
Antioxidant Activity, α-Glucosidase Inhibition and UHPLC–ESI–MS/MS Profile of Shmar (Arbutus pavarii Pamp)


Abstract
:1. Introduction
2. Results and Discussion
2.1. Total Phenolic Content (TPC)
2.2. Total Flavonoid Content (TFC)
2.3. 2,2-diphenyl-1-picrylhydrazyl (DPPH) Radical Scavenging Activity
2.4. Ferric Reducing Antioxidant Power (FRAP)
2.5. α-Glucosidase Inhibitory Activity
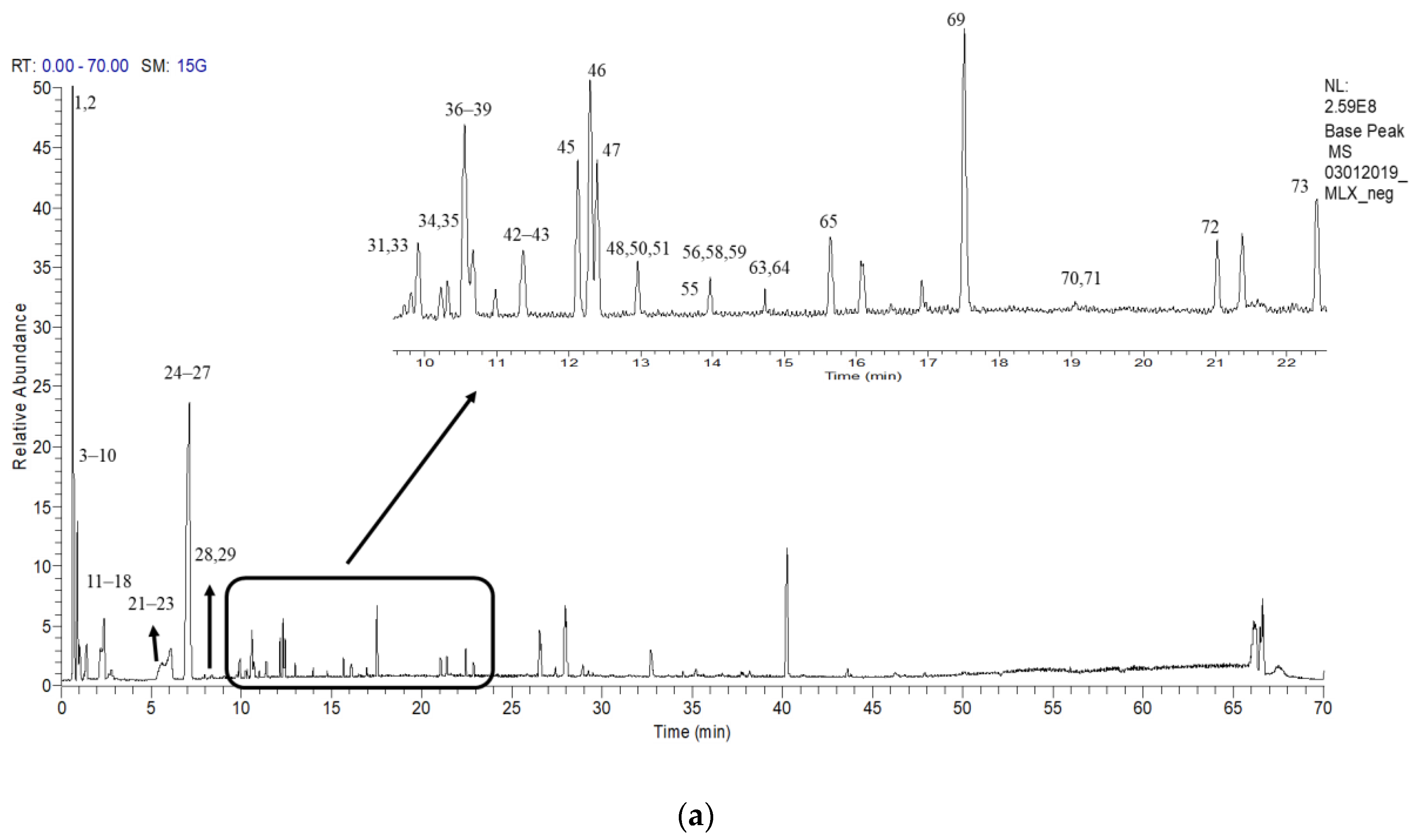
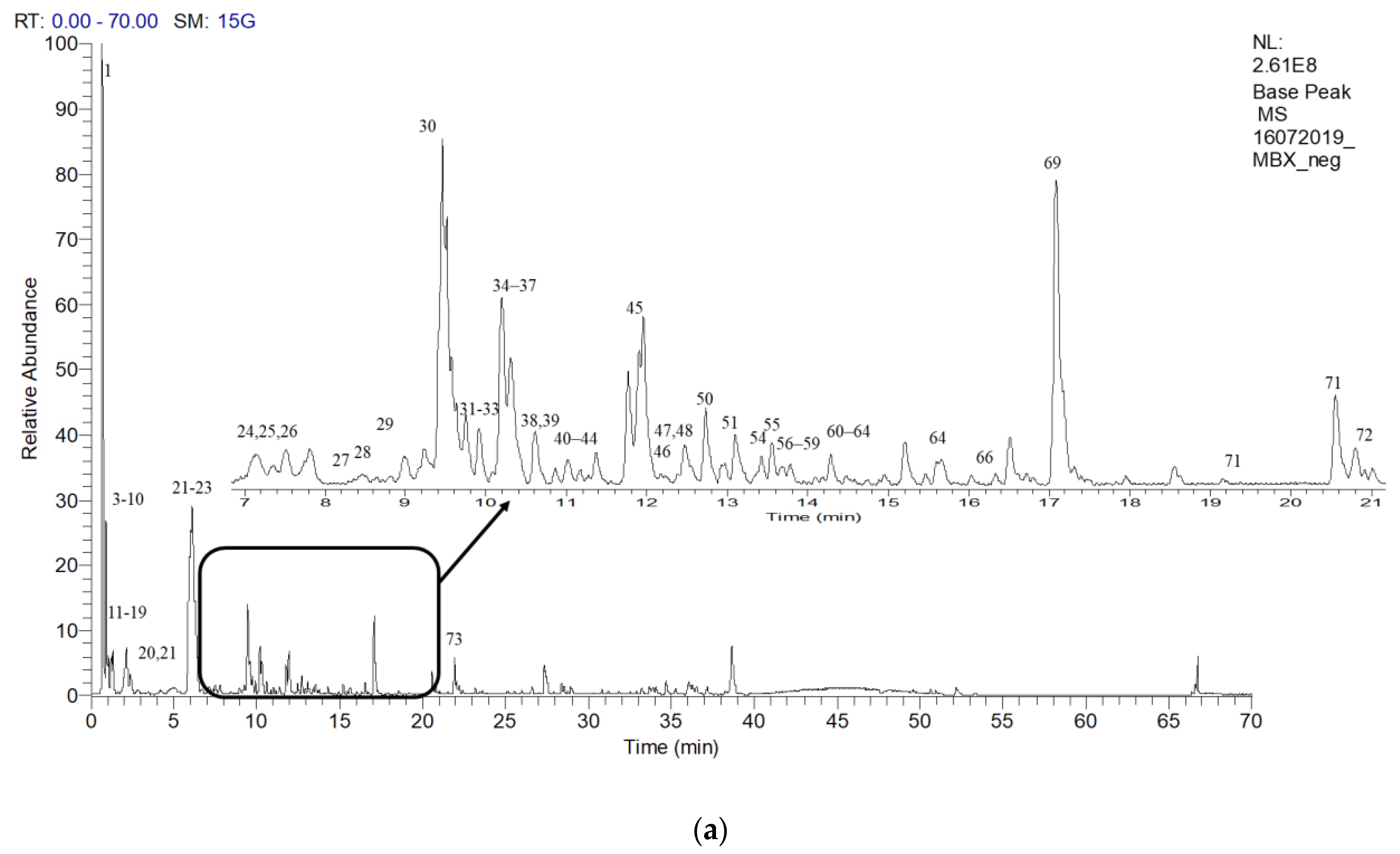
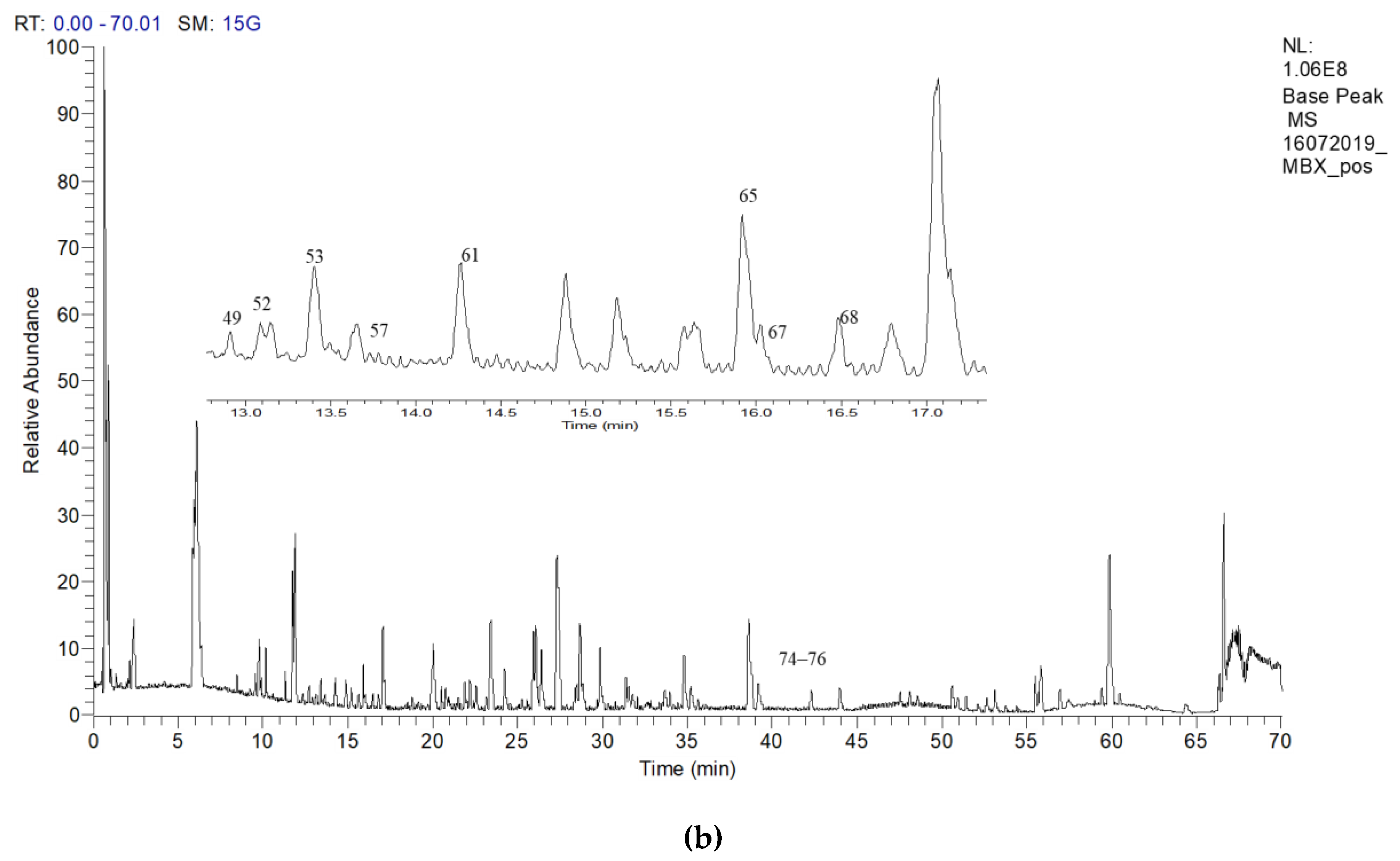
2.6. Putative LCMS Profiles of A. pavarii Crude Leaf and Stem Bark Methanol Extracts
2.6.1. Identification of Phenolic Acid and Phenolic Acid Glycoside Derivatives
2.6.2. Identification of Flavonoids and Derivatives
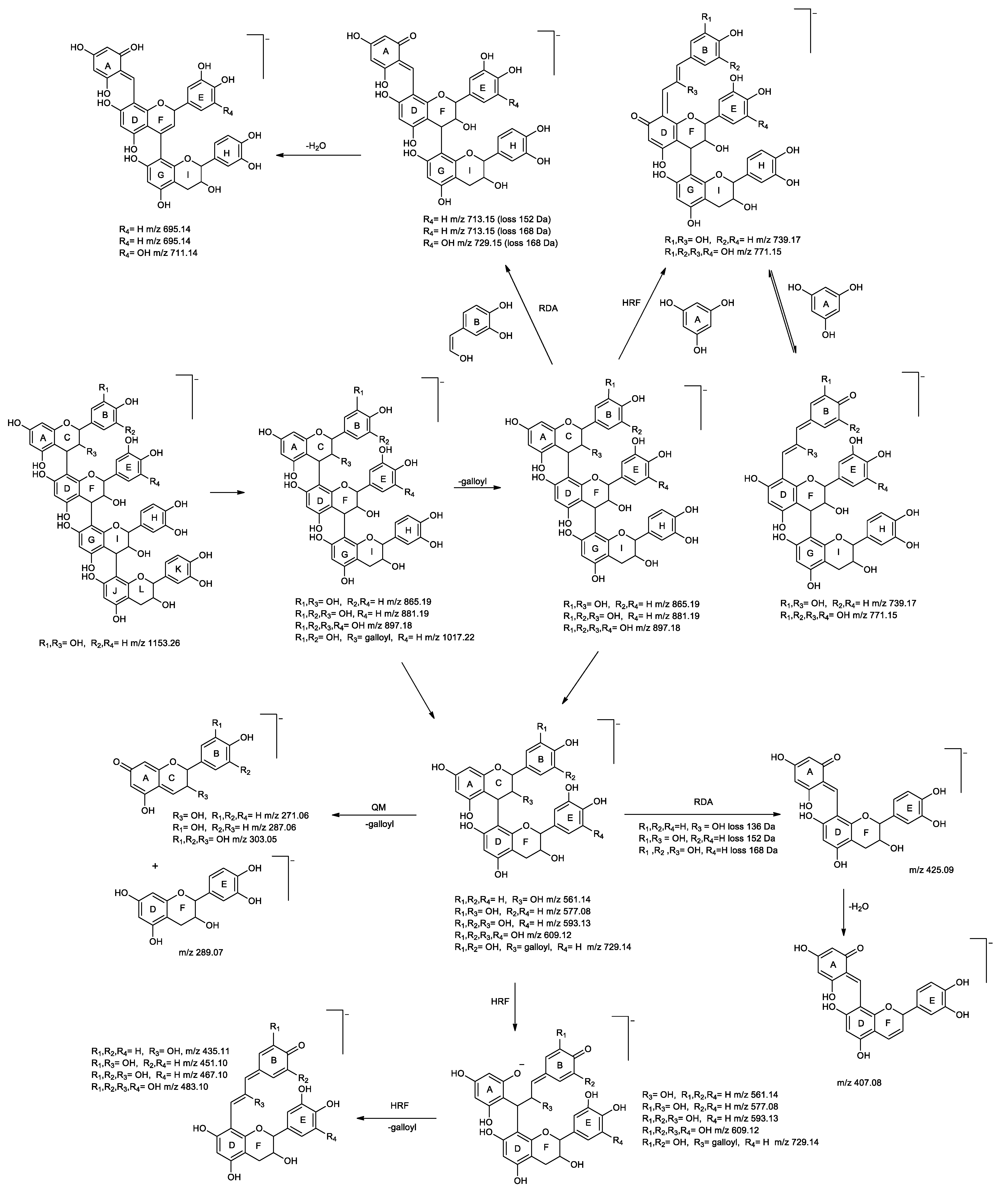
Identification of Flavan-3-ols and Derivatives
Identification of Anthocyanins and Derivatives
Identification of Flavonol
2.6.3. Identification of Triterpenoid Derivatives
2.6.4. Identification of Fatty Acid Derivatives and Other Compounds
2.6.5. Identification of Other Compounds
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Plant Materials
3.3. Preparation of Leaf and Stem Bark Extracts and Their Respective Fractions
3.4. Total Phenolic Content (TPC) Assay
3.5. Total Flavonoid Content (TFC) Assay
3.6. Free Radical Scavenging (DPPH) Assay
3.7. Ferric Reducing Antioxidant Power (FRAP) Assay
3.8. α-Glucosidase Inhibitory Assay
3.9. UHPLC–MS/MS Analysis
3.10. Data Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Chaturvedi, D.; Singh, K.; Singh, V.K. Therapeutic and pharmacological aspects of photodynamic product chlorophyllin. Eur. J. Biol. Res. 2019, 9, 164–176. [Google Scholar]
- Joshi, B.K.; Shrestha, R.; Gautam, I.P.; Poudel, A.P.; Gotame, T.P. Neglected and Underutilized Species (NUS), and Future Smart Food (FSF) in Nepal; National Agriculture Genetic Resources Center (NAGRC, National Genebank): Kathmandu, Nepal, 2019. [Google Scholar]
- Amin, M.U.; Khurram, M.; Khattak, B.; Khan, J. Antibiotic additive and synergistic action of rutin, morin and quercetin against methicillin resistant Staphylococcus aureus. BMC Complement Altern. Med. 2015, 15, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riani, M.K.L.; Anwar, E.; Nurhayati, T. Antioxidant and anti-collagenase activity of sargassum plagyophyllum extract as an anti-wrinkle cosmetic ingredient. Pharmacogn. J. 2018, 10, 932–936. [Google Scholar]
- Zhang, Y.J.; Gan, R.Y.; Li, S.; Zhou, Y.; Li, A.N.; Xu, D.P.; Li, H.B. Antioxidant phytochemicals for the prevention and treatment of chronic diseases. Molecules 2015, 20, 21138–21156. [Google Scholar] [CrossRef] [PubMed]
- Lourenço, S.C.; Moldão-Martins, M.; Alves, V.D. Antioxidants of natural plant origins: From sources to food industry applications. Molecules 2019, 24, 4132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinaffi, A.C.D.C.; Sampaio, G.R.; Soares, M.J.; Shahidi, F.; de Camargo, A.C.; Torres, E.A. Insoluble-Bound Polyphenols Released from Guarana Powder: Inhibition of Alpha-Glucosidase and Proanthocyanidin Profile. Molecules 2020, 25, 679. [Google Scholar] [CrossRef] [Green Version]
- Lobo, V.; Patil, A.; Phatak, A.; Chandra, N. Free radicals, antioxidants and functional foods: Impact on human health. Pharmacogn. Rev. 2010, 4, 118. [Google Scholar] [CrossRef] [Green Version]
- Tan, Y.; Chang, S.K.; Zhang, Y. Comparison of α-amylase, α-glucosidase and lipase inhibitory activity of the phenolic substances in two black legumes of different genera. Food Chem. 2017, 214, 259–268. [Google Scholar] [CrossRef] [Green Version]
- Malik, A.; Jamil, U.; Butt, T.T.; Waquar, S.; Gan, S.H.; Shafique, H.; Jafar, T.H. In silico and in vitro studies of lupeol and iso-orientin as potential antidiabetic agents in a rat model. Drug Des. Dev. Ther. 2019, 13, 1501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alkhalidy, H.; Wang, Y.; Liu, D. Dietary flavonoids in the prevention of T2D: An overview. Nutrients 2018, 10, 438. [Google Scholar] [CrossRef] [Green Version]
- Tundis, R.; Loizzo, M.R.; Menichini, F. Natural products as α-amylase and α-glucosidase inhibitors and their hypoglycaemic potential in the treatment of diabetes: An update. Mini-Rev. Med. 2010, 10, 315–331. [Google Scholar]
- Hegazy, A.; Boulos, L.; Kabiel, H.; Sharashy, O. Vegetation and species altitudinal distribution in Al-Jabal Al-Akhdar landscape, Libya. Pak. J. Bot. 2011, 43, 1885–1898. [Google Scholar]
- Alghazeer, R.; Abourghiba, T.; Ibrahim, A.; Zreba, E. Bioactive properties of some selected Libyan plants. J. Med. Plant Res. 2016, 10, 67–76. [Google Scholar]
