
Transcriptomes of Saussurea (Asteraceae) Provide Insights into High-Altitude Adaptation

 , , ,
, , ,  ,
,
Abstract
:1. Introduction
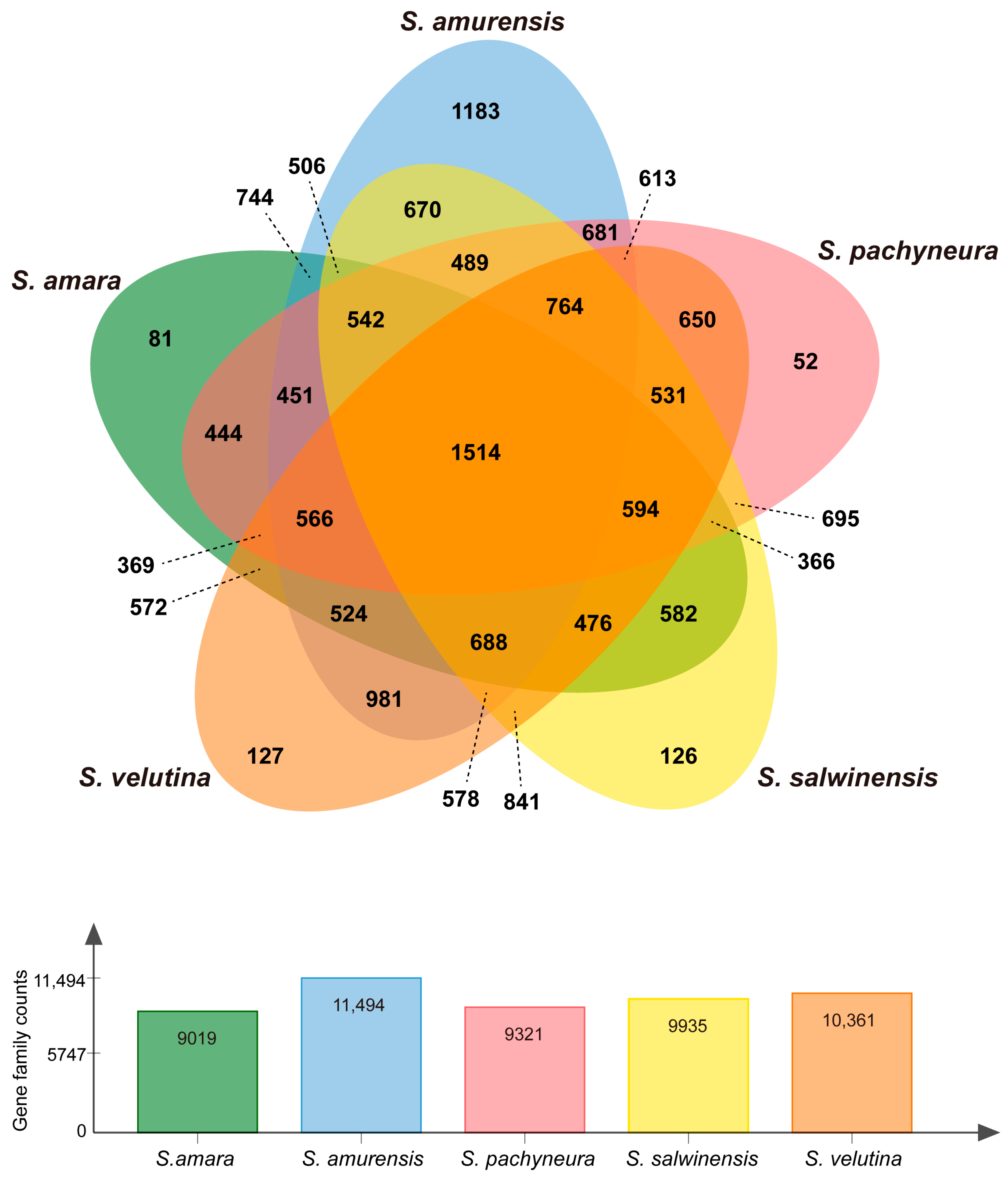
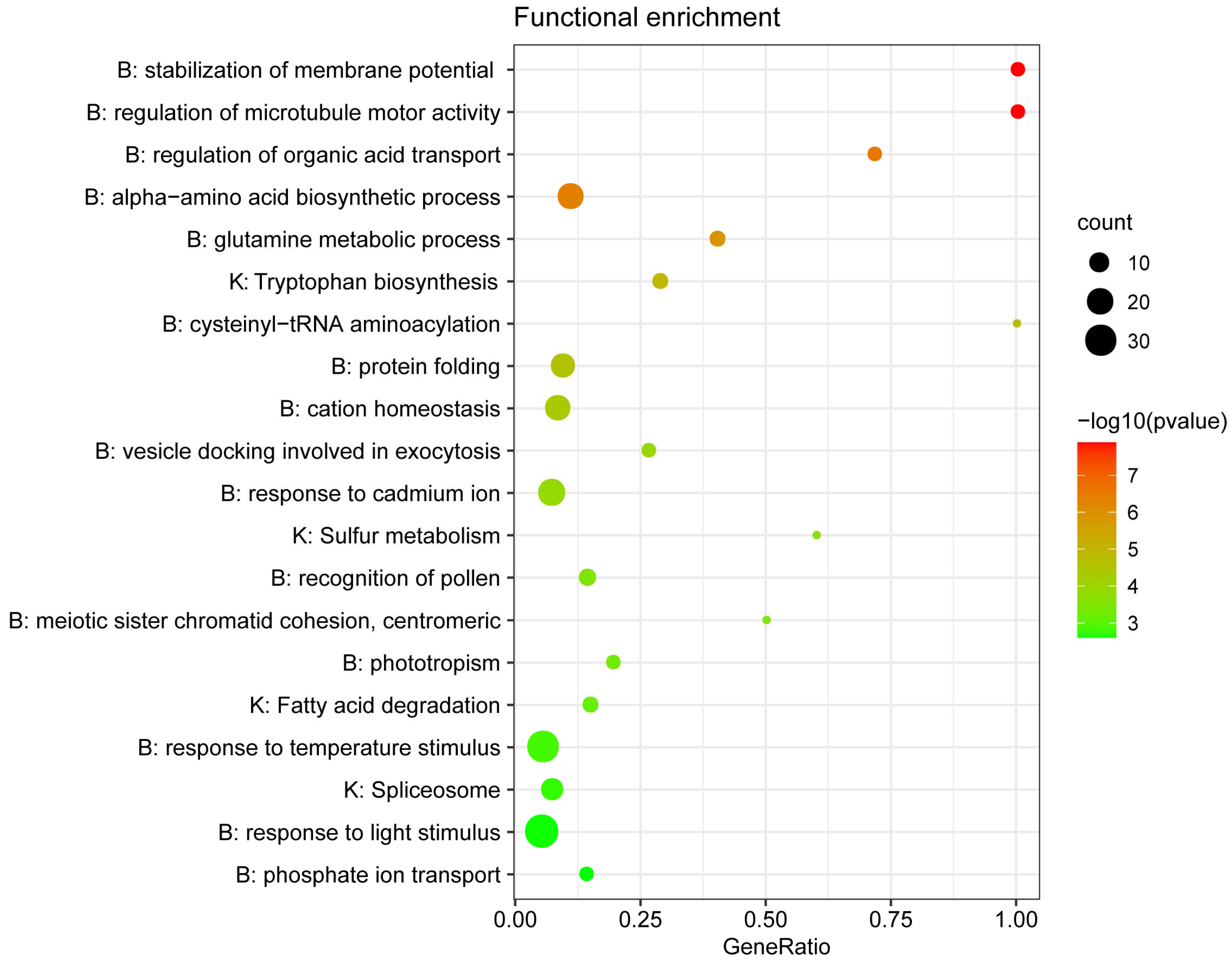
2. Results
2.1. De novo Assembly and Annotation of Unigenes
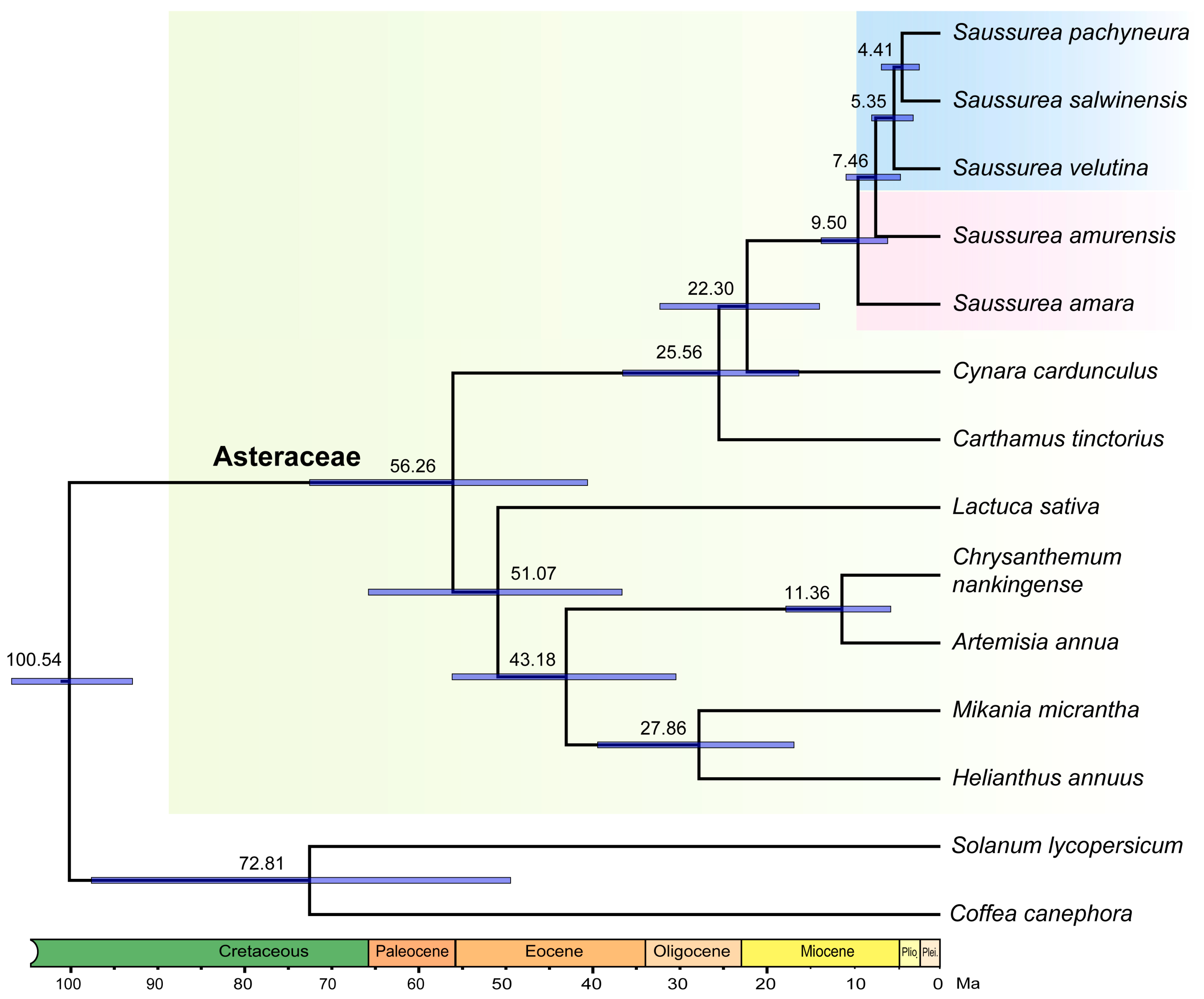
2.2. Estimation of Divergence Time
2.3. Evolutionary Analysis
2.4. Genes under Positive Selection
3. Discussion
4. Materials and Methods
4.1. Sample Collection and Transcriptome Sequencing
4.2. Transcriptome Assembly and Annotation
4.3. Estimation of Divergence Time
4.4. Evolutionary Analysis
4.5. Phylogenetic Tests of Positive Selection
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yang, J.; Jin, Z.B.; Chen, J.; Huang, X.F.; Li, X.M.; Liang, Y.B.; Mao, J.Y.; Chen, X.; Zheng, Z.; Bakshi, A.; et al. Genetic signatures of high-altitude adaptation in Tibetans. Proc. Natl. Acad. Sci. USA 2017, 114, 4189–4194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huerta-Sanchez, E.; Jin, X.; Bianba, A.Z.; Peter, B.M.; Vinckenbosch, N.; Liang, Y.; Yi, X.; He, M.; Somel, M.; Ni, P.; et al. Altitude adaptation in Tibetans caused by introgression of Denisovan-like DNA. Nature 2014, 512, 194–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bigham, A.W. Genetics of human origin and evolution: High-altitude adaptations. Curr. Opin. Genet. Dev. 2016, 41, 8–13. [Google Scholar] [CrossRef] [Green Version]
- Guo, W.; Xin, M.; Wang, Z.; Yao, Y.; Hu, Z.; Song, W.; Yu, K.; Chen, Y.; Wang, X.; Guan, P.; et al. Origin and adaptation to high altitude of Tibetan semi-wild wheat. Nat. Commun. 2020, 11, 5085. [Google Scholar] [CrossRef]
- Zeng, X.; Long, H.; Wang, Z.; Zhao, S.; Tang, Y.; Huang, Z.; Wang, Y.; Xu, Q.; Mao, L.; Deng, G.; et al. The draft genome of Tibetan hulless barley reveals adaptive patterns to the high stressful Tibetan Plateau. Proc. Natl. Acad. Sci. USA 2015, 112, 1095–1100. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Qiao, Q.; Novikova, P.Y.; Wang, Q.; Yue, J.; Guan, Y.; Ming, S.; Liu, T.; De, J.; Liu, Y.; et al. Genome of Crucihimalaya himalaica, a close relative of Arabidopsis, shows ecological adaptation to high altitude. Proc. Natl. Acad. Sci. USA 2019, 116, 7137–7146. