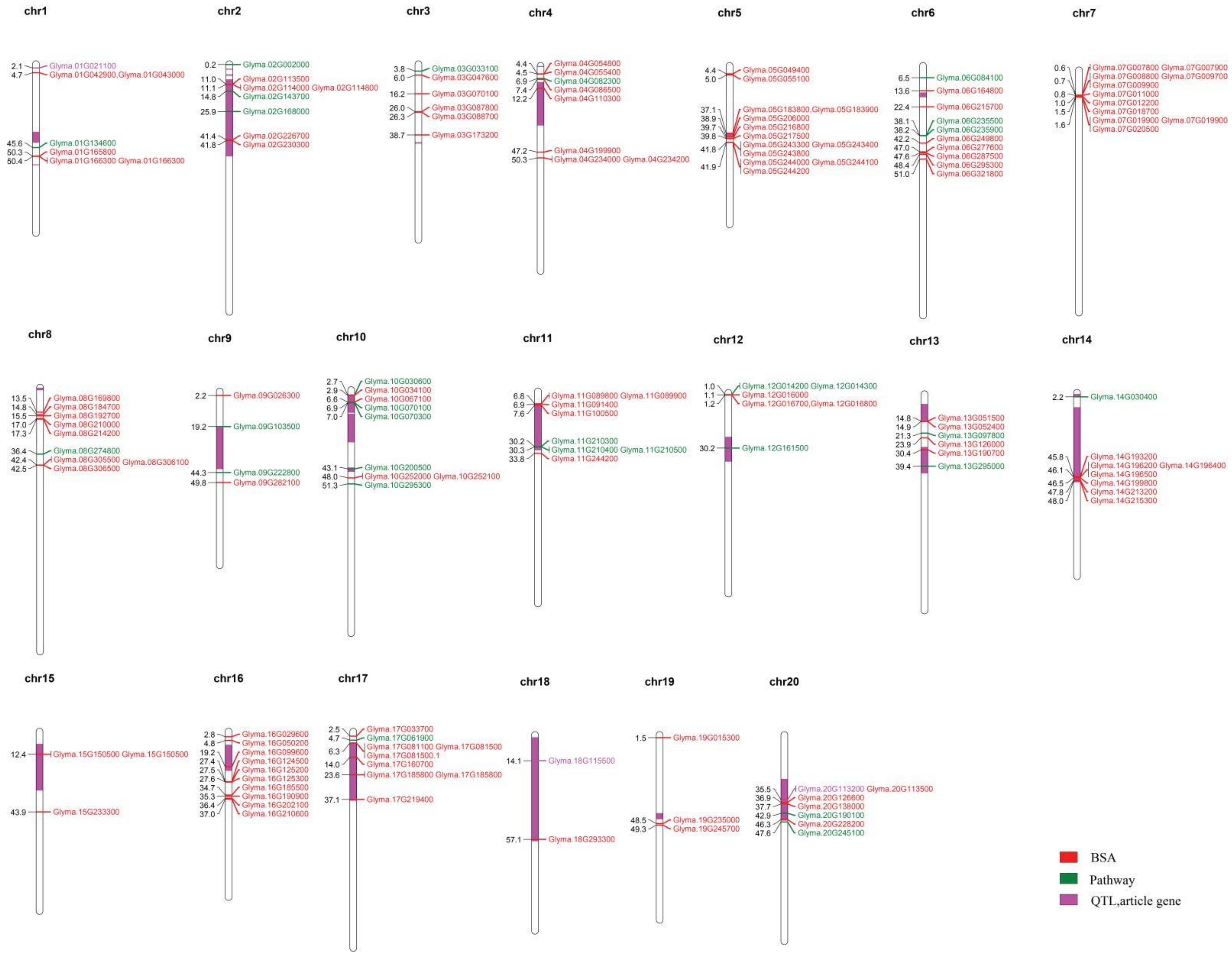
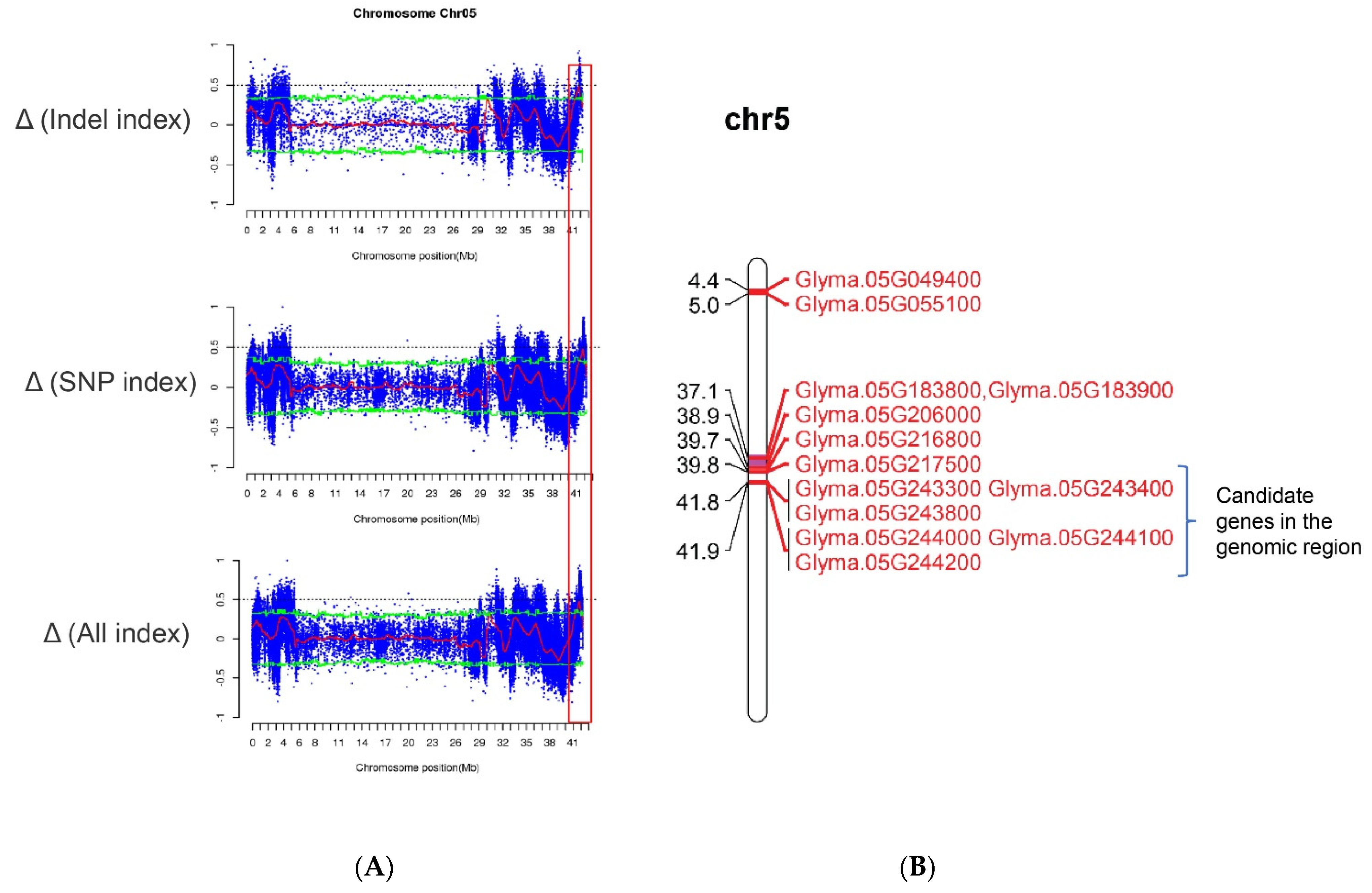
2.4. Identification of Candidate Genes
2.5. Gene Identification by Sliding Window Analysis
2.6. GO Enrichment Analysis of Candidate Genes
3. Discussion
3.1. Soybean Tocopherol Profiling and WGR Coupled with BSA-Seq
3.2. BSA-Seq Combined with Haplotype Analysis Can Quickly Identify Candidate Genes
3.3. Uridine-Diphosphate-Glycosyltransferase (UGT) May Be Involved in Tocopherol Accumulation
3.4. Lipoxygenases May Be Involved in Tocopherol Accumulation
3.5. Tocopherol and Lipid (PUFA) Interaction
3.6. Tocopherol and ABA Interaction
4. Materials and Methods
4.1. Plant Materials and Tocopherol Phenotyping
4.2. Genomic DNA Isolation
4.3. Construction of Genomic DNA Bulks for Sequencing
4.4. Sequencing Data Quality Analysis
4.5. Sequence Alignment
4.6. Variant (SNPs and InDels) Detection and Annotation
4.7. Variant (SNP/InDel-Index) Association Analysis
4.8. Sliding Window Analysis
4.9. Candidate Gene Identification
4.10. GO Analysis of Predicted Candidate Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fritsche, S.; Wang, X.; Li, J.; Stich, B.; Kopisch-Obuch, F.J.; Endrigkeit, J.; Leckband, G.; Dreyer, F.; Friedt, W.; Meng, J.; et al. A candidate gene-based association study of tocopherol content and composition in rapeseed (Brassica napus). Front. Plant Sci. 2012, 3, 129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Liu, H.; Han, Y.; Wu, X.; Teng, W.; Liu, G.; Li, W. Identification of QTL underlying vitamin E contents in soybean seed among multiple environments. Theor. Appl. Genet. 2010, 120, 1405–1413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, A.K.; Muthusamy, V.; Zunjare, R.U.; Chauhan, H.S.; Sharma, P.K.; Bhat, J.S.; Guleria, S.K.; Saha, S.; Hossain, F. Genetic variability-, genotype× environment interactions-and combining ability-analyses of kernel tocopherols among maize genotypes possessing novel allele of γ-tocopherol methyl transferase (ZmVTE4). J. Cereal Sci. 2019, 86, 1–8. [Google Scholar] [CrossRef]
- Ma, J.; Qiu, D.; Gao, H.; Wen, H.; Wu, Y.; Pang, Y.; Wang, X.; Qin, Y. Over-expression of a γ-tocopherol methyltransferase gene in vitamin E pathway confers PEG-simulated drought tolerance in alfalfa. BMC Plant Biol. 2020, 20, 226. [Google Scholar] [CrossRef]
- Mene-Saffrane, L.; Pellaud, S. Current strategies for vitamin E biofortification of crops. Curr. Opin. Biotechnol. 2017, 44, 189–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinutha, T.; Bansal, N.; Kumari, K.; Prashat, R.; Sreevathsa, R.; Krishnan, V.; Kumari, S.; Dahuja, A.; Lal, S.; Sachdev, A.; et al. Comparative analysis of tocopherol biosynthesis genes and its transcriptional regulation in soybean seeds. J. Agric. Food Chem. 2017, 65, 11054–11064. [Google Scholar] [CrossRef]