- Tenuta, M.C.; Tundis, R.; Xiao, J.; Loizzo, M.R.; Dugay, A.; Deguin, B. Arbutus species (Ericaceae) as source of valuable bioactive products. Crit. Rev. Food Sci. 2019, 59, 864–881. [Google Scholar] [CrossRef] [PubMed]
- Buzgaia, N.; Awin, T.; Elabbar, F.; Abdusalam, K.; Lee, S.Y.; Rukayadi, Y.; Abas, F.; Shaari, K. Antibacterial Activity of Arbutus pavarii Pamp against Methicillin-Resistant Staphylococcus aureus (MRSA) and UHPLC-MS/MS Profile of the Bioactive Fraction. Plants 2020, 9, 1539. [Google Scholar] [CrossRef]
- El Shibani, F.A.E.S. A Pharmacognostical Study of Arbutus pavarii Pampan. Family Ericaceae and Sarcopoterium spinosum L. Family Rosaceae Growing in Libya. Ph.D. Thesis, Cairo University, Cairo, Egypt, 2017. [Google Scholar]
- de Camargo, A.C.; Favero, B.T.; Morzelle, M.C.; Franchin, M.; Alvarez-Parrilla, E.; de la Rosa, L.A.; Geraldi, M.V.; Marostica Junior, M.R.; Shahidi, F.; Schwember, A.R. Is chickpea a potential substitute for soybean? Phenolic bioactives and potential health benefits. Int. J. Mol. Sci. 2019, 20, 2644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Camargo, A.C.; Biasoto, A.C.T.; Schwember, A.R.; Granato, D.; Rasera, G.B.; Franchin, M.; Rosalen, P.L.; Alencar, S.M.; Shahidi, F. Should we ban total phenolics and antioxidant screening methods? The link between antioxidant potential and activation of NF-κB using phenolic compounds from grape by-products. Food Chem. 2019, 290, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Falcão, H.G.; Silva, M.B.R.; de Camargo, A.C.; Shahidi, F.; Franchin, M.; Rosalen, P.L.; Alencar, S.M.; Kurozawa, L.E.; Ida, E.I. Optimizing the potential bioactivity of isoflavones from soybeans via ultrasound pretreatment: Antioxidant potential and NF--κB activation. J. Food Biochem. 2019, 43, e13018. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Mediani, A.; Nur Ashikin, A.H.; Azliana, A.B.S.; Abas, F. Antioxidant and α-glucosidase inhibitory activities of the leaf and stem of selected traditional medicinal plants. Int. Food Res. J. 2014, 21, 165–172. [Google Scholar]
- Truong, D.H.; Nguyen, D.H.; Ta, N.T.A.; Bui, A.V.; Do, T.H.; Nguyen, H.C. Evaluation of the use of different solvents for phytochemical constituents, antioxidants, and in vitro anti-inflammatory activities of Severinia buxifolia. J. Food Qual. 2019, 2019, 8178294. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.O.; Jeong, S.W.; Lee, C.Y. Antioxidant capacity of phenolic phytochemicals from various cultivars of plums. Food Chem. 2003, 81, 321–326. [Google Scholar] [CrossRef]
- Deng, Y.; Zhao, Y.; Padilla-Zakour, O.; Yang, G. Polyphenols, antioxidant and antimicrobial activities of leaf and bark extracts of Solidago canadensis L. Ind. Crops Prod. 2019, 74, 803–809. [Google Scholar] [CrossRef]
- Balasundram, N.; Sundram, K.; Samman, S. Phenolic compounds in plants and agri-industrial by-products: Antioxidant activity, occurrence, and potential uses. Food Chem. 2006, 99, 191–203. [Google Scholar] [CrossRef]
- Alam, M.A.; Zaidul, I.S.M.; Ghafoor, K.; Sahena, F.; Hakim, M.A.; Rafii, M.Y.; Abir, H.M.; Bostanudin, M.F.; Perumal, V.; Khatib, A. In vitro antioxidant and, α-glucosidase inhibitory activities and comprehensive metabolite profiling of methanol extract and its fractions from Clinacanthus nutans. BMC Compl. Altern. Med. 2017, 17, 181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apak, R.; Güclü, K.; Özyürek, M.; Celik, S.E. Mechanism of antioxidant capacity assays and the CUPRAC (cupric ion reducing antioxidant capacity) assay. Microchim. Acta 2008, 160, 413–419. [Google Scholar] [CrossRef]
- Aruoma, O.I. Methodological considerations for characterizing potential antioxidant actions of bioactive components in plant foods. Mutat. Res. 2003, 523, 9–20. [Google Scholar] [CrossRef]
- Noreen, H.; Semmar, N.; Farman, M.; McCullagh, J.S. Measurement of total phenolic content and antioxidant activity of aerial parts of medicinal plant Coronopus didymus. Asian Pac. J. Trop. Med. 2017, 10, 792–801. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Ra, J.H.; Jee, Y.; Kim, J.S. Impact of different partitioned solvents on chemical composition and bioavailability of Sasa quelpaertensis Nakai leaf extract. J. Food Drug Anal. 2017, 25, 316–326. [Google Scholar] [CrossRef] [Green Version]
- Kadum, H.; Hamid, A.A.; Abas, F.; Ramli, N.S.; Mohammed, A.K.S.; Muhialdin, B.J.; Jaafar, A.H. Bioactive Compounds Responsible for Antioxidant Activity of Different Varieties of Date (Phoenix dactylifera L.) Elucidated by 1H-NMR Based Metabolomics. Int. J. Food Prop. 2019, 22, 462–476. [Google Scholar] [CrossRef] [Green Version]