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.B.; Fu, T.T.; Jin, J.Q.; Murphy, R.W.; Hillis, D.M.; Zhang, Y.P.; Che, J. Species groups distributed across elevational gradients reveal convergent and continuous genetic adaptation to high elevations. Proc Natl. Acad. Sci. USA 2018, 115, E10634–E10641. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Tian, Y.; Yan, L.; Zhang, G.; Wang, X.; Zeng, Y.; Zhang, J.; Ma, X.; Tan, Y.; Long, N.; et al. Genome of Plant Maca (Lepidium meyenii) Illuminates Genomic Basis for High-Altitude Adaptation in the Central Andes. Mol. Plant. 2016, 9, 1066–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witt, K.E.; Huerta-Sanchez, E. Convergent evolution in human and domesticate adaptation to high-altitude environments. Philos. Trans. R. Soc. London. Ser. B Biol. Sci. 2019, 374, 20180235. [Google Scholar] [CrossRef]
- Monge, C.; Leon-Velarde, F. Physiological adaptation to high altitude: Oxygen transport in mammals and birds. Physiol. Rev. 1991, 71, 1135–1172. [Google Scholar] [CrossRef]
- Hao, Y.; Xiong, Y.; Cheng, Y.; Song, G.; Jia, C.; Qu, Y.; Lei, F. Comparative transcriptomics of 3 high-altitude passerine birds and their low-altitude relatives. Proc. Natl. Acad. Sci. USA 2019, 116, 11851–11856. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Sun, Y.; Landis, J.B.; Zhang, J.; Yang, L.; Lin, N.; Zhang, H.; Guo, R.; Li, L.; Zhang, Y.; et al. Genomic insights into adaptation to heterogeneous environments for the ancient relictual Circaeaster agrestis (Circaeasteraceae, Ranunculales). New Phytol. 2020, 228, 285–301. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef]
- Cheviron, Z.A.; Brumfield, R.T. Genomic insights into adaptation to high-altitude environments. Heredity 2012, 108, 354–361. [Google Scholar] [CrossRef] [Green Version]
- Stark, R.; Grzelak, M.; Hadfield, J. RNA sequencing: The teenage years. Nat. Rev. Genet. 2019, 20, 631–656. [Google Scholar] [CrossRef] [PubMed]