- Hwang, J.E.; Ahn, J.-W.; Kwon, S.J.; Kim, J.B.; Kim, S.H.; Kang, S.Y.; Kim, D.S. Selection and molecular characterization of a high tocopherol accumulation rice mutant line induced by gamma irradiation. Mol. Biol. Rep. 2014, 41, 7671–7681. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Quinn, P.J. The location and function of vitamin E in membranes. Mol. Membr. Biol. 2000, 17, 143–156. [Google Scholar] [CrossRef]
- Hussain, N.; Irshad, F.; Jabeen, Z.; Shamsi, I.H.; Li, Z.; Jiang, L. Biosynthesis, structural, and functional attributes of tocopherols in planta; past, present, and future perspectives. J. Agric. Food Chem. 2013, 61, 6137–6149. [Google Scholar] [CrossRef]
- Jo, Y.; Hyun, T.K. Genome-wide identification of antioxidant component biosynthetic enzymes: Comprehensive analysis of ascorbic acid and tocochromanols biosynthetic genes in rice. Comput. Biol. Chem. 2011, 35, 261–268. [Google Scholar] [CrossRef]
- Ruiz-Sola, M. Carotenoid biosynthesis in Arabidopsis: A colorful pathway. Arab. Book 2012, 10, e0158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Wood, D.; Wang, H.H.; Jones, J.P. Predicting potential impacts of China’s retaliatory tariffs on the US farm sector. Choices 2018, 33, 1–6. [Google Scholar] [CrossRef]
- Garg, M.; Sharma, N.; Sharma, S.; Kapoor, P.; Kumar, A.; Chunduri, V.; Arora, P. Biofortified crops generated by breeding, agronomy, and transgenic approaches are improving lives of millions of people around the world. Front. Nutr. 2018, 5, 12. [Google Scholar] [CrossRef]
- Azam, M.; Zhang, S.; Qi, J.; Abdelghany, A.M.; Shaibu, A.S.; Ghosh, S.; Feng, Y.; Huai, Y.; Gebregziabher, B.S.; Li, J.; et al. Profiling and associations of seed nutritional characteristics in Chinese and USA soybean cultivars. J. Food Compos. Anal. 2021, 98, 103803. [Google Scholar] [CrossRef]
- Agyenim-Boateng, G.K.; Zhang, S.; Islam, S.; Gu, Y.; Li, B.; Azam, M.; Abdelghany, A.M.; Qi, J.; Ghosh, S.; Schaibu, A.S.; et al. Profiling of naturally occurring folates in a diverse soybean germplasm by HPLC-MS/MS. Food Chem. 2022, 384, 132520. [Google Scholar] [CrossRef]
- Sui, M.; Jing, Y.; Li, H.; Zhan, Y.; Luo, J.; Teng, W.; Qiu, L.; Zheng, H.; Li, W.; Zhao, X.; et al. Identification of loci and candidate genes analyses for tocopherol concentration of soybean seed. Front. Plant Sci. 2020, 11, 539460. [Google Scholar] [CrossRef]
- Seguin, P.; Tremblay, G.; Pageau, D.; Liu, W. Soybean tocopherol concentrations are affected by crop management. J. Agric. Food Chem. 2010, 58, 5495–5501. [Google Scholar] [CrossRef]
- Michelmore, R.W.; Paran, I.; Kesseli, R.V. Identification of markers linked to disease-resistance genes by bulked segregant analysis: A rapid method to detect markers in specific genomic regions by using segregating populations. Proc. Natl. Acad. Sci. USA. 1991, 88, 9828–9832. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Tian, X.; Wang, Z.; Wei, X.; Zhao, Y.; Su, H.; Zhao, X.; Tian, B.; Yuan, Y.; Zhang, X.-W. Fine mapping and candidate gene identification of a white flower gene BrWF3 in Chinese cabbage (Brassica rapa L. ssp. pekinensis). Front. Plant Sci. 2021, 12, 832. [Google Scholar] [CrossRef]
