
Genetic Diversity and Structure of Rear Edge Populations of Sorbus aucuparia (Rosaceae) in the Hyrcanian Forest

 , and
, and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Sampling, DNA Extraction, and SSR Amplification
2.2. Genetic Diversity
2.3. Genetic Structure
2.4. Mantel Test
2.5. Spatial Autocorrelation
3. Results
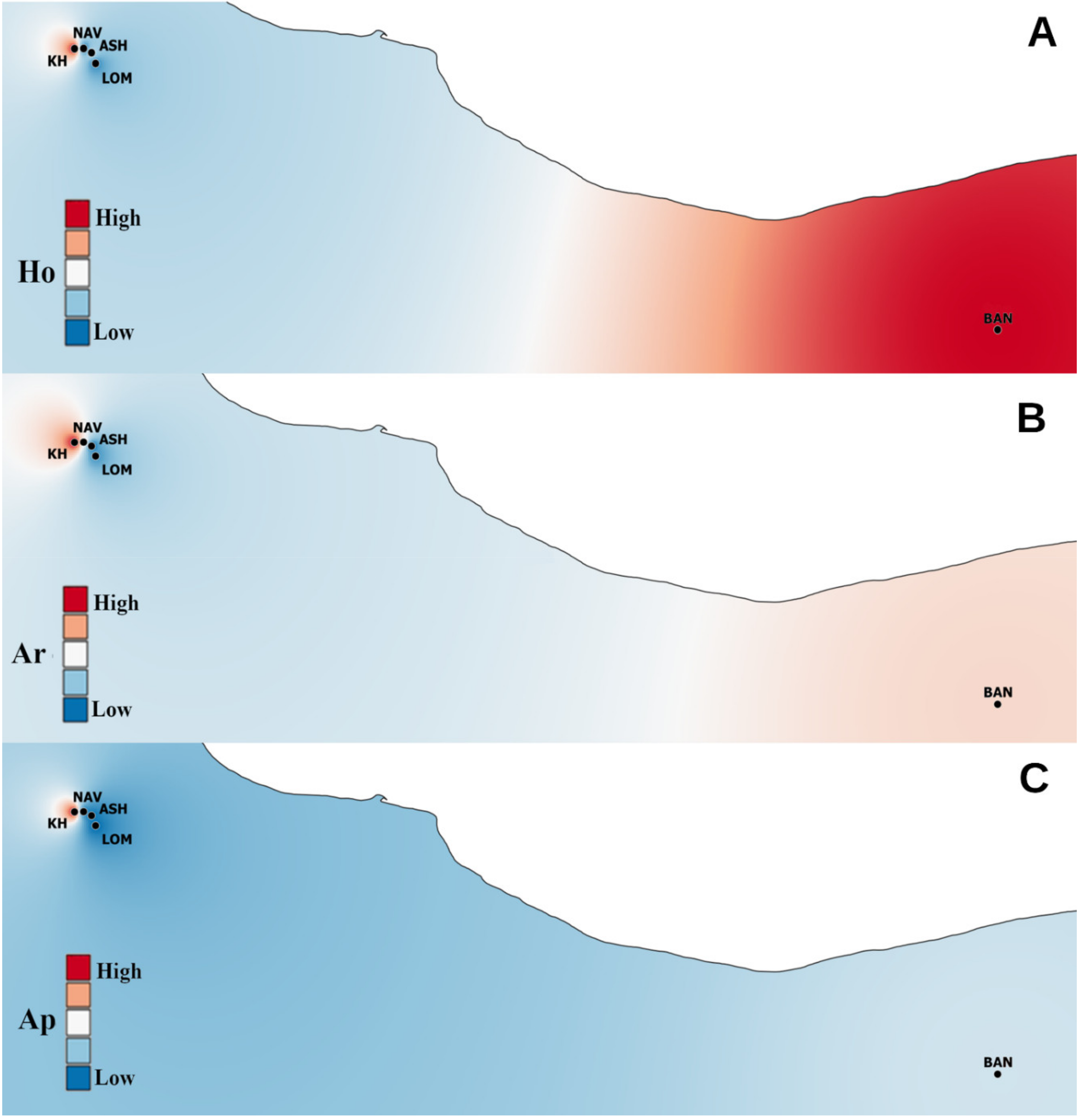
3.1. Genetic Diversity
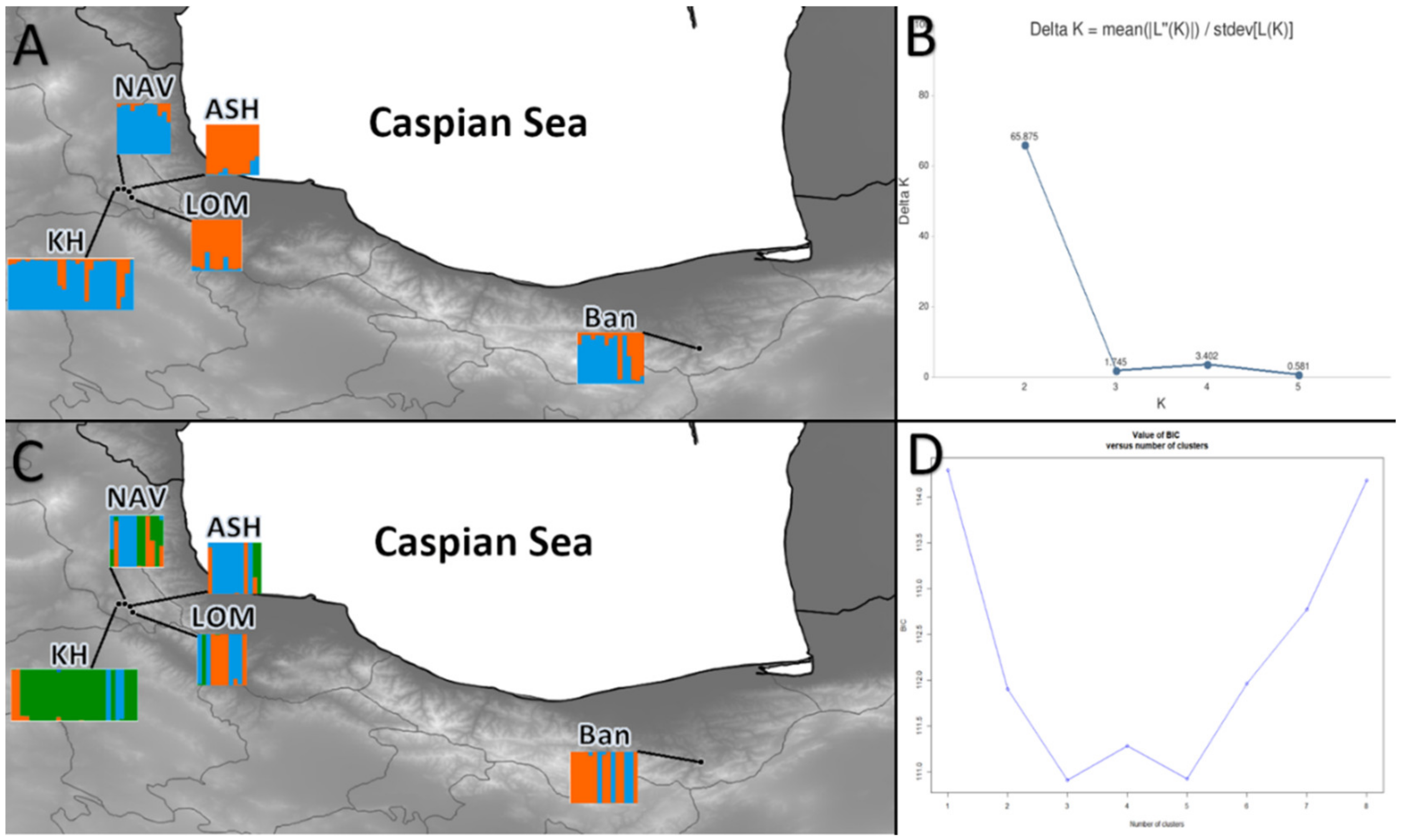
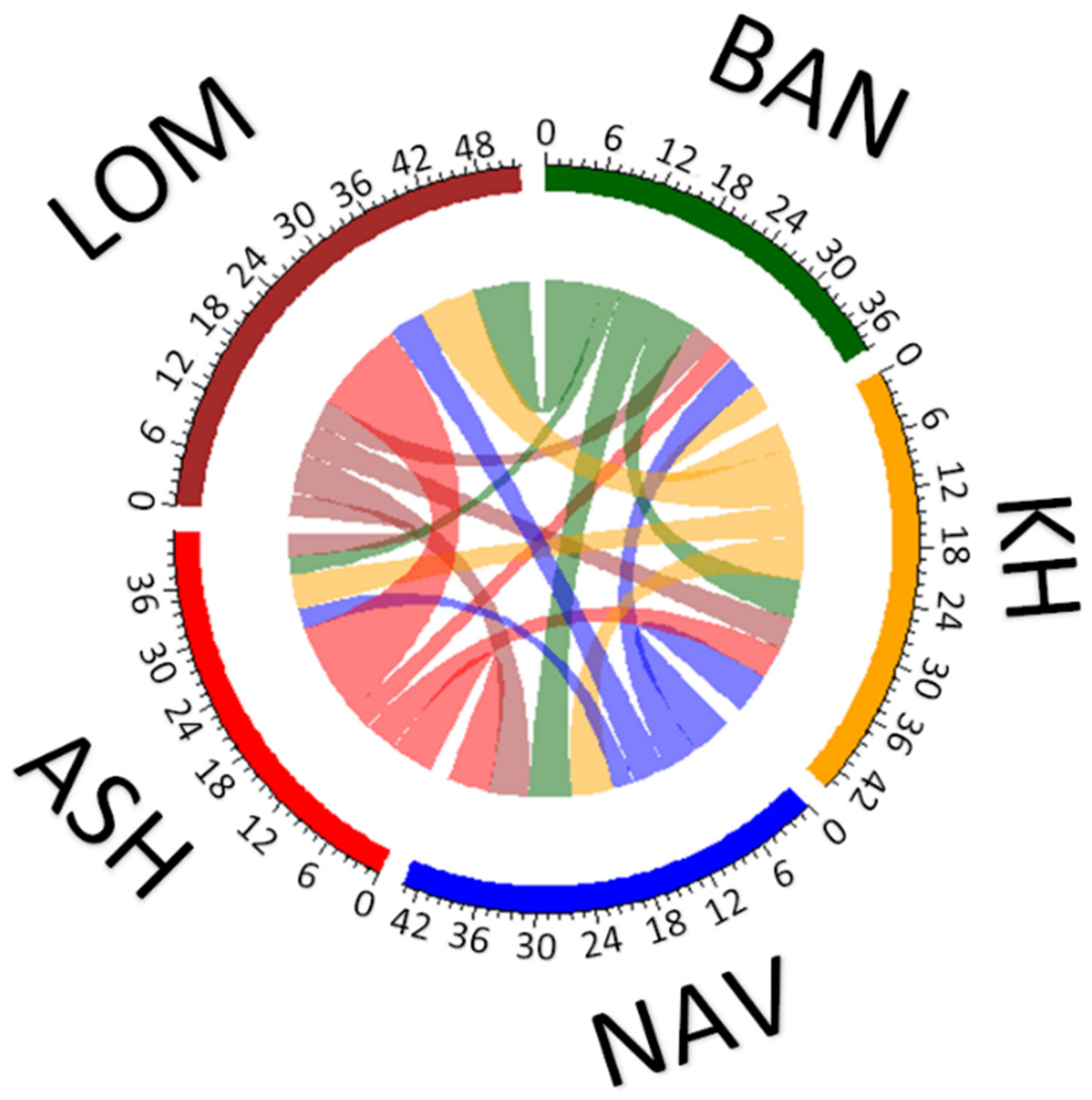
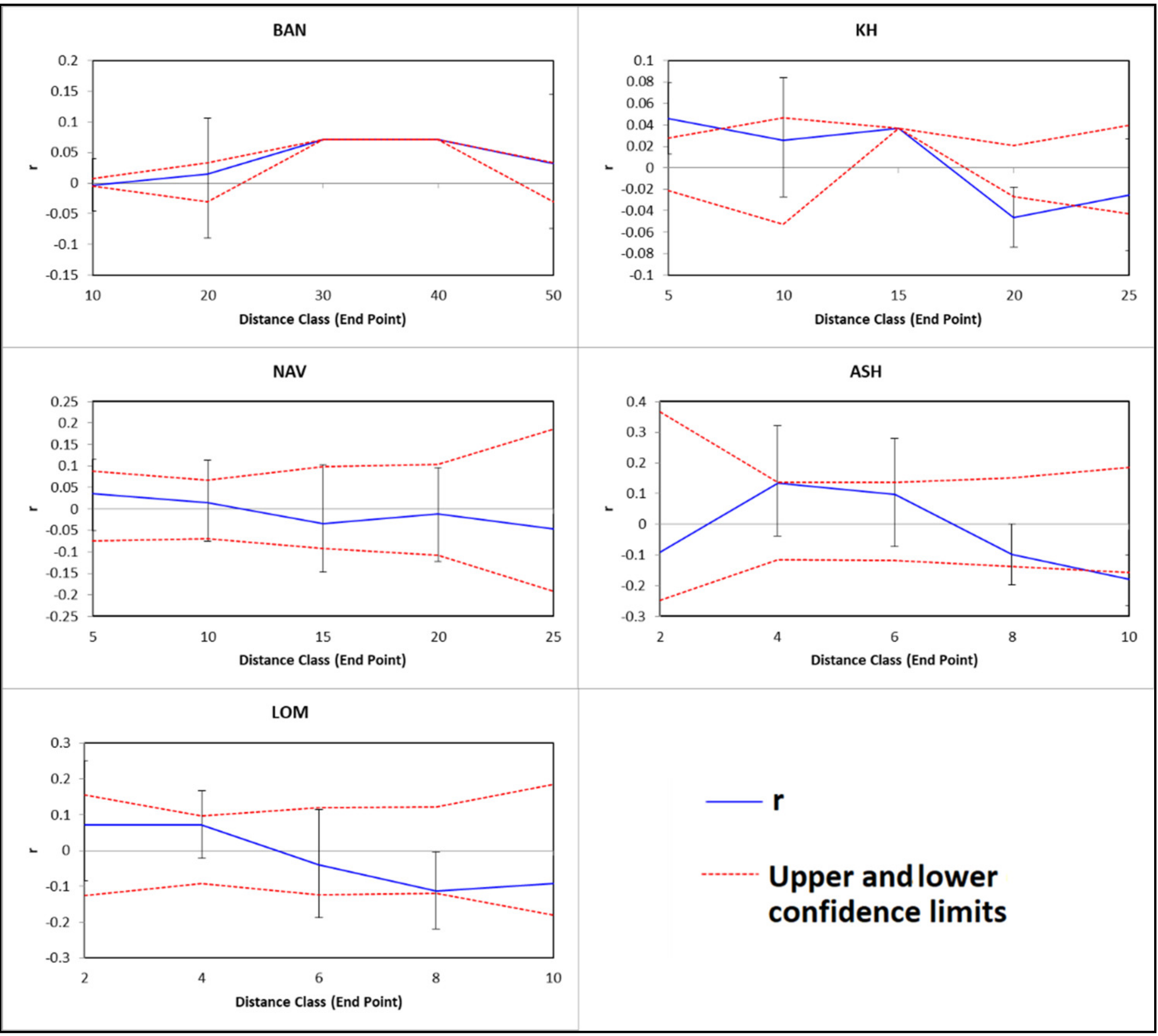
3.2. Spatial Genetic Structure and Gene Flow
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bayat, M.; Burkhart, H.; Namiranian, M.; Hamidi, S.K.; Heidari, S.; Hassani, M. Assessing biotic and abiotic effects on biodiversity index using machine learning. Forests 2021, 12, 461. [Google Scholar] [CrossRef]
- Leestmans, R. Le refuge caspiens et son importance en biogéographie. Linneana Belg. 2005, 20, 97–102. [Google Scholar]
- Zohary, M. Geobotanical Foundations of the Middle East; Gustav Fischer Verlag Press: Stuttgart, Germany; Swets & Zeitlinger: Amsterdam, The Netherlands, 1973; Volume 1. [Google Scholar]
- Kozlowski, G.; Gibbs, D.; Huan, F.; Frey, D.; Gratzfeld, J. Conservation of threatened relict trees through living ex situ collections: Lessons from the global survey of the genus Zelkova (Ulmaceae). Biodivers. Conserv. 2012, 21, 671–685. [Google Scholar] [CrossRef]