- Todd, E.V.; Black, M.A.; Gemmell, N.J. The power and promise of RNA-seq in ecology and evolution. Mol. Ecol. 2016, 25, 1224–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spicer, R.A.; Farnsworth, A.; Su, T. Cenozoic topography, monsoons and biodiversity conservation within the Tibetan Region: An evolving story. Plant. Divers. 2020, 42, 229–254. [Google Scholar] [CrossRef]
- Favre, A.; Packert, M.; Pauls, S.U.; Jahnig, S.C.; Uhl, D.; Michalak, I.; Muellner-Riehl, A.N. The role of the uplift of the Qinghai-Tibetan Plateau for the evolution of Tibetan biotas. Biol. Rev. Camb. Philos. Soc. 2015, 90, 236–253. [Google Scholar] [CrossRef]
- Mosbrugger, V.; Favre, A.; Muellner-Riehl, A.N.; Päckert, M.; Mulch, A. Cenozoic Evolution of Geobiodiversity in the Tibeto-Himalayan Region. In Mountains, Climate and Biodiversity; Hoorn, C., Perrigo, A., Antonelli, A., Eds.; Wiley-Blackwell: London, UK, 2018; Volume 28, pp. 429–448. [Google Scholar]
- Wen, J.; Zhang, J.Q.; Nie, Z.L.; Zhong, Y.; Sun, H. Evolutionary diversifications of plants on the Qinghai-Tibetan Plateau. Front. Genet. 2014, 5, 4. [Google Scholar] [CrossRef] [Green Version]
- Xing, Y.; Ree, R.H. Uplift-driven diversification in the Hengduan Mountains, a temperate biodiversity hotspot. Proc. Natl. Acad. Sci. USA 2017, 114, E3444–E3451. [Google Scholar] [CrossRef] [Green Version]
- Qiao, Q.; Huang, Y.; Qi, J.; Qu, M.; Jiang, C.; Lin, P.; Li, R.; Song, L.; Yonezawa, T.; Hasegawa, M.; et al. The genome and transcriptome of Trichormus sp. NMC-1: Insights into adaptation to extreme environments on the Qinghai-Tibet Plateau. Sci. Rep. 2016, 6, 29404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, D.L.; Niu, Y.; Song, B.; Chen, J.G.; Li, Z.M.; Yang, Y.; Sun, H. Woolly and overlapping leaves dampen temperature fluctuations in reproductive organ of an alpine Himalayan forb. J. Plant. Ecol. 2015, 8, 159–165. [Google Scholar] [CrossRef] [Green Version]
- Song, B.; Stöcklin, J.; Peng, D.; Gao, Y.; Sun, H. The bracts of the alpine ‘glasshouse’ plant Rheum alexandrae (Polygonaceae) enhance reproductive fitness of its pollinating seed-consuming mutualist. Bot. J. Linn. Soc. 2015, 179, 349–359. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Sun, H. The Bracts of Saussurea velutina (Asteraceae) Protect Inflorescences from Fluctuating Weather at High Elevations of the Hengduan Mountains, Southwestern China. Arct. Antarct. Alp. Res. 2009, 41, 515–521. [Google Scholar] [CrossRef]