- Haase, N.J.; Beissinger, T.; Hirsch, C.N.; Vaillancourt, B.; Deshpande, S.; Barry, K.; Buell, C.R.; Kaeppler, S.M.; De Leon, N. Shared genomic regions between derivatives of a large segregating population of maize identified using bulked segregant analysis sequencing and traditional linkage analysis. G3 Genes Genomes Genet. 2015, 5, 1593–1602. [Google Scholar] [CrossRef] [Green Version]
- Feng, C.; Bluhm, B.; Shi, A.; Correll, J.C. Development of molecular markers linked to three spinach downy mildew resistance loci. Euphytica 2018, 214, 174. [Google Scholar] [CrossRef]
- Li, Z.; Xu, Y. Bulk segregation analysis in NGS era: A review for its teenage years. Plant J. 2021, 109, 1355–1374. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Bi, B.; Xu, X.; Li, B.; Tian, S.; Wang, J.; Zhang, H.; Wang, G.; Han, Y.; McElroy, J.S. Rapid identification of a candidate nicosulfuron sensitivity gene (Nss) in maize (Zea mays L.) via combining bulked segregant analysis and RNA-seq. Theor. Appl. Genet. 2019, 132, 1351–1361. [Google Scholar] [CrossRef] [PubMed]
- Takagi, H.; Uemura, A.; Yaegashi, H.; Tamiru, M.; Abe, A.; Mitsuoka, C.; Utsushi, H.; Natsume, S.; Kanzaki, H.; Matsumura, H.; et al. MutMap-Gap: Whole-genome resequencing of mutant F2 progeny bulk combined with de novo assembly of gap regions identifies the rice blast resistance gene Pii. New Phytol. 2013, 200, 276–283. [Google Scholar] [CrossRef]
- Wang, J.-Y.; Chen, J.-D.; Wang, S.-L.; Chen, L.; Ma, C.-L.; Yao, M.-Z. Repressed gene expression of photosynthetic antenna proteins associated with yellow leaf variation as revealed by bulked segregant RNA-seq in tea plant Camellia sinensis. J. Agric. Food Chem. 2020, 68, 8068–8079. [Google Scholar] [CrossRef]
- Zou, C.; Wang, P.; Xu, Y. Bulked sample analysis in genetics, genomics and crop improvement. Plant Biotechnol. J. 2016, 14, 1941–1955. [Google Scholar] [CrossRef] [Green Version]
- Feng, X.; Li, X.; Yang, X.; Zhu, P. Fine mapping and identification of the leaf shape gene BoFL in ornamental kale. Theor. Appl. Genet. 2020, 133, 1303–1312. [Google Scholar] [CrossRef]
- Whipple, C.J.; Kebrom, T.H.; Weber, A.L.; Yang, F.; Hall, D.; Meeley, R.; Schmidt, R.; Doebley, J.; Brutnell, T.P.; Jackson, D.P. Grassy tillers1 promotes apical dominance in maize and responds to shade signals in the grasses. Proc. Natl. Acad. Sci. USA 2011, 108, E506–E512. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Fan, S.; Yu, F.; Chen, Y.; Zhang, S.; Han, F.; Yan, S.; Wang, L.; Sun, J. High-resolution mapping of QTL for fatty acid composition in soybean using specific-locus amplified fragment sequencing. Theor. Appl. Genet. 2017, 130, 1467–1479. [Google Scholar] [CrossRef] [Green Version]
- Pei, R.; Zhang, J.; Tian, L.; Zhang, S.; Han, F.; Yan, S.; Wang, L.; Li, B.; Sun, J. Identification of novel QTL associated with soybean isoflavone content. Crop J. 2018, 6, 244–252. [Google Scholar] [CrossRef]