- Kozlowski, G.; Song, Y.-G.; Bétrisey, S.; Alvarado, E.V.; Bétrisey, S. Wingnuts (Pterocarya) & Walnut Family: Relict Trees: Linking the Past, Present and Future; Natural History Museum: Fribourg, Switzerland, 2018. [Google Scholar]
- Hultén, E.; Fries, M. Atlas of North European Vascular Plants. North of the Tropic of Cancer; Koeltz Scientific Books: Königstein, Germany, 1986. [Google Scholar]
- Raspé, O.; Findlay, C.; Jacquemart, A. Sorbus aucuparia L.: Biological flora of the British Isles, list br. Vasc. Pl. 232, 1. J. Ecol. 2000, 88, 910–930. [Google Scholar] [CrossRef]
- Żywiec, M.; Ledwoń, M. Spatial and temporal patterns of rowan (Sorbus aucuparia L.) regeneration in west Carpathian subalpine spruce forest. Plant Ecol. 2008, 194, 283–291. [Google Scholar] [CrossRef]
- Sabeti, H. Forest, Trees and Shrubs of Iran; Yazd University Publishers: Yazd, Iran, 1997; 810p. [Google Scholar]
- McInerny, G.; Turner, J.; Wong, H.; Travis, J.; Benton, T. How range shifts induced by climate change affect neutral evolution. Proc. R. Soc. B Biol. Sci. 2009, 276, 1527–1534. [Google Scholar] [CrossRef]
- Wróblewska, A.; Mirski, P. From past to future: Impact of climate change on range shifts and genetic diversity patterns of circumboreal plants. Reg. Environ. Change 2018, 18, 409–424. [Google Scholar] [CrossRef]