- Brown, A.; Anderson, D.; Racicot, K.; Pilkenton, S.J.; Apostolidis, E. Evaluation of phenolic phytochemical enriched commercial plant extracts on the in vitro inhibition of α-glucosidase. Front. Nutr. 2017, 4, 56. [Google Scholar] [CrossRef] [Green Version]
- Ganzera, M.; Sturm, S. Recent advances on HPLC/MS in medicinal plant analysis—An update covering 2011–2016. J. Pharm. Biomed. Anal. 2018, 147, 211–233. [Google Scholar] [CrossRef]
- Mendes, L.; de Freitas, V.; Baptista, P.; Carvalho, M. Comparative antihemolytic and radical scavenging activities of strawberry tree (Arbutus unedo L.) leaf and fruit. Food Chem. Toxicol. 2011, 49, 2285–2291. [Google Scholar] [CrossRef]
- Abu-Reidah, I.M.; Ali-Shtayeh, M.S.; Jamous, R.M.; Arráez-Román, D.; Segura-Carretero, A. HPLC–DAD–ESI–MS/MS screening of bioactive components from Rhus coriaria L. (Sumac) fruits. Food Chem. 2015, 166, 179–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Seeram, N.P. Liquid chromatography coupled with time-of-flight tandem mass spectrometry for comprehensive phenolic characterization of pomegranate fruit and flower extracts used as ingredients in botanical dietary supplements. J. Sep. Sci. 2018, 41, 3022–3033. [Google Scholar] [CrossRef]
- Karar, M.; Kuhnert, N. UPLC-ESI-Q-TOF-MS/MS characterization of phenolics from Crataegus monogyna and Crataegus laevigata (Hawthorn) leaf, fruits and their herbal derived drops (Crataegutt Tropfen). J. Chem. Biol. Therap. 2015, 1, 102. [Google Scholar]
- Morales-Soto, A.; Gómez-Caravaca, A.M.; García-Salas, P.; Segura-Carretero, A.; Fernández-Gutiérrez, A. High-performance liquid chromatography coupled to diode array and electrospray time-of-flight mass spectrometry detectors for a comprehensive characterization of phenolic and other polar compounds in three pepper (Capsicum annuum L.) samples. Food Res. Int. 2013, 51, 977–984. [Google Scholar] [CrossRef]
- Gutiérrez-Grijalva, E.P.; Picos-Salas, M.A.; Leyva-López, N.; Criollo-Mendoza, M.S.; Vazquez-Olivo, G.; Heredia, J.B. Flavonoids and phenolic acids from oregano: Occurrence, biological activity and health benefits. Plants 2018, 7, 2. [Google Scholar] [CrossRef] [Green Version]
- Nijveldt, R.J.; Van Nood, E.L.S.; Van Hoorn, D.E.; Boelens, P.G.; Van Norren, K.; Van Leeuwen, P.A. Flavonoids: A review of probable mechanisms of action and potential applications. Am. J. Clin. Nutr. 2001, 74, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Thilakarathna, S.H.; Wang, Y.; Rupasinghe, H.V.; Ghanam, K. Apple peel flavonoid-and triterpene-enriched extracts differentially affect cholesterol homeostasis in hamsters. J. Funct. Foods. 2012, 4, 963–971. [Google Scholar] [CrossRef]
- Tungmunnithum, D.; Thongboonyou, A.; Pholboon, A.; Yangsabai, A. Flavonoids and other phenolic compounds from medicinal plants for pharmaceutical and medical aspects: An overview. Medicines 2018, 5, 93. [Google Scholar] [CrossRef]
- Singh, A.; Kumar, S.; Kumar, B. LC-MS Identification of Proanthocyanidins in Bark and Fruit of six Terminalia species. Nat. Prod. Commun. 2018, 13, 1934578X1801300511. [Google Scholar] [CrossRef] [Green Version]
- Stöggl, W.M.; Huck, C.W.; Bonn, G.K. Structural elucidation of catechin and epicatechin in sorrel leaf extracts using liquid--chromatography coupled to diode array--, fluorescence--, and mass spectrometric detection. J. Sep. Sci. 2004, 27, 524–528. [Google Scholar] [CrossRef]
- Yuzuak, S.; Ballington, J.; Xie, D.Y. HPLC-qTOF-MS/MS-Based Profiling of Flavan-3-ols and Dimeric Proanthocyanidins in Berries of Two Muscadine Grape Hybrids FLH 13-11 and FLH 17-66. Metabolites 2018, 8, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeszka-Skowron, M.; Zgoła-Grześkowiak, A.; Frankowski, R. Cistus incanus a promising herbal tea rich in bioactive compounds: LC–MS/MS determination of catechins, flavonols, phenolic acids and alkaloids—A comparison with Camellia sinensis, Rooibos and Hoan Ngoc herbal tea. J. Food Compost. Anal. 2018, 74, 71–81. [Google Scholar] [CrossRef]
- Di Lecce, G.; Arranz, S.; Jáuregui, O.; Tresserra-Rimbau, A.; Quifer-Rada, P.; Lamuela-Raventós, R.M. Phenolic profiling of the skin, pulp and seeds of Albariño grapes using hybrid quadrupole time-of-flight and triple-quadrupole mass spectrometry. Food Chem. 2014, 145, 874–882. [Google Scholar] [CrossRef]