- Jo, H.; Kim, M.; Cho, H.; Ha, B.-K.; Kang, S.; Song, J.T.; Lee, J.-D. Identification of a potential gene for elevating ω-3 concentration and its efficiency for improving the ω-6/ω-3 ratio in soybean. J. Agric. Food Chem. 2021, 69, 3836–3847. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murithi, H.; Beed, F.; Tukamuhabwa, P.; Thomma, B.; Joosten, M. Soybean production in eastern and southern Africa and threat of yield loss due to soybean rust caused by Phakopsorapachyrhizi. Plant Pathol. 2016, 65, 176–188. [Google Scholar] [CrossRef]
- Raederstorff, D.; Wyss, A.; Calder, P.C.; Weber, P.; Eggersdorfer, M. Vitamin E function and requirements in relation to PUFA. Br. J. Nutr. 2015, 114, 1113–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Traber, M.G.; Stevens, J.F. Medicine. Vitamins C and E: Beneficial effects from a mechanistic perspective. Free Radic. Biol. Med. 2011, 51, 1000–1013. [Google Scholar] [CrossRef] [Green Version]
- Fekih, R.; Takagi, H.; Tamiru, M.; Abe, A.; Natsume, S.; Yaegashi, H.; Sharma, S.; Sharma, S.; Kanzaki, H.; Matsumura, H.; et al. MutMap+: Genetic mapping and mutant identification without crossing in rice. PLoS ONE 2013, 8, e68529. [Google Scholar] [CrossRef]
- Song, J.; Li, Z.; Liu, Z.; Guo, Y.; Qiu, L.-J. Next-generation sequencing from bulked-segregant analysis accelerates the simultaneous identification of two qualitative genes in soybean. Front. Plant Sci. 2017, 8, 919. [Google Scholar] [CrossRef] [Green Version]
- Metzker, M.L. Sequencing technologies—the next generation. Nat. Rev. Genet. 2010, 11, 31–46. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Chen, P.; Zhao, Q.; Cai, G.; Hu, Y.; Xiang, Y.; Yang, Q.; Wang, Y.; Zhou, Y. Co-location of QTL for Sclerotinia stem rot resistance and flowering time in Brassica napus. Crop J. 2019, 7, 227–237. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, X.; Pan, Q.; Li, P.; Liu, Y.; Lu, X.; Zhong, W.; Li, M.; Han, L.; Li, J.; et al. QTG-Seq accelerates QTL fine mapping through QTL partitioning and whole-genome sequencing of bulked segregant samples. Mol. Plant 2019, 12, 426–437. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S.; Zhang, S.; Azam, M.; Qi, J.; Abdelghany, A.M.; Shaibu, A.S.; Gebregziabher, B.S.; Feng, Y.; Huai, Y.; Htway, H.T.P.; et al. Seed tocopherol assessment and geographical distribution of 1151 Chinese soybean accessions from diverse ecoregions. J. Food Compos. Anal. 2021, 100, 103932. [Google Scholar] [CrossRef]
- Ghosh, S.; Zhang, S.; Azam, M.; Gebregziabher, B.S.; Abdelghany, A.M.; Shaibu, A.S.; Qi, J.; Feng, Y.; Agyenim-Boateng, K.; Liu, Y.; et al. Natural variation of seed tocopherol composition in diverse world soybean accessions from maturity group 0 to VI grown in China. Plants 2022, 11, 206. [Google Scholar] [CrossRef] [PubMed]
- Shaw, E.J.; Kakuda, Y.; Rajcan, I. Effect of genotype, environment, and genotype× environment interaction on tocopherol accumulation in soybean seed. Crop Sci. 2016, 56, 40–50. [Google Scholar] [CrossRef]