- Jump, A.S.; Hunt, J.M.; Penuelas, J. Rapid climate change-related growth decline at the southern range edge of Fagus sylvatica. Glob. Change Biol. 2006, 12, 2163–2174. [Google Scholar] [CrossRef]
- Lesica, P.; Allendorf, F.W. When are peripheral populations valuable for conservation? Conserv. Biol. 1995, 9, 753–760. [Google Scholar] [CrossRef]
- Hampe, A.; Petit, R.J. Conserving biodiversity under climate change: The rear edge matters. Ecol. Lett. 2005, 8, 461–467. [Google Scholar] [CrossRef]
- Arnaud-Haond, S.; Teixeira, S.; Massa, S.I.; Billot, C.; Saenger, P.; Coupland, G.; Duarte, C.M.; Serrao, E. Genetic structure at range edge: Low diversity and high inbreeding in southeast Asian mangrove (Avicennia marina) populations. Mol. Ecol. 2006, 15, 3515–3525. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, A.A.; Blows, M.W. Species borders: Ecological and evolutionary perspectives. Trends Ecol. Evol. 1994, 9, 223–227. [Google Scholar] [CrossRef]
- Bradshaw, A.D. The croonian lecture, 1991. Genostasis and the limits to evolution. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 1991, 333, 289–305. [Google Scholar]
- Sáenz-Romero, C.; O’Neill, G.; Aitken, S.N.; Lindig-Cisneros, R. Assisted migration field tests in Canada and Mexico: Lessons, limitations, and challenges. Forests 2021, 12, 9. [Google Scholar] [CrossRef]