- Sun, H.; Niu, Y.; Chen, Y.S.; Song, B.; Liu, C.Q.; Peng, D.L.; Chen, J.G.; Yang, Y. Survival and reproduction of plant species in the Qinghai-Tibet Plateau. J. Syst. Evol. 2014, 52, 378–396. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Körner, C.; Sun, H. The Ecological Significance of Pubescence in Saussurea medusa, a High-Elevation Himalayan “Woolly Plant”. Arct. Antarct. Alp. Res. 2008, 40, 250–255. [Google Scholar] [CrossRef] [Green Version]
- Shi, Z.; von Raab-Straube, E. Cardueae. In Flora of China; Wu, Z.Y., Raven, P.H., Hong, D.Y., Eds.; Science Press & Missouri Botanical Garden Press: Beijing, China; St. Louis, MI, USA, 2011; Volume 20–21, pp. 42–194. [Google Scholar]
- Chen, Y.S. Asteraceae II Saussurea. In Flora of Pan-Himalaya; Hong, D.-Y., Sun, H., Watson, M., Wen, J., Zhang, X.-C., Eds.; Science Press: Beijing, China, 2015; Volume 48. [Google Scholar]
- Wang, Y.J.; Susanna, A.; Von Raab-Straube, E.; Milne, R.; Liu, J.Q. Island-like radiation of Saussurea (Asteraceae: Cardueae) triggered by uplifts of the Qinghai-Tibetan Plateau. Biol. J. Linn. Soc. 2009, 97, 893–903. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Deng, T.; Moore, M.J.; Ji, Y.; Lin, N.; Zhang, H.; Meng, A.; Wang, H.; Sun, Y.; Sun, H. Plastome phylogenomics of Saussurea (Asteraceae: Cardueae). BMC Plant. Biol. 2019, 19, 290. [Google Scholar] [CrossRef]
- Xu, L.S.; Herrando-Moraira, S.; Susanna, A.; Galbany-Casals, M.; Chen, Y.S. Phylogeny, origin and dispersal of Saussurea (Asteraceae) based on chloroplast genome data. Mol. Phylogenet. Evol. 2019, 141, 106613. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Landis, J.B.; Sun, Y.; Zhang, H.; Feng, T.; Lin, N.; Tiamiyu, B.B.; Huang, X.; Deng, T.; Wang, H.; et al. Insights into the drivers of radiating diversification in biodiversity hotspots using Saussurea (Asteraceae) as a case. bioRxiv 2021. [Google Scholar] [CrossRef]