- Meech, R.; Hu, D.G.; McKinnon, R.A.; Mubarokah, S.N.; Haines, A.Z.; Nair, P.C.; Rowland, A.; Mackenzie, P.I. The UDP-glycosyltransferase (UGT) superfamily: New members, new functions, and novel paradigms. Physiol. Rev. 2019, 99, 1153–1222. [Google Scholar] [CrossRef]
- Zhou, Y.; Fu, W.-B.; Si, F.-L.; Yan, Z.-T.; Zhang, Y.-J.; He, Q.-Y.; Chen, B. UDP-glycosyltransferase genes and their association and mutations associated with pyrethroid resistance in Anopheles sinensis (Diptera: Culicidae). Malar. J. 2019, 18, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luque, T.; O’Reilly, D.R. Functional and phylogenetic analyses of a putative Drosophila melanogaster UDP-glycosyltransferase gene. Insect Biochem. Mol. Biol. 2002, 32, 1597–1604. [Google Scholar] [CrossRef]
- Dixon, R.A.; Paiva, N.L. Stress-induced phenylpropanoid metabolism. Plant Cell 1995, 7, 1085–1097. [Google Scholar] [CrossRef]
- Peng, M.; Shahzad, R.; Gul, A.; Subthain, H.; Shen, S.; Lei, L.; Zheng, Z.; Zhou, J.; Lu, D.; Wang, S.; et al. Differentially evolved glucosyltransferases determine natural variation of rice flavone accumulation and UV-tolerance. Nat. Commun. 2017, 8, 1975. [Google Scholar] [CrossRef]
- Dong, N.-Q.; Sun, Y.; Guo, T.; Shi, C.-L.; Zhang, Y.-M.; Kan, Y.; Xiang, Y.-H.; Zhang, H.; Yang, Y.-B.; Li, Y.-C.; et al. UDP-glucosyltransferase regulates grain size and abiotic stress tolerance associated with metabolic flux redirection in rice. Nat. Commun. 2020, 11, 2629. [Google Scholar] [CrossRef]
- Fraser, C.M.; Chapple, C. The phenylpropanoid pathway in Arabidopsis. Arabidopsis Book 2011, 9, e0152. [Google Scholar] [CrossRef] [Green Version]
- Xie, M.; Zhang, J.; Tschaplinski, T.J.; Tuskan, G.A.; Chen, J.-G.; Muchero, W. Regulation of lignin biosynthesis and its role in growth-defensetradeoffs. Front. Plant Sci. 2018, 9, 1427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, V.; Rani, A.; Jha, P.; Hussain, L.; Pal, V.; Petwal, V.; Kumar, P.; Dwivedi, J. Lipoxygenase and tocopherol profiling of soybean genotypes exposed to electron beam irradiation. J. Am. Oil Chem. Soc. 2017, 94, 457–463. [Google Scholar] [CrossRef]
- Dixit, A.K.; Kumar, V.; Rani, A.; Manjaya, J.; Bhatnagar, D. Effect of gamma irradiation on lipoxygenases, trypsin inhibitor, raffinose family oligosaccharides and nutritional factors of different seed coat colored soybean (Glycine max L.). Radiat. Phys. Chem. 2011, 80, 597–603. [Google Scholar] [CrossRef]
- Bhardwaj, P.K.; Kaur, J.; Sobti, R.C.; Ahuja, P.S.; Kumar, S. Lipoxygenase in Caragana jubata responds to low temperature, abscisic acid, methyl jasmonate and salicylic acid. Gene 2011, 483, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Viswanath, K.K.; Varakumar, P.; Pamuru, R.R.; Basha, S.J.; Mehta, S.; Rao, A.D. Plant lipoxygenases and their role in plant physiology. J. Plant Biol. 2020, 63, 83–95. [Google Scholar] [CrossRef]
- Elisia, I.; Young, J.W.; Yuan, Y.V.; Kitts, D.D. Association between tocopherol isoform composition and lipid oxidation in selected multiple edible oils. Food Res. Int. 2013, 52, 508–514. [Google Scholar] [CrossRef]