- Yosefzadeh, H.; Tabari, M.; Akbarinia, M.; Akbarian, M.R.; Bussotti, F. Morphological plasticity of Parrotia persica leaves in eastern Hyrcanian forests (Iran) is related to altitude. Nord. J. Bot. 2010, 28, 344–349. [Google Scholar] [CrossRef]
- Gaitán-Espitia, J.D.; Hobday, A.J. Evolutionary principles and genetic considerations for guiding conservation interventions under climate change. Glob. Change Biol. 2021, 27, 475–488. [Google Scholar] [CrossRef]
- Rischkowsky, B.; Pilling, D. The State of the World’s Animal Genetic Resources for Food and Agriculture; Food & Agriculture Org.: Rome, Italy, 2007. [Google Scholar]
- Robertson, A.; Newton, A.; Ennos, R. Breeding systems and continuing evolution in the endemic Sorbus taxa on Arran. Heredity 2004, 93, 487–495. [Google Scholar] [CrossRef]
- Hoebee, S.E.; Menn, C.; Rotach, P.; Finkeldey, R.; Holderegger, R. Spatial genetic structure of Sorbus torminalis: The extent of clonal reproduction in natural stands of a rare tree species with a scattered distribution. For. Ecol. Manag. 2006, 226, 1–8. [Google Scholar] [CrossRef]
- Angelone, S.; Hilfiker, K.; Holderegger, R.; Bergamini, A.; Hoebee, S. Regional population dynamics define the local genetic structure in Sorbus torminalis. Mol. Ecol. 2007, 16, 1291–1301. [Google Scholar] [CrossRef]