- Barres, L.; Sanmartín, I.; Anderson, C.L.; Susanna, A.; Buerki, S.; Galbany-Casals, M.; Vilatersana, R. Reconstructing the evolution and biogeographic history of tribe Cardueae (Compositae). Am. J. Bot. 2013, 100, 867–882. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Shi, Y.; Yang, S. Advances and challenges in uncovering cold tolerance regulatory mechanisms in plants. New Phytol. 2019, 222, 1690–1704. [Google Scholar] [CrossRef] [Green Version]
- Chinnusamy, V.; Zhu, J.-K.; Sunkar, R. Gene Regulation during Cold Stress Acclimation in Plants. In Plant Stress Tolerance: Methods and Protocols; Sunkar, R., Ed.; Humana Press: Totowa, NJ, USA, 2010; pp. 39–55. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Wang, X.; Ban, Q.; Zhu, X.; Jiang, C.; Wei, C.; Bennetzen, J.L. Comparative transcriptomic analysis reveals gene expression associated with cold adaptation in the tea plant Camellia sinensis. BMC Genom. 2019, 20, 624. [Google Scholar] [CrossRef] [Green Version]
- Crickard, J.B.; Moevus, C.J.; Kwon, Y.; Sung, P.; Greene, E.C. Rad54 Drives ATP Hydrolysis-Dependent DNA Sequence Alignment during Homologous Recombination. Cell 2020, 181, 1380–1394.e1318. [Google Scholar] [CrossRef]
- Gruber, H.; Heijde, M.; Heller, W.; Albert, A.; Seidlitz, H.K.; Ulm, R. Negative feedback regulation of UV-B-induced photomorphogenesis and stress acclimation in Arabidopsis. Proc. Natl. Acad. Sci. USA 2010, 107, 20132–20137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, N.Q.; Lin, H.X. Contribution of phenylpropanoid metabolism to plant development and plant-environment interactions. J. Integr. Plant. Biol. 2021, 63, 180–209. [Google Scholar] [CrossRef]
- Xie, D.F.; Yu, Y.; Wen, J.; Huang, J.; Chen, J.P.; Li, J.; Zhou, S.D.; He, X.J. Phylogeny and highland adaptation of Chinese species in Allium section Daghestanica (Amaryllidaceae) revealed by transcriptome sequencing. Mol. Phylogenet. Evol. 2020, 146, 106737. [Google Scholar] [CrossRef]
- Forde, B.G.; Lea, P.J. Glutamate in plants: Metabolism, regulation, and signalling. J. Exp. Bot. 2007, 58, 2339–2358. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.G.; Yang, Y.; Stocklin, J.; Cavieres, L.A.; Peng, D.L.; Li, Z.M.; Sun, H. Soil nutrient availability determines the facilitative effects of cushion plants on other plant species at high elevations in the south-eastern Himalayas. Plant. Ecol. Divers. 2015, 8, 199–210. [Google Scholar] [CrossRef]