- Harayama, T.; Shimizu, T. Roles of polyunsaturated fatty acids, from mediators to membranes. J. Lipid Res. 2020, 61, 1150–1160. [Google Scholar] [CrossRef]
- Sattler, S.E.; Gilliland, L.U.; Magallanes-Lundback, M.; Pollard, M.; DellaPenna, D. Vitamin E is essential for seed longevity and for preventing lipid peroxidation during germination. Plant Cell 2004, 16, 1419–1432. [Google Scholar] [CrossRef] [Green Version]
- Verma, V.; Ravindran, P.; Kumar, P.P. Plant hormone-mediated regulation of stress responses. BMC Plant Biol. 2016, 16, 86. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.C.; Luan, S. ABA signal transduction at the crossroad of biotic and abiotic stress responses. Plant Cell Environ. 2012, 35, 53–60. [Google Scholar] [CrossRef]
- Fritsche, S.; Wang, X.; Jung, C. Recent advances in our understanding of tocopherol biosynthesis in plants: An overview of key genes, functions, and breeding of vitamin E improved crops. Antioxidants 2017, 6, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhary, N.; Khurana, P. Vitamin E biosynthesis genes in rice: Molecular characterization, expression profiling and comparative phylogenetic analysis. Plant Sci. 2009, 177, 479–491. [Google Scholar] [CrossRef]
- Fleta-Soriano, E.; Munné-Bosch, S. Enhanced plastochromanol-8 accumulation during reiterated drought in maize (Zea mays L.). Plant Physiol. Biochem. 2017, 112, 283–289. [Google Scholar] [CrossRef]
- Shinozaki, K.; Yamaguchi-Shinozaki, K. Gene networks involved in drought stress response and tolerance. J. Exp. Bot. 2007, 58, 221–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghassemian, M.; Lutes, J.; Chang, H.-S.; Lange, I.; Chen, W.; Zhu, T.; Wang, X.; Lange, B.M. Abscisic acid-induced modulation of metabolic and redox control pathways in Arabidopsis thaliana. Phytochemistry 2008, 69, 2899–2911. [Google Scholar] [CrossRef] [PubMed]
- Dwiyanti, M.S.; Ujiie, A.; Le, T.B.T.; Yamada, T.; Kitamura, K. Genetic analysis of high α-tocopherol content in soybean seeds. Breeding Science 2007, 57, 23–28. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Takagi, H.; Abe, A.; Yoshida, K.; Kosugi, S.; Natsume, S.; Mitsuoka, C.; Uemura, A.; Utsushi, H.; Tamiru, M.; Takuno, S.; et al. QTL-seq: Rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. Plant J. 2013, 74, 174–183. [Google Scholar] [CrossRef]
- Du, H.; Wen, C.; Zhang, X.; Xu, X.; Yang, J.; Chen, B.; Geng, S. Identification of a major QTL (qRRs-10.1) that confers resistance to Ralstonia solanacearum in pepper (Capsicum annuum) using SLAF-BSA and QTL mapping. Int. J. Mol. Sci. 2019, 20, 5887. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample a | Reference Genome | Number of Plants Bulked | Raw Bases (bp) | Clean Bases (bp) | Effective Rate (%) | Error Rate (%) | Q20 (%) | Q30 (%) | GC Content b (%) |
|---|---|---|---|---|---|---|---|---|---|