- Rasmussen, K.K.; Kollmann, J. Low genetic diversity in small peripheral populations of a rare European tree (Sorbus torminalis) dominated by clonal reproduction. Conserv. Genet. 2008, 9, 1533–1539. [Google Scholar] [CrossRef]
- Jankowska-Wroblewska, S.; Meyza, K.; Sztupecka, E.; Kubera, L.; Burczyk, J. Clonal structure and high genetic diversity at peripheral populations of Sorbus torminalis (L.) Crantz. iForest-Biogeosciences For. 2016, 9, 892. [Google Scholar] [CrossRef]
- Kavaliauskas, D.; Šeho, M.; Baier, R.; Fussi, B. Genetic variability to assist in the delineation of provenance regions and selection of seed stands and gene conservation units of wild service tree (Sorbus torminalis (L.) Crantz) in southern Germany. Eur. J. For. Res. 2021, 140, 1–15. [Google Scholar] [CrossRef]
- Liu, C.; Dou, Y.; Guan, X.; Fu, Q.; Zhang, Z.; Hu, Z.; Zheng, J.; Lu, Y.; Li, W. De novo transcriptomic analysis and development of est-ssrs for Sorbus pohuashanensis (Hance) hedl. PLoS ONE 2017, 12. [Google Scholar] [CrossRef]
- Abid, M.; Scheffran, J.; Schneider, U.A.; Ashfaq, M. Farmers’ perceptions of and adaptation strategies to climate change and their determinants: The case of Punjab province, Pakistan. Earth Syst. Dyn. 2015, 6, 225–243. [Google Scholar] [CrossRef]
- Ellis, J.; Burke, J. Est-SSRs as a resource for population genetic analyses. Heredity 2007, 99, 125–132. [Google Scholar] [CrossRef]