- Qiao, Q.; Wang, Q.; Han, X.; Guan, Y.; Sun, H.; Zhong, Y.; Huang, J.; Zhang, T. Transcriptome sequencing of Crucihimalaya himalaica (Brassicaceae) reveals how Arabidopsis close relative adapt to the Qinghai-Tibet Plateau. Sci. Rep. 2016, 6, 21729. [Google Scholar] [CrossRef] [Green Version]
- Capaldi, F.R.; Gratao, P.L.; Reis, A.R.; Lima, L.W.; Azevedo, R.A. Sulfur Metabolism and Stress Defense Responses in Plants. Trop. Plant. Biol. 2015, 8, 60–73. [Google Scholar] [CrossRef] [Green Version]
- Pandey, A.K.; Gautam, A. Stress responsive gene regulation in relation to hydrogen sulfide in plants under abiotic stress. Physiol. Plant. 2020, 168, 511–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Grabherr, M.; Haas, B.; Yassour, M.; Levin, J.; Thompson, D.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Trinity: Reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith-Unna, R.; Boursnell, C.; Patro, R.; Hibberd, J.M.; Kelly, S. TransRate: Reference-free quality assessment of de novo transcriptome assemblies. Genome Res. 2016, 26, 1134–1144. [Google Scholar] [CrossRef] [Green Version]
- Simao, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [Green Version]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conesa, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [Green Version]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Capella-Gutierrez, S.; Silla-Martinez, J.M.; Gabaldon, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.; Wong, T.; Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Suleski, M.; Hedges, S.B. TimeTree: A Resource for Timelines, Timetrees, and Divergence Times. Mol. Biol Evol 2017, 34, 1812–1819. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Dong, Z.; Fang, L.; Luo, Y.; Wei, Z.; Guo, H.; Zhang, G.; Gu, Y.Q.; Coleman-Derr, D.; Xia, Q.; et al. OrthoVenn2: A web server for whole-genome comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res. 2019, 47, W52–W58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Nielsen, R.; Yang, Z. Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Mol. Biol. Evol. 2005, 22, 2472–2479. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