- Munekata, P.E.S.; Franco, D.; Trindade, M.A.; Lorenzo, J.M. Characterization of phenolic composition in chestnut leaf and beer residue by LC–DAD–ESI–MS. LWT-Food Sci. Technol. 2016, 68, 52–58. [Google Scholar] [CrossRef]
- Ismail, B.B.; Pu, Y.; Guo, M.; Ma, X.; Liu, D. LC-MS/QTOF identification of phytochemicals and the effects of solvents on phenolic constituents and antioxidant activity of baobab (Adansonia digitata) fruit pulp. Food Chem. 2019, 277, 279–288. [Google Scholar] [CrossRef]
- Wang, C.; Li, Q.; Han, G.; Zou, L.; Lv, L.; Zhou, Q.; Li, N. LC MS/MS for simultaneous determination of four major active catechins of tea polyphenols in rat plasma and its application to pharmacokinetics. Chin. Herb. Med. 2010, 2, 289–296. [Google Scholar]
- Gođevac, D.; Jadranin, M.; Aljančić, I.; Vajs, V.; Tešević, V.; Milosavljević, S. Application of Spectroscopic Methods and Hyphenated Techniques to the Analysis of Complex Plant Extracts. In Medicinal and Aromatic Plants of the World; Springer: Dordrecht, The Netherlands, 2015; pp. 61–85. [Google Scholar]
- Bystrom, L.M.; Lewis, B.A.; Brown, D.L.; Rodriguez, E.; Obendorf, R.L. Characterisation of phenolics by LC–UV/Vis, LC–MS/MS and sugars by GC in Melicoccus bijugatus Jacq. ‘Montgomery’fruits. Food Chem. 2008, 111, 1017–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedrich, W.; Eberhardt, A.; Galensa, R. Investigation of proanthocyanidins by HPLC with electrospray ionization mass spectrometry. Eur. Food Res. Technol. 2000, 211, 56–64. [Google Scholar] [CrossRef]
- Gu, L.; Kelm, M.A.; Hammerstone, J.F.; Beecher, G.; Holden, J.; Haytowitz, D.; Prior, R.L. Screening of foods containing proanthocyanidins and their structural characterization using LC-MS/MS and thiolytic degradation. J. Agric. Food Chem. 2003, 51, 7513–7521. [Google Scholar] [CrossRef]
- Delcambre, A.; André, Y.; Saucier, C. Sequencing of red wine proanthocyanidins by UHPLC-ESI-Q-TOF. J. Appl. Bioanal. 2015, 1, 46–54. [Google Scholar] [CrossRef]
- Verardo, V.; Bonoli, M.; Marconi, E.; Caboni, M.F. Determination of free flavan-3-ol content in barley (Hordeum vulgare L.) air-classified flours: Comparative study of HPLC-DAD/MS and spectrophotometric determinations. J. Agric. Food Chem. 2008, 56, 6944–6948. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, R.; Jayasinghe, L.; Kuhnert, N. Identification and characterization of proanthocyanidins of 16 members of the Rhododendron genus (Ericaceae) by tandem LC–MS. J. Mass Spectrom. 2012, 47, 502–515. [Google Scholar] [CrossRef] [PubMed]
- Prasain, J.K.; Peng, N.; Dai, Y.; Moore, R.; Arabshahi, A.; Wilson, L.; Kim, H.; Watts, R.L. Liquid chromatography tandem mass spectrometry identification of proanthocyanidins in rat plasma after oral administration of grape seed extract. Phytomedicine 2009, 16, 233–243. [Google Scholar] [CrossRef] [Green Version]
- Pallauf, K.; Rivas-Gonzalo, J.C.; Del Castillo, M.D.; Cano, M.P.; de Pascual-Teresa, S. Characterization of the antioxidant composition of strawberry tree (Arbutus unedo L.) fruits. J. Food Compos. Anal. 2008, 21, 273–281. [Google Scholar] [CrossRef] [Green Version]
- Rue, E.A.; Rush, M.D.; Van Breemen, R.B. Procyanidins: A comprehensive review encompassing structure elucidation via mass spectrometry. Phytochem. Rev. 2018, 17, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Rockenbach, I.I.; Jungfer, E.; Ritter, C.; Santiago-Schübel, B.; Thiele, B.; Fett, R.; Galensa, R. Characterization of flavan-3-ols in seeds of grape pomace by CE, HPLC-DAD-MSn and LC-ESI-FTICR-MS. Food Res. Int. 2012, 48, 848–855. [Google Scholar] [CrossRef] [Green Version]
- Hamed, A.I.; Al--Ayed, A.S.; Moldoch, J.; Piacente, S.; Oleszek, W.; Stochmal, A. Profiles analysis of proanthocyanidins in the argun nut (Medemia argun—An ancient Egyptian palm) by LC–ESI–MS/MS. J. Mass Spectrom. 2014, 49, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Nawrot-Hadzik, I.; Ślusarczyk, S.; Granica, S.; Hadzik, J.; Matkowski, A. Phytochemical Diversity in Rhizomes of Three Reynoutria Species and their Antioxidant Activity Correlations Elucidated by LC–ESI–MS/MS Analysis. Molecules 2019, 24, 1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Downey, M.O.; Rochfort, S. Simultaneous separation by reversed-phase high-performance liquid chromatography and mass spectral identification of anthocyanins and flavonols in Shiraz grape skin. J. Chromatogr. A 2008, 1201, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Trikas, E.; Papi, R.; Kyriakidis, D.; Zachariadis, G. A sensitive LC-MS method for anthocyanins and comparison of byproducts and equivalent wine content. Separations 2016, 3, 18. [Google Scholar] [CrossRef]