- Doyle, J.J.; Doyle, J.L. Isolation of plant DNA from fresh tissue. Focus 1990, 12, 39–40. [Google Scholar]
- Dellaporta, S.L.; Wood, J.; Hicks, J.B. A plant DNA minipreparation: Version ii. Plant Mol. Biol. Rep. 1983, 1, 19–21. [Google Scholar] [CrossRef]
- Janfaza, S.; Nasr, S.M.H.; Yousefzadeh, H.; Botta, R. Phylogenetic relationships of the genus Castanea based on chloroplast rbcl with focusing Iranian chestnut. JBES 2015, 5, 312–323. [Google Scholar]
- Bassam, B.J.; Caetano-Anollés, G.; Gresshoff, P.M. Fast and sensitive silver staining of DNA in polyacrylamide gels. Anal. Biochem. 1991, 196, 80–83. [Google Scholar] [CrossRef]
- Van Oosterhout, C.; Hutchinson, W.F.; Wills, D.P.; Shipley, P. Micro-checker: Software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes 2004, 4, 535–538. [Google Scholar] [CrossRef]
- Chapuis, M.-P.; Estoup, A. Microsatellite null alleles and estimation of population differentiation. Mol. Biol. Evol. 2007, 24, 621–631. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. Genalex 6: Genetic analysis in excel. Population genetic software for teaching and research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar] [CrossRef]
- Chybicki, I.J.; INEST 2.2. The User Manual. 2017. Available online: https://www.ukw.edu.pl/pracownicy/strona/igor_chybicki/software_ukw (accessed on 12 October 2018).