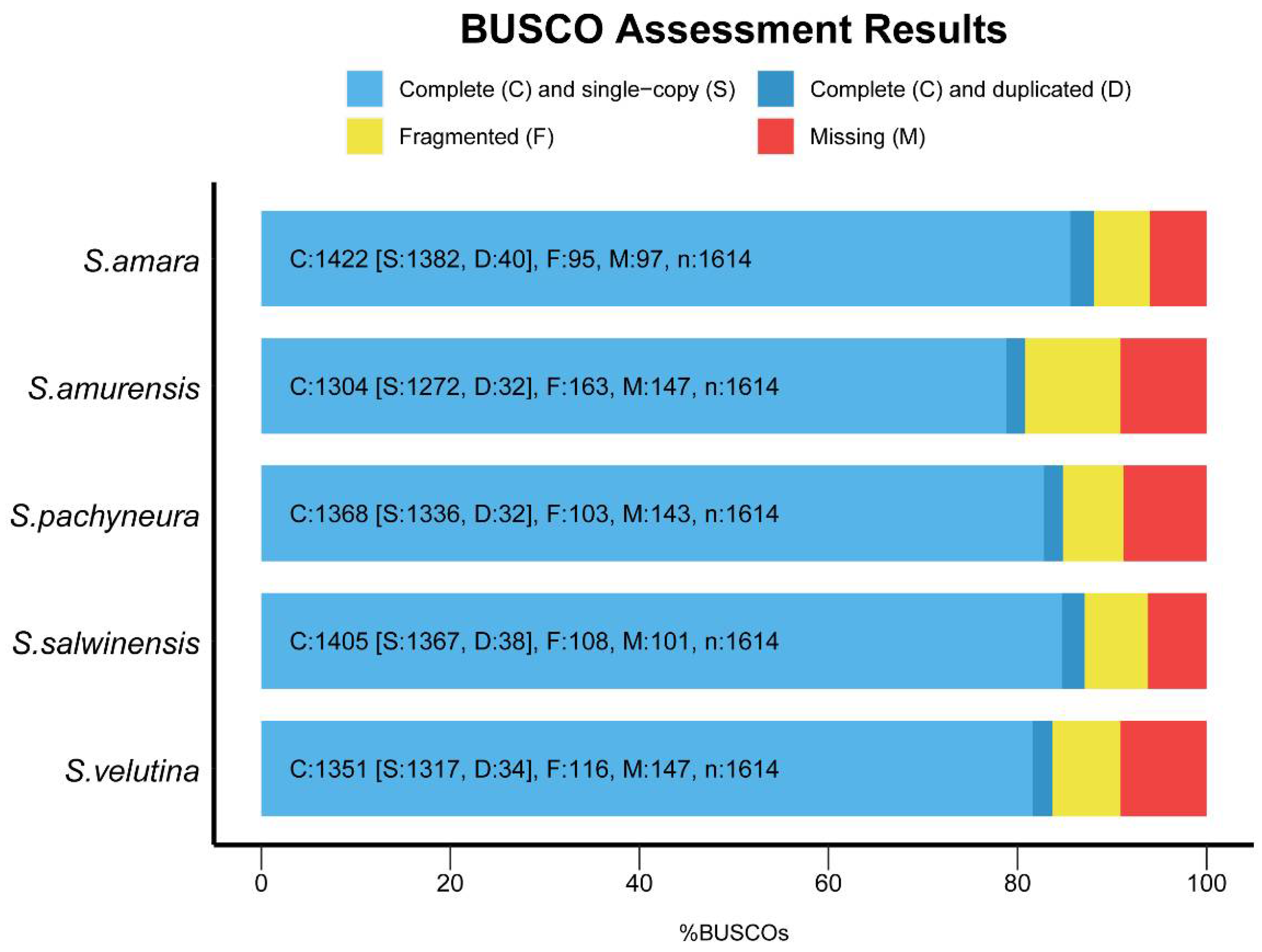
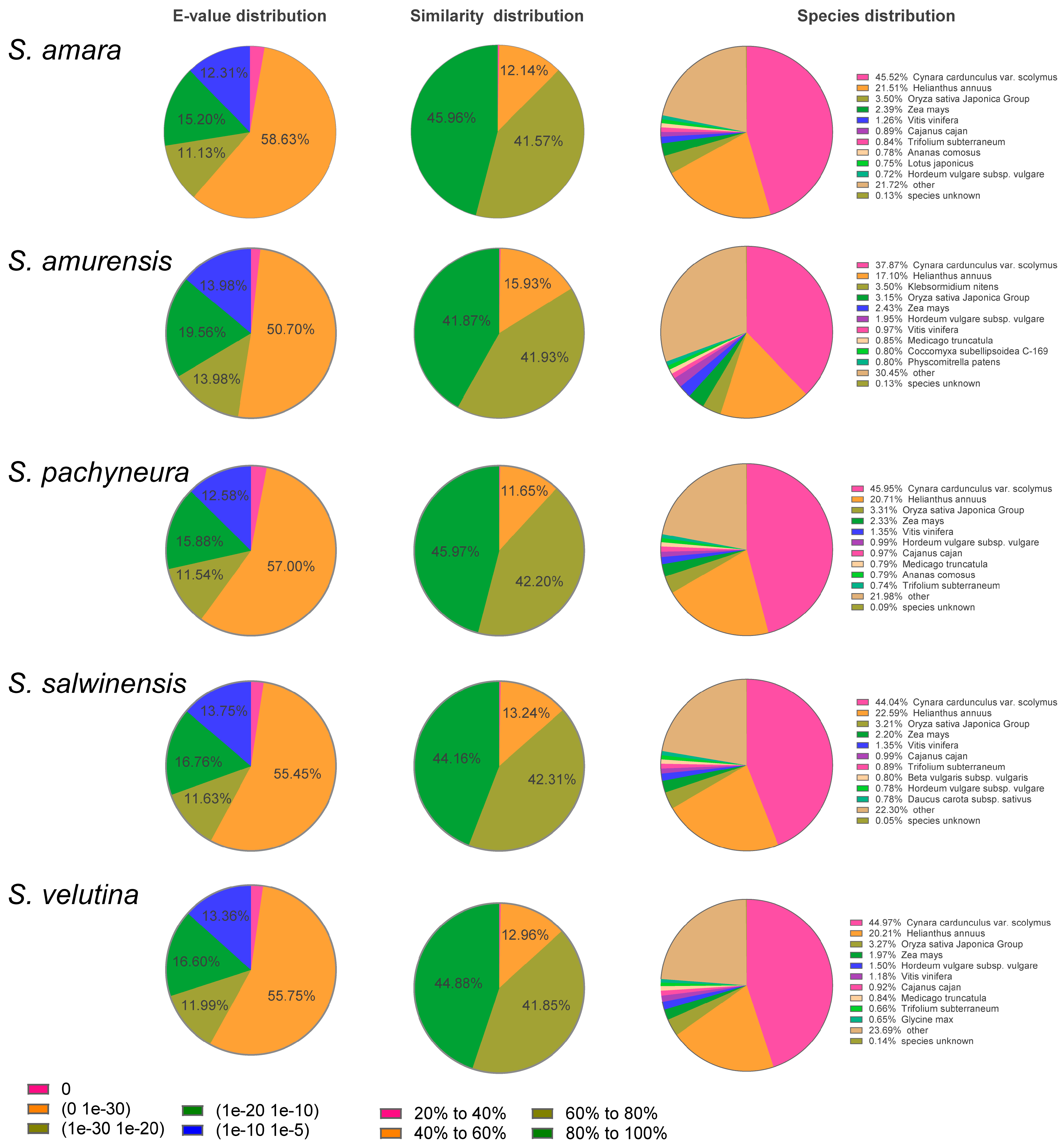
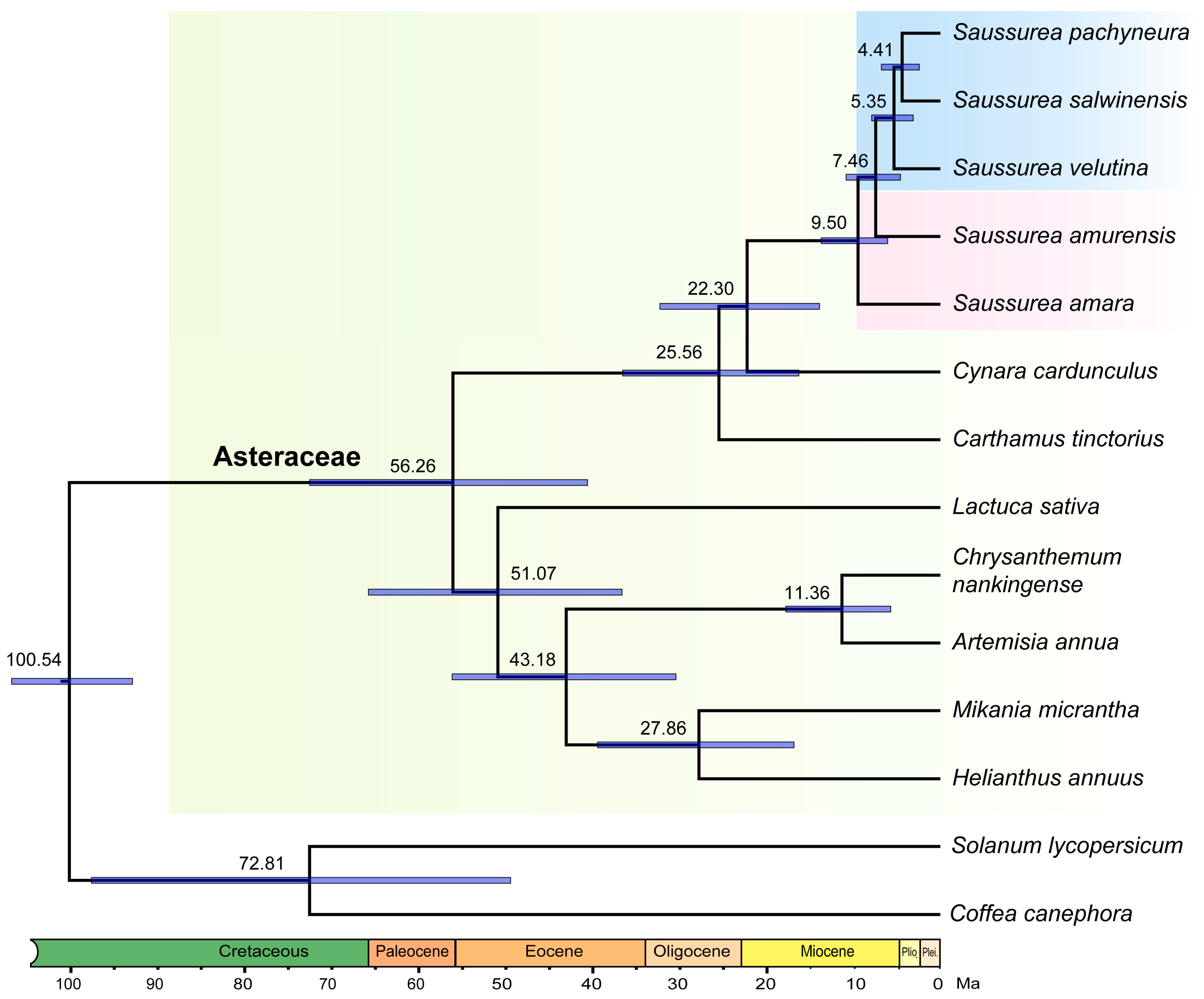
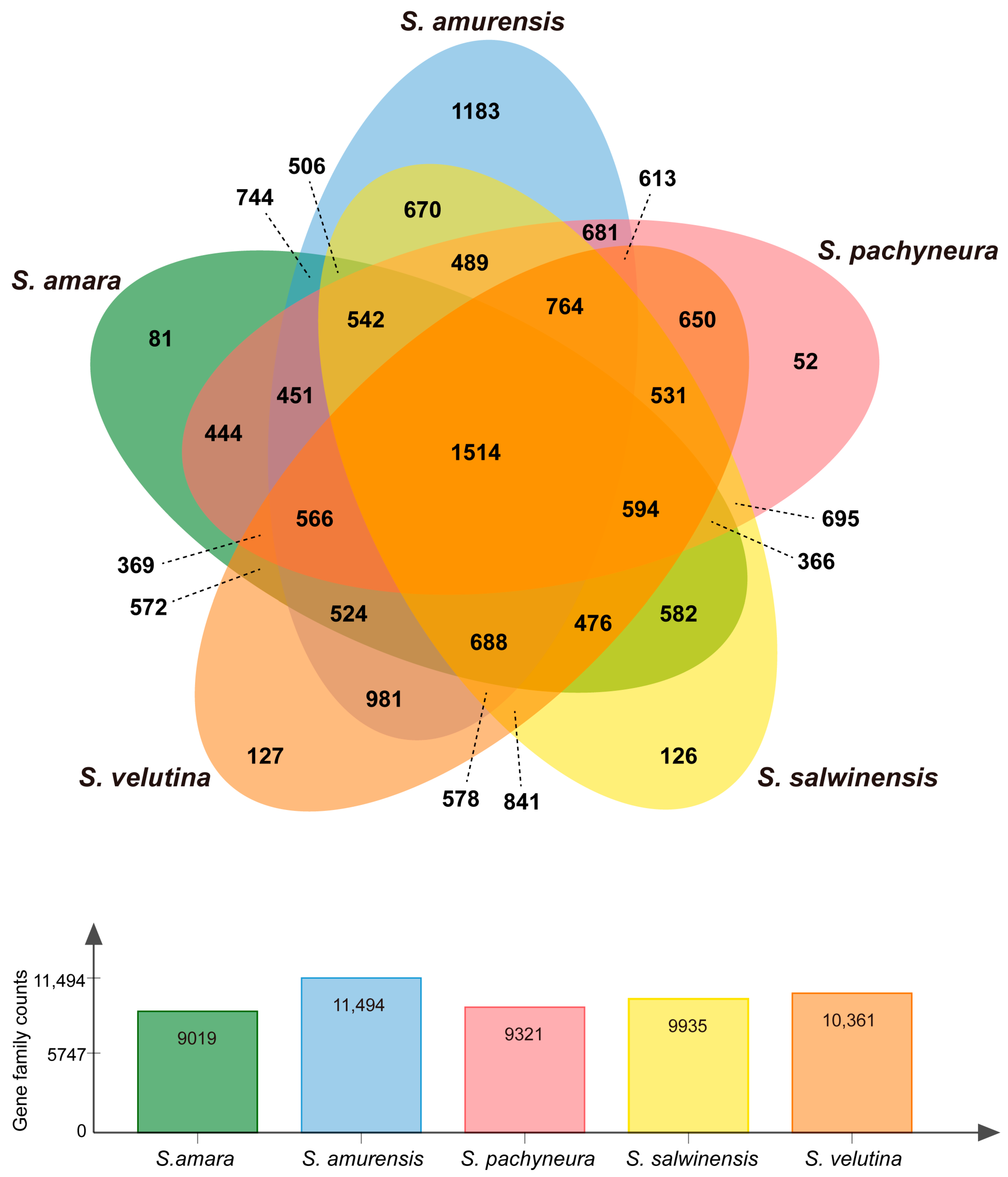
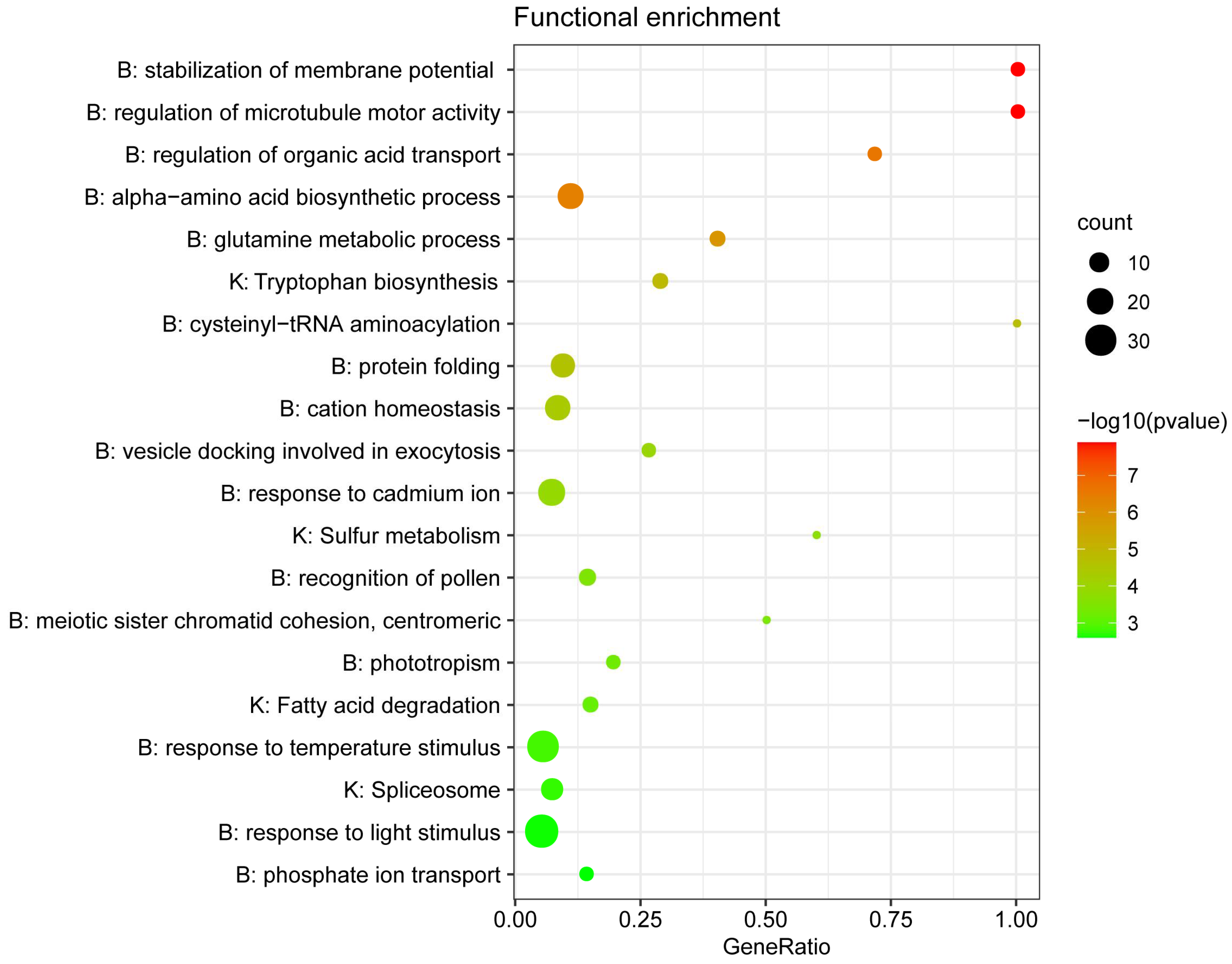

{kind=link}
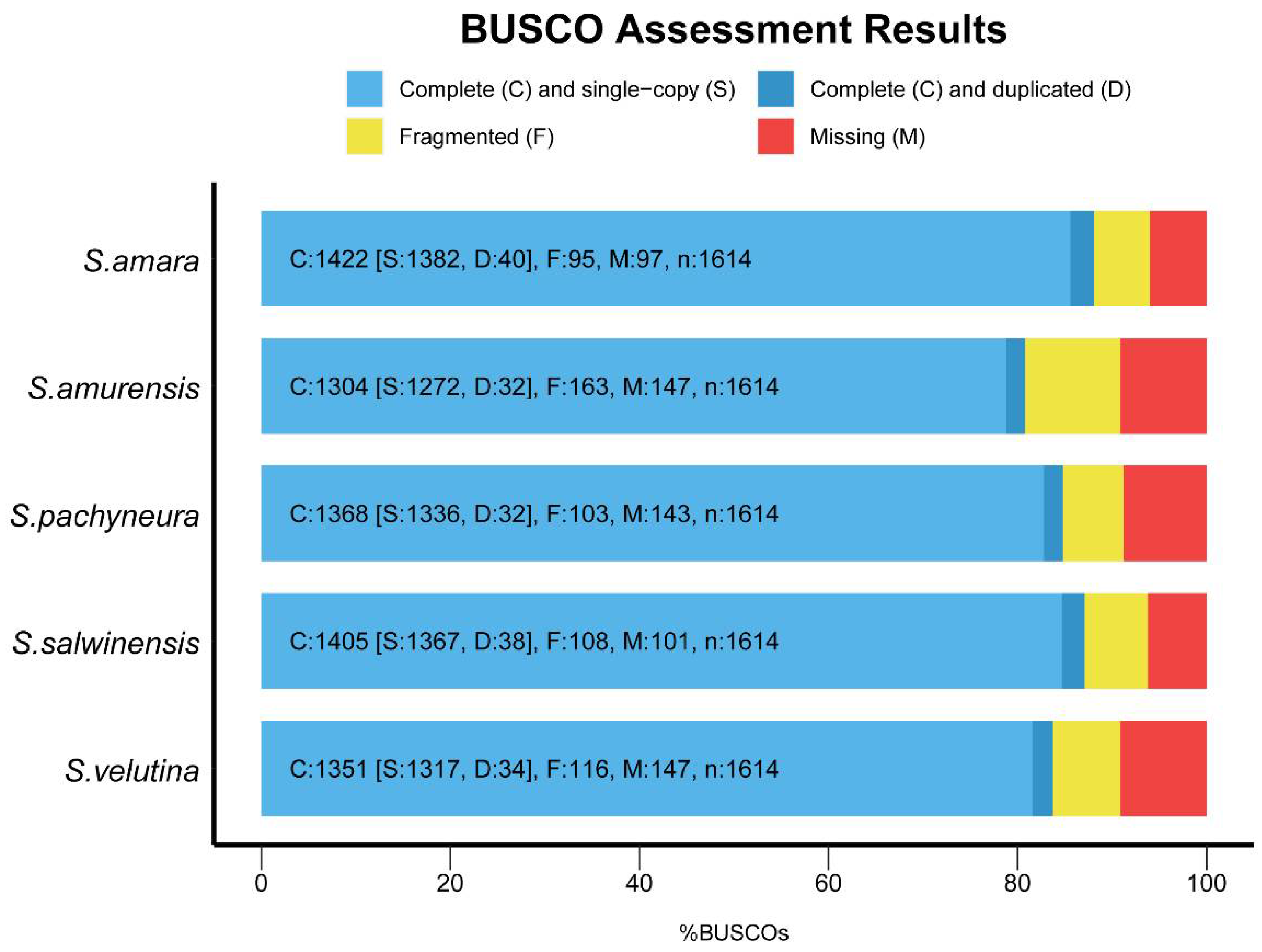
{kind=link}
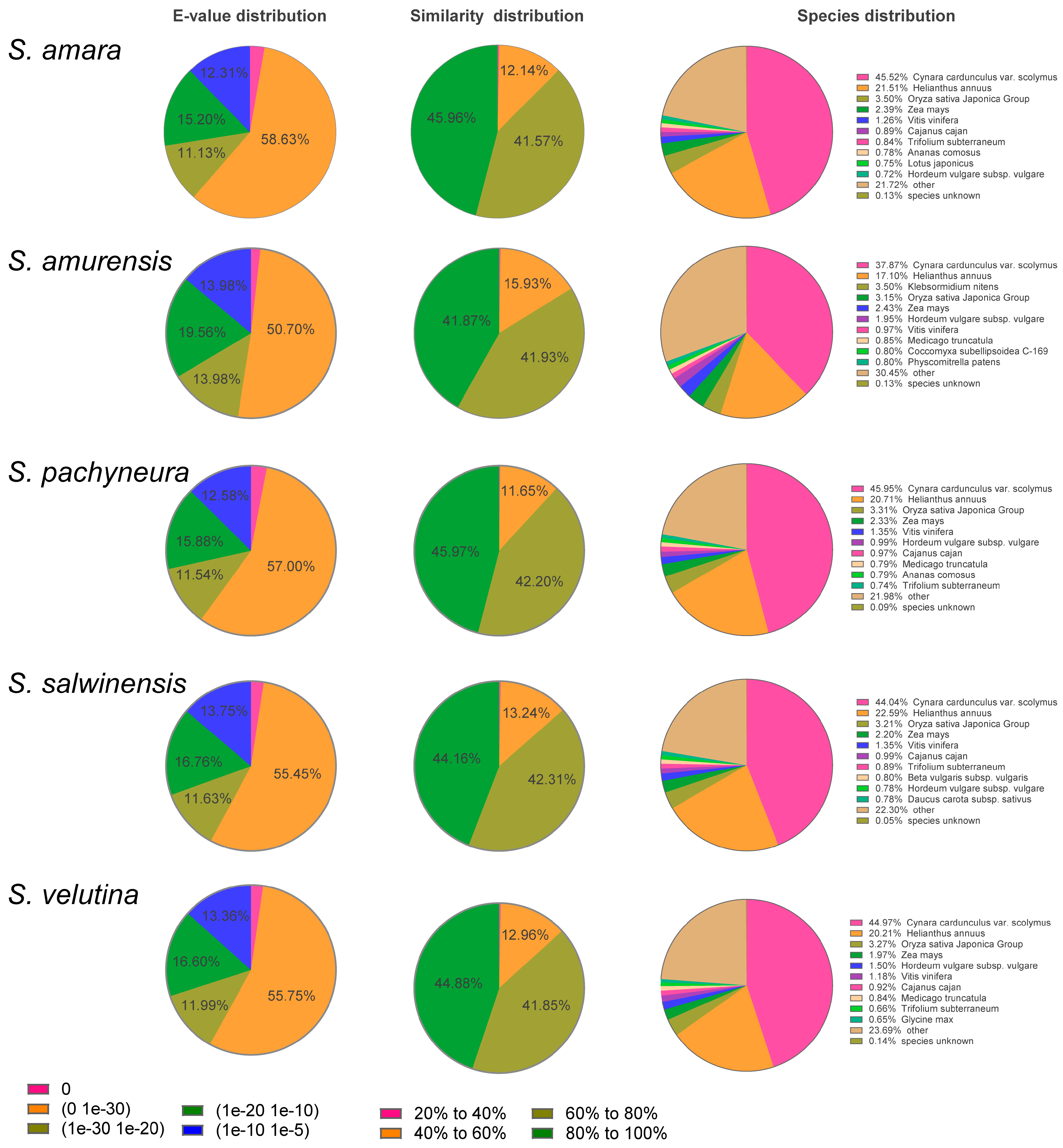
{kind=link}
{kind=link}
{kind=link}
| Species | Type | Total Sequence Number | Total Sequence Base (bp) | Percent GC (%) | Largest(bp) | Average (bp) | N50 (bp) |
|---|---|---|---|---|---|---|---|
| S. amara | Unigenes | 92,247 | 70,437,963 | 39.69 | 15,736 | 763.58 | 1334 |
| Transcripts | 185,952 | 179,868,433 | 39.95 | 15,736 | 967.28 | 1562 | |
| S. amurensis | Unigenes | 147,303 | 84,653,535 | 44.78 | 14,904 | 574.69 | 787 |
| Transcripts | 249,898 | 161,547,729 | 43.63 | 14,904 | 646.45 | 899 | |
| S. pachyneura | Unigenes | 91,980 | 65,877,634 | 40.21 | 13,829 | 716.22 | 1196 |
| Transcripts | 176,265 | 151,495,643 | 40.44 | 13,829 | 859.48 | 1354 | |
| S. salwinensis | Unigenes | 120,849 | 87,535,736 | 39.30 | 16,755 | 724.34 | 1154 |
| Transcripts | 270,778 | 237,345,971 | 39.83 | 16,755 | 876.53 | 1349 | |
| S. velutina | Unigenes | 106,591 | 78,896,059 | 39.81 | 15,517 | 740.18 | 1186 |
| Transcripts | 228,331 | 209,268,034 | 39.97 | 15,517 | 916.51 | 1422 |
| Species | ORF (%) | NR (%) | SWISS-PROT (%) | PFAM (%) | COG (%) | KEGG (%) | GO (%) |
|---|---|---|---|---|---|---|---|
| S. amara | 16,851 (18.27) | 21,395 (23.19) | 12,683 (13.75) | 9826 (10.65) | 10,440 (11.32) | 8110 (8.79) | 12,543 (13.60) |
| S. amurensis | 28,182 (19.13) | 31,705 (21.52) | 22,320 (15.15) | 15,886 (10.78) | 21,901 (14.87) | 14,179 (9.63) | 22,081 (14.99) |
| S. pachyneura | 15,906 (17.29) | 21,642 (23.53) | 12,819 (13.94) | 9474 (10.30) | 10,726 (11.66) | 8204 (8.92) | 12,674 (13.78) |
| S. salwinensis | 19,979 (16.53) | 26,176 (31.66) | 14,722 (12.18) | 11,017 (9.12) | 11,890 (9.84) | 9479 (7.84) | 14,577 (12.06) |
| S. velutina | 19,978 (18.74) | 24,748 (23.22) | 15,182 (14.24) | 11,268 (10.57) | 12,359 (11.59) | 10,008 (9.39) | 15,021 (14.09) |