- Kajdžanoska, M.; Gjamovski, V.; Stefova, M. HPLC–DAD–ESI–MSn identification of phenolic compounds in cultivated strawberries from Macedonia. Maced. J. Chem. Chem. Eng. 2010, 29, 181–194. [Google Scholar] [CrossRef] [Green Version]
- Veberic, R.; Slatnar, A.; Bizjak, J.; Stampar, F.; Mikulic-Petkovsek, M. Anthocyanin composition of different wild and cultivated berry species. LWT-Food Sci. Technol. 2015, 60, 509–517. [Google Scholar] [CrossRef]
- Stein-Chisholm, R.; Beaulieu, J.; Grimm, C.; Lloyd, S. LC–MS/MS and UPLC–UV evaluation of anthocyanins and anthocyanidins during rabbiteye blueberry juice processing. Beverages 2017, 3, 56. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Liang, F.; Bin, Y.; Li, P.; Duan, C. Screening non-colored phenolics in red wines using liquid chromatography/ultraviolet and mass spectrometry/mass spectrometry libraries. Molecules 2007, 12, 679–693. [Google Scholar] [CrossRef] [Green Version]
- Hvattum, E.; Ekeberg, D. Study of the collision-induced radical cleavage of flavonoid glycosides using negative electrospray ionization tandem quadrupole mass spectrometry. J. Mass Spectrom. 2003, 38, 43–49. [Google Scholar] [CrossRef]
- Singh, A.P.; Wang, Y.; Olson, R.M.; Luthria, D.; Banuelos, G.S.; Pasakdee, S.; Vorsa, N.; Wilson, T. LC-MS-MS analysis and the antioxidant activity of flavonoids from eggplant skins grown in organic and conventional environments. J. Food Sci. Nutr. 2017, 8, 869. [Google Scholar] [CrossRef] [Green Version]
- Fan, L.; Tong, Q.; Dong, W.; Yang, G.; Hou, X.; Xiong, W.; Wang, W. Tissue distribution, excretion, and metabolic profile of dihydromyricetin, a flavonoid from vine tea (Ampelopsis grossedentata) after oral administration in rats. J. Agric. Food Chem. 2017, 65, 4597–4604. [Google Scholar] [CrossRef]
- Engström, M.T.; Pälijärvi, M.; Salminen, J.P. Rapid fingerprint analysis of plant extracts for ellagitannins, gallic acid, and quinic acid derivatives and quercetin-, kaempferol-and myricetin-based flavonol glycosides by UPLC-QqQ-MS/MS. J. Agric. Food Chem. 2015, 63, 4068–4079. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.Z.; Wu, W.; Jiao, L.L.; Yang, P.F.; Guo, M.Q. Analysis of flavonoids in lotus (Nelumbo nucifera) leaf and their antioxidant activity using macroporous resin chromatography coupled with LC-MS/MS and antioxidant biochemical assays. Molecules 2015, 20, 10553–10565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez-Rabaneda, F.; Jauregui, O.; Lamuela-Raventós, R.M.; Viladomat, F.; Bastida, J.; Codina, C. Qualitative analysis of phenolic compounds in apple pomace using liquid chromatography coupled to mass spectrometry in tandem mode. Rapid Commun. Mass Spectrom. 2004, 18, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Chandra, P.; Bajpai, V.; Singh, A.; Srivastava, M.; Mishra, D.K.; Kumar, B. Rapid qualitative and quantitative analysis of bioactive compounds from Phyllanthus amarus using LC/MS/MS techniques. Ind. Crops Prod. 2015, 69, 143–152. [Google Scholar] [CrossRef]
- Hossain, M.B.; Rai, D.K.; Brunton, N.P.; Martin-Diana, A.B.; Barry-Ryan, C. Characterization of phenolic composition in Lamiaceae spices by LC–ESI–MS/MS. J. Agric. Food Chem. 2010, 58, 10576–10581. [Google Scholar] [CrossRef]
- Budzikiewicz, H.; Wilson, J.; Djerassi, C. Mass spectrometry in structural and stereochemical problems. XXXII. 1 Pentacyclic triterpenes. J. Am. Chem. Soc. 1963, 85, 3688–3699. [Google Scholar] [CrossRef]
- Huang, L.; Chen, T.; Ye, Z.; Chen, G. Use of liquid chromatography–atmospheric pressure chemical ionization–ion trap mass spectrometry for identification of oleanolic acid and ursolic acid in Anoectochilus roxburghii (wall.) Lindl. Int. J. Mass Spectrom. 2007, 42, 910–917. [Google Scholar] [CrossRef]
- Kosyakov, D.S.; Ul’Yanovskii, N.V.; Falev, D.I. Determination of triterpenoids from birch bark by liquid chromatography-tandem mass spectrometry. J. Anal. Chem. 2014, 69, 1264–1269. [Google Scholar] [CrossRef]
- Naumoska, K.; Vovk, I. Analysis of triterpenoids and phytosterols in vegetables by thin-layer chromatography coupled to tandem mass spectrometry. J. Chromatogr. A. 2015, 1381, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Sánchez, C.; Lozano-Sánchez, J.; Gabaldón-Hernández, J.A.; Segura-Carretero, A.; Fernández-Gutiérrez, A. RP-HPLC–ESI–QTOF/MS2 based strategy for the comprehensive metabolite profiling of Sclerocarya Birrea (Marula) bark. Ind. Crops Prod. 2015, 71, 214–234. [Google Scholar] [CrossRef]