| VE-Low | Williams 82 | 49 | 57,765,692,100 | 57,645,759,600 | 99.79 | 0.03 | 97.66 | 93.37 | 36.25 |
| VE-High | Williams 82 | 49 | 53,145,146,700 | 53,070,136,800 | 99.86 | 0.03 | 97.41 | 92.77 | 36.52 |
| Sample | Mapped Reads | Total Reads | Mapping Rate (%) | Average depth (X) | Coverage at Least 1× (%) | Coverage at Least 4× (%) |
|---|---|---|---|---|---|---|
| VE-High | 349,651,428 | 353,800,912 | 98.83 | 41.52 | 99.61 | 99.04 |
| VE-Low | 379,650,505 | 384,305,064 | 98.79 | 44.57 | 99.65 | 99.11 |
| Category | SNP | InDel |
|---|---|---|
| Intergenic | 2,701,815 | 404,485 |
| Upstream | 207,958 | 57,177 |
| Downstream | 180,387 | 44,948 |
| Upstream/Downstream | 10,174 | 2853 |
| Intronic | 326,872 | 69,637 |
| Stop-gain | 1614 | 126 |
| Stop-loss | 315 | 27 |
| Frameshift deletion | - | 1902 |
| Frameshift insertion | - | 1744 |
| Non-frameshift deletion Exonic | - | 1513 |
| Non-frameshift insertion | - | 1323 |
| Synonymous | 55,156 | - |
| Non-synonymous | 74,303 | - |
| Splicing | 774 | 266 |
| Insertion | - | 282,838 |
| Deletion | - | 304,169 |
| Transitions, conversions | 2,325,983 | - |
| Transversions, change | 1,233,385 | - |
| Conversion to transversal ratio | 1885 | - |
| Total | 3,559,368 | 587,007 |
| Position | Chromosome | Ref | Alt | Effect | Gene ID | Ortholog in Arabidopsis | Description |
|---|---|---|---|---|---|---|---|
| 39758623 | Chr05 | A | G | Nonsynonymous | Glyma.05G217500 | AT1G64450.1 | Glycine-rich protein family |
| 41807338 | Chr05 | T | G | Nonsynonymous | Glyma.05G243400 | AT1G18070.2 | Translation elongation factor EF1A/initiation factor IF2gamma family protein |
| 48428377 | Chr06 | C | T | Nonsynonymous | Glyma.06G295300 | AT4G28140.1 | Integrase-type DNA-binding superfamily protein |
| 17002951 | Chr08 | A | G | Nonsynonymous | Glyma.08G210000 | AT1G15420.1 | Repeat-containing protein |
| 14945609 | Chr13 | A | C | Nonsynonymous | Glyma.13G052400 | - | - |
| 46481282 | Chr14 | C | G | Nonsynonymous | Glyma.14G199800 | AT1G13960.1 | WRKY DNA-binding protein 4 |
| 35333785 | Chr16 | C | T | Nonsynonymous | Glyma.16G190900 | AT2G34930.1 | Disease resistance family protein/LRR family protein |
| 36350176 | Chr16 | C | G | Nonsynonymous | Glyma.16G202100 | AT4G05200.1 | Cysteine-rich RLK (Receptor-like protein kinase) 25 |
| 36970367 | Chr16 | G | C | Nonsynonymous | Glyma.16G210600 | AT5G36930.2 | Disease resistance protein (TIR-NBS-LRR class) family |
| 6309627 | Chr17 | C | T | Nonsynonymous | Glyma.17G081100 | AT2G03820.1 | Nonsense-mediated mRNA decay NMD3 family protein |
| 6333162 | Chr17 | A | T | Stop-gain | Glyma.17G081500 | - | - |
| 14819413 | Chr08 | GCAGT | - | Frameshift Deletion | Glyma.08G184700 | AT5G24750.1 | UDP-Glycosyltransferase superfamily protein |
| 43894622 | Chr15 | A | - | Frameshift Deletion | Glyma.15G233300 | AT1G55020.1 | Lipoxygenase 1; Linoleate 9S-lipoxygenase/Linoleate 9-lipoxygenase |
| GO ID | Term | Annotated | Count | Expected | p-Value | Genes |
|---|---|---|---|---|---|---|