| Species | GO Term | GO Term Definition | p-Value |
|---|---|---|---|
| S. pachyneura | GO:0016705 | Oxidoreductase activity, acting on paired donors, with incorporation or reduction of molecular oxygen | 0.0065 |
| GO:0050896 | Response to stimulus | 0.0250 | |
| GO:0016491 | Oxidoreductase activity | 0.0140 | |
| S. salwinensis | GO:0045490 | Pectin catabolic process | 0.0000 |
| GO:0070072 | Vacuolar proton-transporting V-type ATPase complex assembly | 0.0001 | |
| GO:0005615 | Extracellular space | 0.0001 | |
| GO:0009908 | Flower development | 0.0038 | |
| GO:0005976 | Polysaccharide metabolic process | 0.0360 | |
| S. velutina | GO:0016787 | Hydrolase activity | 0.0045 |
| GO:0016491 | Oxidoreductase activity | 0.0230 | |
| GO:0016209 | Antioxidant activity | 0.0210 | |
| GO:0006725 | Cellular aromatic compound metabolic process | 0.0054 | |
| GO:0050896 | Response to stimulus | 0.0120 | |
| GO:0006869 | Lipid transport | 0.0020 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Sun, Y.; Landis, J.B.; Shen, J.; Zhang, H.; Kuang, T.; Sun, W.; Sun, J.; Tiamiyu, B.B.; Deng, T.; et al. Transcriptomes of Saussurea (Asteraceae) Provide Insights into High-Altitude Adaptation. Plants 2021, 10, 1715. https://doi.org/10.3390/plants10081715
Zhang X, Sun Y, Landis JB, Shen J, Zhang H, Kuang T, Sun W, Sun J, Tiamiyu BB, Deng T, et al. Transcriptomes of Saussurea (Asteraceae) Provide Insights into High-Altitude Adaptation. Plants. 2021; 10(8):1715. https://doi.org/10.3390/plants10081715
Chicago/Turabian StyleZhang, Xu, Yanxia Sun, Jacob B. Landis, Jun Shen, Huajie Zhang, Tianhui Kuang, Wenguang Sun, Jiao Sun, Bashir B. Tiamiyu, Tao Deng, and et al. 2021. "Transcriptomes of Saussurea (Asteraceae) Provide Insights into High-Altitude Adaptation" Plants 10, no. 8: 1715. https://doi.org/10.3390/plants10081715
APA StyleZhang, X., Sun, Y., Landis, J. B., Shen, J., Zhang, H., Kuang, T., Sun, W., Sun, J., Tiamiyu, B. B., Deng, T., Sun, H., & Wang, H. (2021). Transcriptomes of Saussurea (Asteraceae) Provide Insights into High-Altitude Adaptation. Plants, 10(8), 1715. https://doi.org/10.3390/plants10081715