- Jiménez-Sánchez, C.; Lozano-Sánchez, J.; Rodríguez-Pérez, C.; Segura-Carretero, A.; Fernández-Gutiérrez, A. Comprehensive, untargeted, and qualitative RP-HPLC-ESI-QTOF/MS2 metabolite profiling of green asparagus (Asparagus officinalis). J. Food Compos. Anal. 2016, 46, 78–87. [Google Scholar] [CrossRef]
- de la Luz Cádiz-Gurrea, M.; Fernández-Arroyo, S.; Joven, J.; Segura-Carretero, A. Comprehensive characterization by UHPLC-ESI-Q-TOF-MS from an Eryngium bourgatii extract and their antioxidant and anti-inflammatory activities. Food Res. Int. 2013, 50, 197–204. [Google Scholar] [CrossRef]

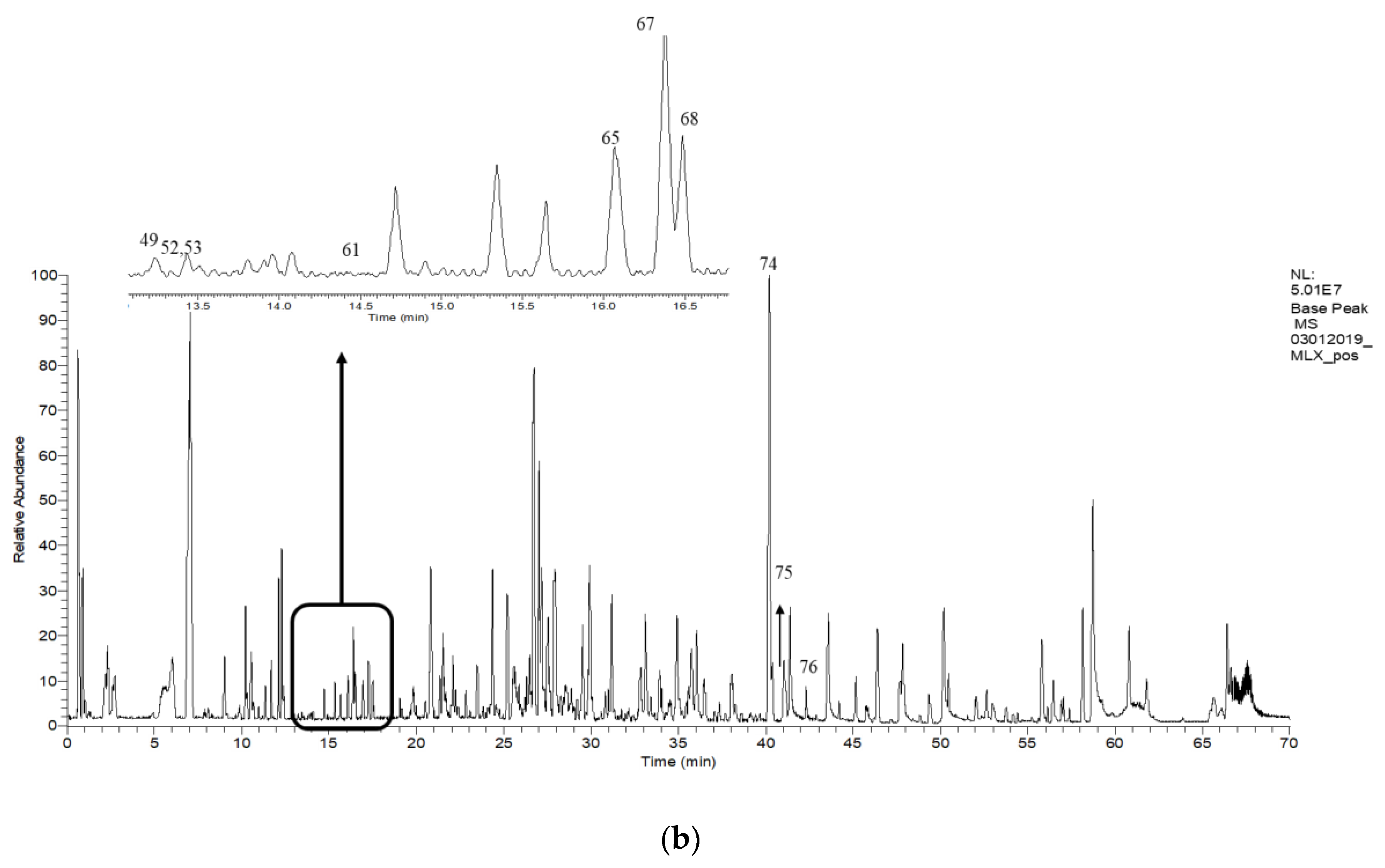
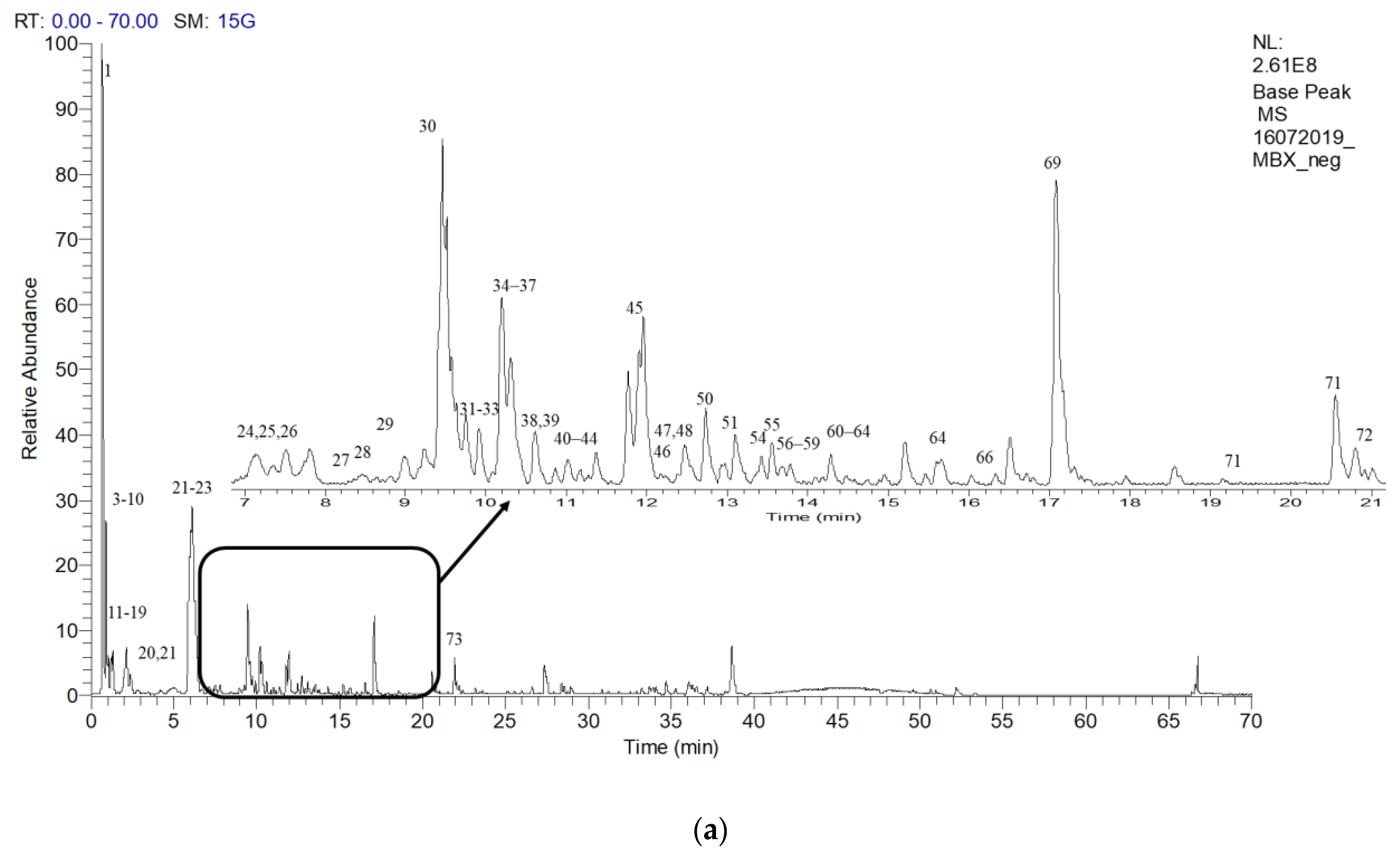



{kind=link}
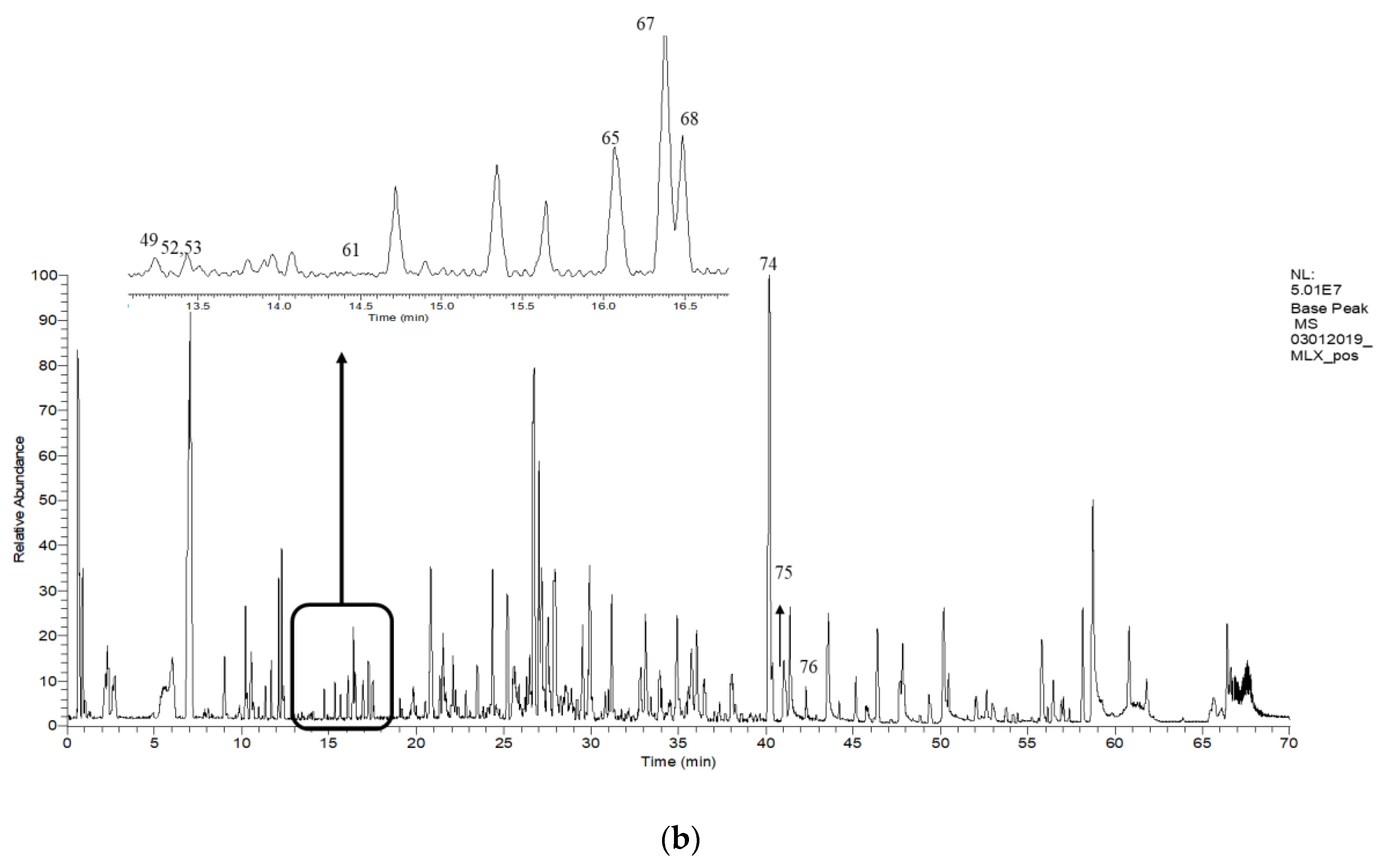{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Extracts/Fraction/Standard | TPC (mg GAE/g Extract) | TFC (mg QE/g Extract) | DPPH IC50 (µg/mL) | FRAP (mmol Fe (II)/g Extract) | α-glycosidase IC50 (µg/mL) |
|---|---|---|---|---|---|
| Leaf | |||||
| EX.MeOH | 886.57 ± 0.93 Aa | 442.06 ± 0.34 Ab | 17.57 ± 0.09 Aa | 82.95 ± 0.13 Aa | 8.75 ± 0.22 Ab |
| F.Hexane | 62.54 ± 0.27 Ba | 58.21 ± 0.06 Ba | 95.82 ± 0.20 Bb | 46.76 ± 0.57 Ba | ND |
| F.Chloroform | 186.94 ± 0.45 Ca | 109.09 ± 0.09 Cb | 39.50 ± 0.01 Ca | 50.43 ± 0.08 Ca | 62.64± 0.01 Ba |
| F.EtOAc | 790.76 ± 0.45 Da | 369.52 ± 0.47 Da | 6.39 ± 0. 30 Da | 86.33 ± 0.68 Da | 4.93 ± 0.09 Ca |
| F.BuOH | 390.47 ± 0.140 Eb | 277.72 ± 0.25 Ea | 27.69 ± 1.22 Eb | 82.17 ± 0.33 Aa | 10.44 ± 0.60 Db |
| Stem Bark | |||||
| EX.MeOH | 795.55 ± 1.45 Ab | 638.93 ± 0.29 Aa | 8.67 ± 0.45 Ab | 78.45 ± 0.61 Ab | 6.78 ± 0.08 Aa |
| F.Hexane | 199.14 ± 0.28 Bb | 38.41 ± 0.25 Bb | 77.07 ± 1.29 Ba | 37.59 ± 0.08 Bb | ND |
| F.Chloroform | 322.68 ± 0.01 Cb | 204.83 ± 0.34 Ca | 49.35 ± 0.01 Ca | 45.03 ± 0.75 Cb | 68.47 ± 0.06 Bb |
| F.EtOAc | 480.21± 0.13 Db | 255.463 ± 0.28 Db | 8.35 ± 0.29 Aa | 77.20 ± 0.32 Db | 5.05 ± 0.01 Ca |
| F.BuOH | 707.61± 0.58 Ea | 213.32 ±0.20 Eb | 8.71 ± 0.06 Aa | 79.19 ± 0.23 Ab | 5.90 ± 0.24 Ca |
| Quercetin | 8.6 ± 0.70 | 12.30 ± 1.40 | |||
| Ascorbic acid | 180.64 ± 0.01 | ||||
| No | Retention Time, Rt (min) | Ionization Mode (+/-) | Experimental Parent Ion (m/z) | Theoretical Parent Ion (m/z) | Error (ppm) | MS/MS Fragment Ions | Compound Identity | Molecular Formula | Detection |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 0.66 | - | 191.0553 | 191.0555 | −1.0468 | 171.03, 127.04, 109.03, 93.03 | Quinic acid | C7H12O6 | L, SB |
| 2 | 0.93 | - | 271.0801 | 271.0817 | −5.9022 | 252.90, 211.02, 108.02 | Arbutin | C12H16O7 | L, SB |
| 3 | 1.00 | - | 331.0671 | 331.0665 | 1.8123 | 271.05, 211.02, 169.01 | Gallic acid glucoside | C13H16O10 | L, SB |
| 4 | 1.05 | - | 331.0675 | 331.0665 | 3.0205 | 271.05, 211.02, 169.01 | Gallic acid glucoside | C13H16O10 | L, SB |
| 5 | 1.24 | - | 331.0672 | 331.0665 | 2.1143 | 271.05, 211.02, 169.01 | Gallic acid glucoside | C13H16O10 | L, SB |
| 6 | 1.25 | - | 169.0134 | 169.0137 | −1.7750 | 125.02 | Gallic acid | C7H6O5 | L, SB |
| 7 | 1.26 | - | 343.0668 | 343.0665 | 0.8744 | 191.06, 169.01 | Galloylquinic acid | C14H16O10 | L, SB |
| 8 | 1.35 | - | 331.0669 | 331.0665 | 1.2082 | 271.05, 211.02, 169.01 | Gallic acid glucoside | C13H16O10 | L, SB |
| 9 | 1.49 | - | 609.1246 | 609.1244 | 0.3283 | 483.10, 441.08, 423.07, 305.07, 177.02, 125.02 | (Epi)gallocat+(epi)gallocat | C30H26O14 | L, SB |
| 10 | 1.96 | - | 315.0721 | 315.0716 | 1.5869 | 153.02, 152.01, 109.03, 108.02 | Protocatechuic acid O-hexoside | C13H16O9 | L, SB |
| 11 | 1.98 | - | 329.0878 | 329.0872 | 1.8232 | 167.03, 152.01, 123.04, 108.02 | Vanillic acid-O-glucoside ester | C14H18O9 | L |
| 12 | 2.04 | - | 329.0875 | 329.0872 | 0.9116 | 167.03, 152.01, 123.04, 108.02 | Vanillic acid-O-glucoside ester | C14H18O9 | L |
| 13 | 2.10 | - | 305.06638 | 305.06613 | 0.81949 | 261.08, 179.03,167.03, 138.03, 137.02, 125.02 | (Epi)gallocatechin | C15H14O7 | L, SB |
| 14 | 2.24 | - | 897.1866 | 897.1878 | −1.3375 | 729.15, 711.14, 593.13, 407.08, 289.07 | (Epi)gallocat+(epi)gallocat+(epi)cat | C45H38O20 | SB |
| 15 | 2.30 | - | 325.0566 | 325.0559 | 2.1534 | 169.01, 125.02 | Galloyl shikimic acid | C14H14O9 | SB |
| 16 | 2.60 | - | 451.1254 | 451.1240 | 1.1083 | 289.07, 245.08, 151.04, 125.02 | (Epi)catechin -3-O-glucoside | C21H24O11 | L, SB |
| 17 | 2.72 | - | 593.1291 | 593.1295 | −0.6743 | 467.10, 441.08, 425.09, 407.08, 289.07, 177.02, 152.02 | (Epi)gallocat+(epi)cat | C30H26O13 | L, SB |
| 18 | 3.04 | - | 359.0981 | 359.0978 | 0.8354 | 197.04, 153.05 | Syringic acid-O-glucoside | C15H20O10 | L, SB |