- Garza, J.; Williamson, E. Detection of reduction in population size using data from microsatellite loci. Mol. Ecol. 2001, 10, 305–318. [Google Scholar] [CrossRef]
- Excoffier, L.; Smouse, P.E.; Quattro, J.M. Analysis of molecular variance inferred from metric distances among DNA haplotypes: Application to human mitochondrial DNA restriction data. Genetics 1992, 131, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Wilson, G.A.; Rannala, B. Bayesian inference of recent migration rates using multilocus genotypes. Genetics 2003, 163, 1177–1191. [Google Scholar] [CrossRef] [PubMed]
- Jombart, T.; Devillard, S.; Balloux, F. Discriminant analysis of principal components: A new method for the analysis of genetically structured populations. BMC Genet. 2010, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Beerli, P. Comparison of bayesian and maximum-likelihood inference of population genetic parameters. Bioinformatics 2006, 22, 341–345. [Google Scholar] [CrossRef]
- Beerli, P.; Felsenstein, J. Maximum-likelihood estimation of migration rates and effective population numbers in two populations using a coalescent approach. Genetics 1999, 152, 763–773. [Google Scholar] [CrossRef]
- Beerli, P.; Palczewski, M. Unified framework to evaluate panmixia and migration direction among multiple sampling locations. Genetics 2010, 185, 313–326. [Google Scholar] [CrossRef]
- McRae, B.H. Isolation by resistance. Evolution 2006, 60, 1551–1561. [Google Scholar] [CrossRef]
- McRae, B.H.; Beier, P. Circuit theory predicts gene flow in plant and animal populations. Proc. Natl. Acad. Sci. USA 2007, 104, 19885–19890. [Google Scholar] [CrossRef]
- Wright, S. Isolation by distance. Genetics 1943, 28, 114. [Google Scholar] [CrossRef]
- Smouse, P.E.; Peakall, R. Spatial autocorrelation analysis of individual multiallele and multilocus genetic structure. Heredity 1999, 82, 561–573. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef]
- Bednorz, L.; Myczko, L.; Kosinski, P. Genetic variability and structure of the wild service tree (Sorbus torminalis (L.) Crantz) in poland. Silvae Genet. 2006, 55, 197–201. [Google Scholar] [CrossRef]
- George, J.-P.; Konrad, H.; Collin, E.; Thevenet, J.; Ballian, D.; Idzojtic, M.; Kamm, U.; Zhelev, P.; Geburek, T. High molecular diversity in the true service tree (Sorbus domestica) despite rareness: Data from Europe with special reference to the Austrian occurrence. Ann. Bot. 2015, 115, 1105–1115. [Google Scholar] [CrossRef]
- Alavi, S.J.; Veiskarami, R.; Esmailzadeh, O.; Gadow, K.V. Analyzing the biological and structural diversity of Hyrcanian forests dominated by Taxus baccata L. Forests 2020, 11, 701. [Google Scholar] [CrossRef]
- Yakimowski, S.B.; Eckert, C.G. Populations do not become less genetically diverse or more differentiated towards the northern limit of the geographical range in clonal Vaccinium stamineum (Ericaceae). New Phytol. 2008, 180, 534–544. [Google Scholar] [CrossRef] [PubMed]
- Assis, J.; Coelho, N.C.; Alberto, F.; Valero, M.; Raimondi, P.; Reed, D.; Serrão, E.A. High and distinct range-edge genetic diversity despite local bottlenecks. PLoS ONE 2013, 8, e68646. [Google Scholar] [CrossRef]
- Kucerova, E.; Clifton, S.W.; Xia, X.-Q.; Long, F.; Porwollik, S.; Fulton, L.; Fronick, C.; Minx, P.; Kyung, K.; Warren, W. Genome sequence of Cronobacter sakazakii baa-894 and comparative genomic hybridization analysis with other Cronobacter species. PLoS ONE 2010, 5, e9556. [Google Scholar] [CrossRef] [PubMed]
- Kottler, E.J.; Dickman, E.E.; Sexton, J.P.; Emery, N.C.; Franks, S.J. Draining the Swamping Hypothesis: Little Evidence that Gene Flow Reduces Fitness at Range Edges. Trends Ecol. Evol. 2021, 36, 533–544. [Google Scholar] [CrossRef]
- Ouborg, N.; Vergeer, P.; Mix, C. The rough edges of the conservation genetics paradigm for plants. J. Ecol. 2006, 94, 1233–1248. [Google Scholar] [CrossRef]