| GO:0033993 | Response to lipid | 565 | 5 | 1.02 | 0.004 | Glyma.04G199900, Glyma.05G243800, Glyma.05G244100, Glyma.06G249800, Glyma.17G219400 |
| GO:0009737 | Response to abscisic acid | 437 | 4 | 0.79 | 0.008 | Glyma.05G243800, Glyma.05G244100, Glyma.06G249800, Glyma.17G219400 |
| GO:0097305 | Response to alcohol | 502 | 4 | 0.91 | 0.013 | Glyma.05G243800, Glyma.05G244100, Glyma.06G249800, Glyma.17G219400 |
| GO:1901700 | Response to oxygen-containing compound | 1104 | 6 | 2.00 | 0.014 | Glyma.03G173200, Glyma.04G199900, Glyma.05G243800, Glyma.05G244100, Glyma.06G249800, Glyma.17G219400 |
| GO:0001101 | Response to acid chemical | 813 | 5 | 1.47 | 0.016 | Glyma.04G199900, Glyma.05G243800, Glyma.05G244100, Glyma.06G249800, Glyma.17G219400 |
| GO:0000041 | Transition metal ion transport | 116 | 2 | 0.21 | 0.019 | Glyma.14G196200, Glyma.17G219400 |
| GO:0009845 | Seed germination | 117 | 2 | 0.21 | 0.019 | Glyma.05G243800, Glyma.05G244100 |
| GO:0090351 | Seedling development | 131 | 2 | 0.24 | 0.024 | Glyma.05G243800, Glyma.05G244100 |
| GO:0042221 | Response to chemical | 2380 | 9 | 4.31 | 0.026 | Glyma.03G173200, Glyma.04G199900, Glyma.05G183800, Glyma.05G243800, Glyma.05G244100, Glyma.06G249800, Glyma.08G169800, Glyma.17G219400, Glyma.18G293300 |
| GO:0071215 | Cellular response to abscisic acid stimulus | 192 | 2 | 0.35 | 0.047 | Glyma.05G243800, Glyma.06G249800 |
| GO:0009719 | Response to endogenous stimulus | 1480 | 6 | 2.68 | 0.050 | Glyma.03G173200, Glyma.04G199900, Glyma.05G243800, Glyma.05G244100, Glyma.06G249800, Glyma.17G219400 |
| GO:0046915 | Transition metal ion transmembrane transporter activity | 103 | 2 | 0.21 | 0.019 | Glyma.14G196200, Glyma.17G219400 |
| GO:0003690 | Double-stranded DNA binding | 150 | 2 | 0.31 | 0.038 | Glyma.01G042900, Glyma.08G306100 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghosh, S.; Zhang, S.; Azam, M.; Agyenim-Boateng, K.G.; Qi, J.; Feng, Y.; Li, Y.; Li, J.; Li, B.; Sun, J. Identification of Genomic Loci and Candidate Genes Related to Seed Tocopherol Content in Soybean. Plants 2022, 11, 1703. https://doi.org/10.3390/plants11131703
Ghosh S, Zhang S, Azam M, Agyenim-Boateng KG, Qi J, Feng Y, Li Y, Li J, Li B, Sun J. Identification of Genomic Loci and Candidate Genes Related to Seed Tocopherol Content in Soybean. Plants. 2022; 11(13):1703. https://doi.org/10.3390/plants11131703
Chicago/Turabian StyleGhosh, Suprio, Shengrui Zhang, Muhammad Azam, Kwadwo Gyapong Agyenim-Boateng, Jie Qi, Yue Feng, Yecheng Li, Jing Li, Bin Li, and Junming Sun. 2022. "Identification of Genomic Loci and Candidate Genes Related to Seed Tocopherol Content in Soybean" Plants 11, no. 13: 1703. https://doi.org/10.3390/plants11131703
APA StyleGhosh, S., Zhang, S., Azam, M., Agyenim-Boateng, K. G., Qi, J., Feng, Y., Li, Y., Li, J., Li, B., & Sun, J. (2022). Identification of Genomic Loci and Candidate Genes Related to Seed Tocopherol Content in Soybean. Plants, 11(13), 1703. https://doi.org/10.3390/plants11131703