| 19 | 3.62 | - | 881.1970 | 881.1929 | 4.6527 | 695.14, 577.14, 425.09, 407.08,303.05, 289.07, 125.02 | (Epi)gallocat+(epi)cat+(epi)cat | C45H38O19 | SB |
| 20 | 5.04 | - | 451.1250 | 451.1240 | 2.2166 | 289.07, 245.08151.04, 125.02 | (Epi)cat-3-O-glucoside | C21H24O11 | SB |
| 21 | 5.66 | - | 593.1314 | 593.1295 | 3.2033 | 467.10, 425.09, 407.08, 289.07, 177.02, 152.02 | (Epi)gallocat+(epi)cat | C30H26O13 | L, SB |
| 22 | 5.94 | - | 577.1348 | 577.1346 | 0.3465 | 451.10, 425.09, 407.08, 289.07, 287.06, 245.08, 125.02 | (Epi)cat+(epi)cat | C30H26O12 | L, SB |
| 23 | 6.81 | - | 451.1241 | 451.1240 | 0.2216 | 289.07, 245.08, 151.04, 125.02 | (Epi)catechin -3-O-glucoside | C21H24O11 | L, SB |
| 24 | 7.01 | - | 289.0714 | 289.0712 | 0.67457 | 271.06, 245.08, 179.03, 165.02, 150.03, 137.02, 125.02 | Catechin | C15H14O6 | L, SB |
| 25 | 7.51 | - | 865.1985 | 865.1979 | 0.6934 | 739.18, 713.15, 695.14, 577.13, 425.09, 407.08, 287.06, 125.02 | (Epi)cat+(epi)cat +(epi)cat | C45H38O18 | L, SB |
| 26 | 7.56 | - | 483.0776 | 483.0774 | 0.4140 | 439.09, 424.54, 331.07, 313.06, 287.08, 271.05, 211.02, 169.01 | Digalloyl glucoside | C20H20O14 | L, SB |
| 27 | 8.08 | - | 1153.2627 | 1153.2613 | 1.2139 | 865.20, 577.14, 407.08, 289.07, 125.02 | (Epi)cat+(epi)cat +(epi)cat +(epi)cat | C60H50O24 | SB |
| 28 | 8.24 | - | 865.1973 | 865.1979 | −0.6934 | 739.17, 713.15, 695.14, 577.13, 425.09, 407.08, 287.06, 125.02 | (Epi)cat+(epi)cat +(epi)cat | C45H38O18 | L, SB |
| 29 | 9.00 | - | 319.0462 | 319.0453 | 2.8209 | 301.04, 193.01, 165.02, 153.02, 151.00, 137.02, 109.03 | Dihydromyricetin | C15H12O8 | L, SB |
| 30 | 9.35 | - | 881.1908 | 881.1929 | −2.3831 | 695.14, 577.14, 425.09, 407.08, 303.05, 289.07, 125.02 | (Epi)gallocat+(epi)cat+(epi)cat | C45H38O19 | SB |
| 31 | 9.84 | - | 577.1356 | 577.1346 | 1.7326 | 451.10, 425.09, 407.08, 289.07, 287.06, 245.08, 125.02 | (Epi)cat+(epi)cat | C30H26O12 | L, SB |
| 32 | 9.86 | - | 289.0717 | 289.0712 | 1.85075 | 271.06, 245.08, 179.03, 165.02, 150.03, 137.02, 125.02 | Epicatechin | C15H14O6 | L, SB |
| 33 | 9.99 | - | 1017.2080 | 1017.2089 | −0.8847 | 865.19, 577.14, 407.08 289.07, 125.02 | (Epi)cat gallate +(epi)cat+(epi)cat | C52H42O22 | SB |
| 34 | 10.30 | - | 1017.2085 | 1017.2089 | −0.3932 | 865.19, 577.14, 407.08 289.07, 125.02 | (Epi)cat gallate +(epi)cat+(epi)cat | C52H42O22 | L, SB |
| 35 | 10.35 | - | 449.1087 | 449.1083 | 0.8906 | 287.06, 269.05, 243.07,151.00,107.01 | Dihydrokaempferol 3- O –glucoside | C21H22O11 | L, SB |
| 36 | 10.42 | - | 635.0889 | 635.0884 | 0.7872 | 465.07, 313.06, 211.02, 169.01 | Tigalloyl glucoside | C27H24O18 | L, SB |
| 37 | 10.45 | - | 1153.2617 | 1153.2613 | 0.3468 | 865.20, 577.14, 407.08, 289.07, 125.02 | (Epi)cat+(epi)cat +(epi)cat +(epi)cat | C60H50O24 | SB |
| 38 | 10.53 | - | 729.1461 | 729.1455 | 0.8228 | 577.14, 559.13, 451.10, 425.09, 407.08, 289.07, 125.02 | (Epi)catechgallate + (epi)cat | C37H30O16 | L, SB |
| 39 | 10.70 | - | 865.1970 | 865.1979 | −1.0402 | 739.17, 713.15, 695.14, 577.14, 425.09, 407.08, 287.06, 125.02 | (Epi)cat+(epi)cat +(epi)cat | C45H38O18 | L, SB |
| 40 | 11.06 | - | 479.0828 | 479.0825 | 0.6261 | 317.03, 316.02, 287.02, 271.02, 179.00, 151.00 | Myricetin 3-O-glucoside | C21H20O13 | SB |
| 41 | 11.21 | - | 1153.2635 | 1153.2613 | 1.9076 | 865.20, 577.14, 407.08, 289.07, 125.02 | (Epi)cat+(epi)cat+(epi)cat+(epi)cat | C60H50O24 | SB |
| 42 | 11.33 | - | 577.1369 | 577.1346 | 3.9852 | 451.10,425.09, 407.08, 289.07, 287.06, 245.05, 125.02 | (Epi)cat+(epi)cat | C30H26O12 | L, SB |
| 43 | 11.42 | - | 729.1481 | 729.1455 | 3.5658 | 577.11, 407.08, 425.09, 289.07, 125.02 | (Epi)catechgallate + (epi)cat | C37H30O16 | L, SB |
| 44 | 11.62 | - | 1017.2098 | 1017.2089 | 0.8847 | 865.19, 577.14, 407.08 289.07, 125.02 | (Epi)catechgallate +(epi)cat+(epi)cat | C52H42O22 | SB |
| 45 | 12.15 | - | 615.0997 | 615.0986 | 1.7883 | 463.09, 300.03, 301.03, 271.02, 179.00, 151.00, 169.01 | Quercetin-O-galloyl-glucoside | C28H24O16 | L, SB |
| 46 | 12.30 | - | 463.0878 | 463.0876 | 0.4318 | 317.03, 316.02, 287.02, 271.02, 179.00, 151.00 | Myricetin 3-O-rhamnoside | C21H20O12 | SB |
| 47 | 12.49 | - | 300.9991 | 300.9984 | 2.3255 | 284.00, 257.00, 229.01, 201.02, 185.02 | Ellagic acid | C14H6O8 | L, SB |
| 48 | 12.91 | - | 609.1456 | 609.1455 | 0.1641 | 301.03, 300.03, 271.02, 255.03 | Rutin (quercetin-3-O-rutinoside) | C27H30O16 | L, SB |
| 49 | 12.96 | + | 465.1027 | 465.1033 | −1.2900 | 303.05, 257.04, 229.05 | Delphinidin-3-O- galacatoside | C21H21O12 | L, SB |