- Michalski, S.G.; Durka, W. High selfing and high inbreeding depression in peripheral populations of juncus atratus. Mol. Ecol. 2007, 16, 4715–4727. [Google Scholar] [CrossRef] [PubMed]
- DeSilva, R.; Dodd, R.S. Patterns of Fine-Scale Spatial Genetic Structure and Pollen Dispersal in Giant Sequoia (Sequoiadendron giganteum). Forests 2021, 12, 61. [Google Scholar] [CrossRef]
- Bacles, C.F.; Jump, A.S. Taking a tree’s perspective on forest fragmentation genetics. Trends Plant Sci. 2011, 16, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Frankham, R. Challenges and opportunities of genetic approaches to biological conservation. Biol. Conserv. 2010, 143, 1919–1927. [Google Scholar] [CrossRef]
- Pannell, J.R.; Dorken, M.E. Colonisation as a common denominator in plant metapopulations and range expansions: Effects on genetic diversity and sexual systems. Landsc. Ecol. 2006, 21, 837–848. [Google Scholar] [CrossRef]
- Jangjoo, M.; Matter, S.F.; Roland, J.; Keyghobadi, N. Connectivity rescues genetic diversity after a demographic bottleneck in a butterfly population network. Proc. Natl. Acad. Sci. USA 2016, 113, 10914–10919. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Population Name | Sample Code | Longitude | Latitude | Altitude | Sample Size | Associate Species |
|---|---|---|---|---|---|---|
| Khalkhal | KH | 373929.7 | 483540.9 | 2100–2500 | 28 | Quercus macranthera-Sorbus graeca |
| Olsehposht | NAV | 373936.3 | 484015.2 | 1650–2100 | 12 | Fagus orientalis, Acer spp. |
| Asbehriseh | ASH | 373741.4 | 484416.2 | 1500–1900 | 12 | Carpinus betulus, Acer spp. |
| Barzekoh | LOM | 373231.5 | 484634.4 | 1450–1850 | 11 | Fagus orientalis, Carpinus orientalis, Acer mazandaranicum |
| Sangedeh | BAN | 360601.6 | 531232.5 | 1900–2400 | 15 | Betula pendula-Acer hyrcanum |
| Pop | Lat | Long | n | A | Ae | Ar | Ap | Null | Ho | He | Fis | M-Ratio |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BAN | 36.06 | 53.124 | 15 | 4.56 | 3.29 | 4.12 | 2.00 | 0.102 | 0.56 | 0.70 | 0.16 | 0.006 |
| KH | 37.393 | 48.354 | 28 | 5.78 | 3.64 | 4.56 | 5.00 | 0.125 | 0.56 | 0.73 | 0.21 | 0.050 |
| NAV | 37.394 | 48.401 | 12 | 4.56 | 3.12 | 4.08 | 0.00 | 0.134 | 0.45 | 0.69 | 0.31 | 0.037 |
| ASH | 37.374 | 48.442 | 12 | 3.67 | 2.79 | 3.48 | 0.00 | 0.082 | 0.49 | 0.61 | 0.17 | 0.004 |
| LOM | 37.323 | 48.463 | 11 | 3.89 | 2.89 | 3.61 | 0.00 | 0.123 | 0.46 | 0.63 | 0.29 | 0.000 |
| BAN | KH | NAV | ASH | LOM | |
|---|---|---|---|---|---|
| BAN | 0.0337 | 0.0326 | 0.0831 | 0.0057 | |
| KH | 0.0330 | 0.0162 | 0.0655 | 0.0411 | |
| NAV | 0.0332 | 0.0126 | 0.0779 | 0.0532 | |
| ASH | 0.0777 | 0.0597 | 0.0732 | 0.0078 | |
| LOM | 0.0064 | 0.0366 | 0.0527 | 0.0108 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yousefzadeh, H.; Raeisi, S.; Esmailzadeh, O.; Jalali, G.; Nasiri, M.; Walas, Ł.; Kozlowski, G. Genetic Diversity and Structure of Rear Edge Populations of Sorbus aucuparia (Rosaceae) in the Hyrcanian Forest. Plants 2021, 10, 1471. https://doi.org/10.3390/plants10071471
Yousefzadeh H, Raeisi S, Esmailzadeh O, Jalali G, Nasiri M, Walas Ł, Kozlowski G. Genetic Diversity and Structure of Rear Edge Populations of Sorbus aucuparia (Rosaceae) in the Hyrcanian Forest. Plants. 2021; 10(7):1471. https://doi.org/10.3390/plants10071471
Chicago/Turabian StyleYousefzadeh, Hamed, Shahla Raeisi, Omid Esmailzadeh, Gholamali Jalali, Malek Nasiri, Łukasz Walas, and Gregor Kozlowski. 2021. "Genetic Diversity and Structure of Rear Edge Populations of Sorbus aucuparia (Rosaceae) in the Hyrcanian Forest" Plants 10, no. 7: 1471. https://doi.org/10.3390/plants10071471
APA StyleYousefzadeh, H., Raeisi, S., Esmailzadeh, O., Jalali, G., Nasiri, M., Walas, Ł., & Kozlowski, G. (2021). Genetic Diversity and Structure of Rear Edge Populations of Sorbus aucuparia (Rosaceae) in the Hyrcanian Forest. Plants, 10(7), 1471. https://doi.org/10.3390/plants10071471