| 50 | 12.97 | - | 441.08258 | 441.0821 | 0.91819 | 289.07, 245.08, 203.07, 169.01 | (Epi)catechin gallate | C22H18O10 | L, SB |
| 51 | 13.02 | - | 463.0885 | 463.0876 | 1.9434 | 301.03, 300.03, 271.07, 255.03 | Quercetin-3-O-glucoside | C21H20O12 | L, SB |
| 52 | 13.18 | + | 465.0996 | 465.1033 | −7.9552 | 303.05, 257.04, 229.05 | Delphinidin-3-O-glucoside | C21H21O12 | L, SB |
| 53 | 13.21 | + | 449.1096 | 449.1083 | 2.8946 | 287.05, 241.00 | Cyanidin-3-O-glucoside | C21H21O11 | L, SB |
| 54 | 13.43 | - | 593.1520 | 593.1506 | 2.3602 | 285.04, 284.03, 255.03, 227.03, 179.00, 151.00 | kaempferol 7-O-rhamnosyl-(1→6)-glucoside | C27H30O15 | SB |
| 55 | 13.52 | - | 729.1456 | 729.1455 | 0.1371 | 577.14, 559.13, 451.11, 425.09, 407.08, 425.09, 289.07, 125.02 | (Epi)catechgallate + (epi)cat | C37H30O16 | L, SB |
| 56 | 13.80 | - | 561.1404 | 561.1396 | 1.4256 | 451.11, 435.11, 289.07, 271.06 | ((Epi)afz + (epi)cat) | C30H25O11 | L, SB |
| 57 | 13.83 | - | 477.1042 | 477.1033 | 1.8863 | 315.05, 314.04, 285.04, 271.02, 243.03 | Isorhamnetin 3-O-glucoside | C22H22O12 | SB |
| 58 | 13.94 | - | 433.0773 | 433.0770 | 0.6927 | 301.03, 300.03, 271.02, 255.03 | Quercetin-3-O-(arabinoside/xyloside) | C20H18O11 | L, SB |
| 59 | 13.98 | - | 447.0936 | 447.0927 | 2.0130 | 285.04, 284.03, 255.03, 227.03 | kaempferol-3-O-glucoside | C21H20O11 | L, SB |
| 60 | 14.43 | - | 317.0310 | 317.0297 | 4.1005 | 289.07, 179.00, 151.00 | Myricetin | C15H10O8 | SB |
| 61 | 14.45 | + | 479.1156 | 479.1189 | −6.8876 | 317.06, 302.03, 274.04, 257.04 | Petunidin-3-O-glucoside | C22H23O12 | L, SB |
| 62 | 14.53 | - | 447.0935 | 447.0927 | 1.7893 | 301.03, 300.03, 271.02, 255.03 | Quercetin-3-O-rhamnoside | C21H20O11 | L, SB |
| 63 | 14.73 | - | 187.0969 | 187.0970 | −0.5344 | 169.09, 143.11, 125.10 | Azelaic acid | C9H16O4 | L, SB |
| 64 | 15.61 | - | 583.1099 | 583.1087 | 2.0579 | 463.09, 301.03, 300.03, 271.03, 255.03 | Quercetin-O-(p-hydroxy)benzoyl-hexoside | C28H24O14 | L, SB |
| 65 | 16.04 | + | 609.1611 | 609.1608 | 0.4924 | 301.07, 286.04, 258.05 | Peonidin-3-O-(p-coumaroyl- glucoside) | C31H29O13 | L, SB |
| 66 | 16.21 | - | 435.1295 | 435.1291 | 0.9192 | 273.08, 167.03, 123.04 | Phloridzin | C21H24O10 | SB |
| 67 | 16.23 | + | 463.1203 | 463.1240 | −7.9892 | 301.07, 286.04, 258.05 | Peonidin-3-O-glucoside | C22H23O11 | L, SB |
| 68 | 16.72 | + | 493.1328 | 493.1346 | −3.6501 | 331.08, 316.05, 315.04, 287.12 | Malvidin-3-O-glucoside | C23H25O12 | L, SB |
| 69 | 17.77 | - | 301.0353 | 301.0348 | 1.6609 | 271.02, 255.03, 179.00, 151.00, 149.02, 121.03, 121.03,107.01 | Quercetin | C15H10O7 | L, SB |
| 70 | 19.79 | - | 327.2179 | 327.2171 | 2.4448 | 229.14, 211.13, 171.10 | Trihydroxy-octadecadienoic acid | C18H32O5 | SB |
| 71 | 20.39 | - | 215.1284 | 215.1283 | 0.4648 | 197.12, 153.13 | Undecanedioic acid | C11H20O4 | L, SB |
| 72 | 21.03 | - | 327.2177 | 327.2171 | 1.8336 | 229.14, 211.13, 171.10 | Trihydroxy-octadecadienoic acid | C18H32O5 | L, SB |
| 73 | 22.43 | - | 329.2333 | 329.2328 | 1.51868 | 229.14, 211.13, 171.10 | Trihydroxy-octadecadienoic acid | C18H34O5 | L, SB |
| 74 | 40.99 | + | 457.3679 | 457.3681 | −0.4372 | 439.35, 411.36, 393.35, 203.18, 189.16 | Betulinic acid | C30H48O3 | L, SB |
| 75 | 41.17 | + | 457.3653 | 457.3681 | −6.1219 | 439.35, 411.36, 393.35, 249.18, 203.18, 133.10 | Ursolic acid | C30H48O3 | L, SB |
| 76 | 42.90 | + | 443.3876 | 443.3889 | −2.9319 | 425.37, 407.36, 191.38 | Betulin | C30H50O2 | L, SB |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buzgaia, N.; Lee, S.Y.; Rukayadi, Y.; Abas, F.; Shaari, K. Antioxidant Activity, α-Glucosidase Inhibition and UHPLC–ESI–MS/MS Profile of Shmar (Arbutus pavarii Pamp). Plants 2021, 10, 1659. https://doi.org/10.3390/plants10081659
Buzgaia N, Lee SY, Rukayadi Y, Abas F, Shaari K. Antioxidant Activity, α-Glucosidase Inhibition and UHPLC–ESI–MS/MS Profile of Shmar (Arbutus pavarii Pamp). Plants. 2021; 10(8):1659. https://doi.org/10.3390/plants10081659
Chicago/Turabian StyleBuzgaia, Nawal, Soo Yee Lee, Yaya Rukayadi, Faridah Abas, and Khozirah Shaari. 2021. "Antioxidant Activity, α-Glucosidase Inhibition and UHPLC–ESI–MS/MS Profile of Shmar (Arbutus pavarii Pamp)" Plants 10, no. 8: 1659. https://doi.org/10.3390/plants10081659
APA StyleBuzgaia, N., Lee, S. Y., Rukayadi, Y., Abas, F., & Shaari, K. (2021). Antioxidant Activity, α-Glucosidase Inhibition and UHPLC–ESI–MS/MS Profile of Shmar (Arbutus pavarii Pamp). Plants, 10(8), 1659. https://doi.org/10.3390/plants10081659