
Development of an Aus-Derived Nested Association Mapping (Aus-NAM) Population in Rice

 ,
,  , , , and
, , , and
Abstract
:1. Introduction
2. Results
2.1. Development of Aus-NAM Population
2.2. Phenotypic Characteristics
2.3. Linkage Map and Projection of Parental Variants
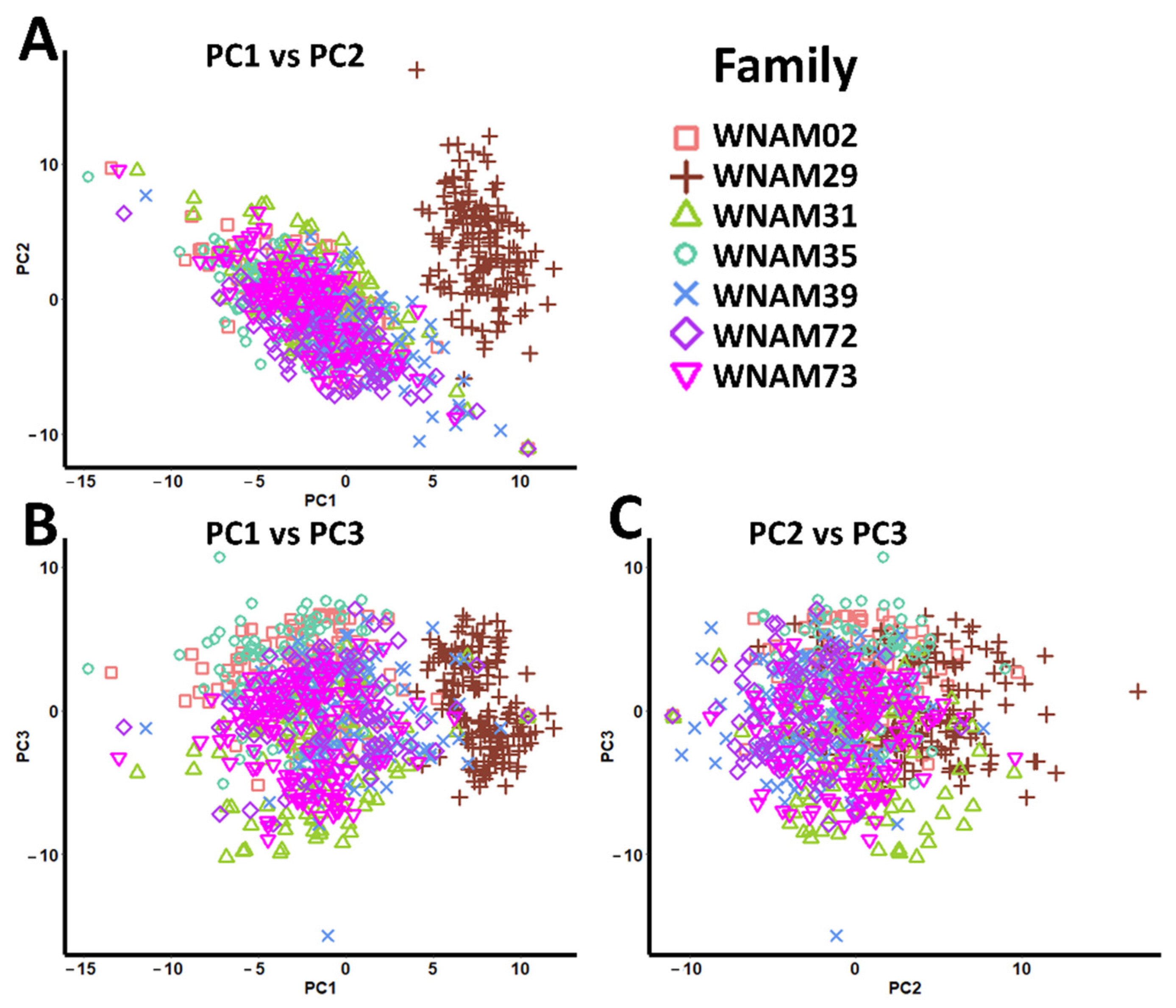
2.4. Population Structure
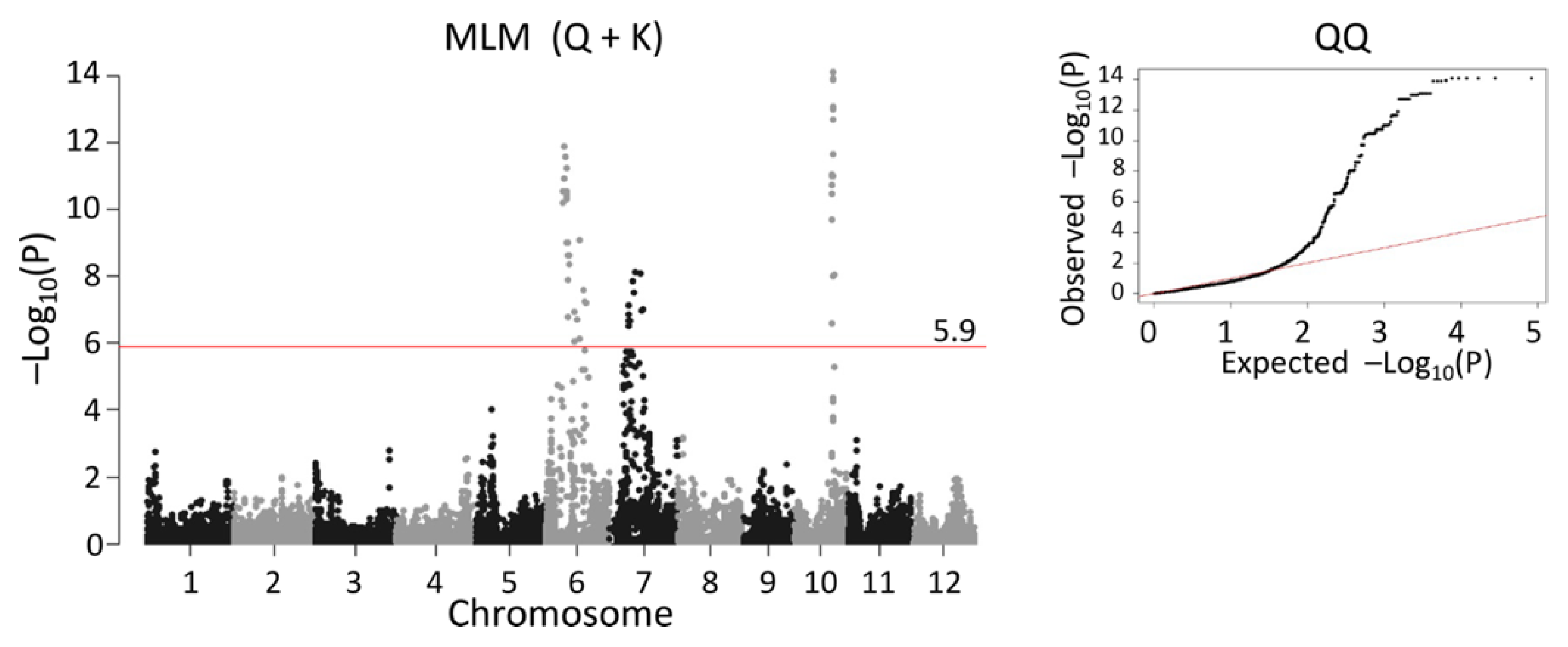
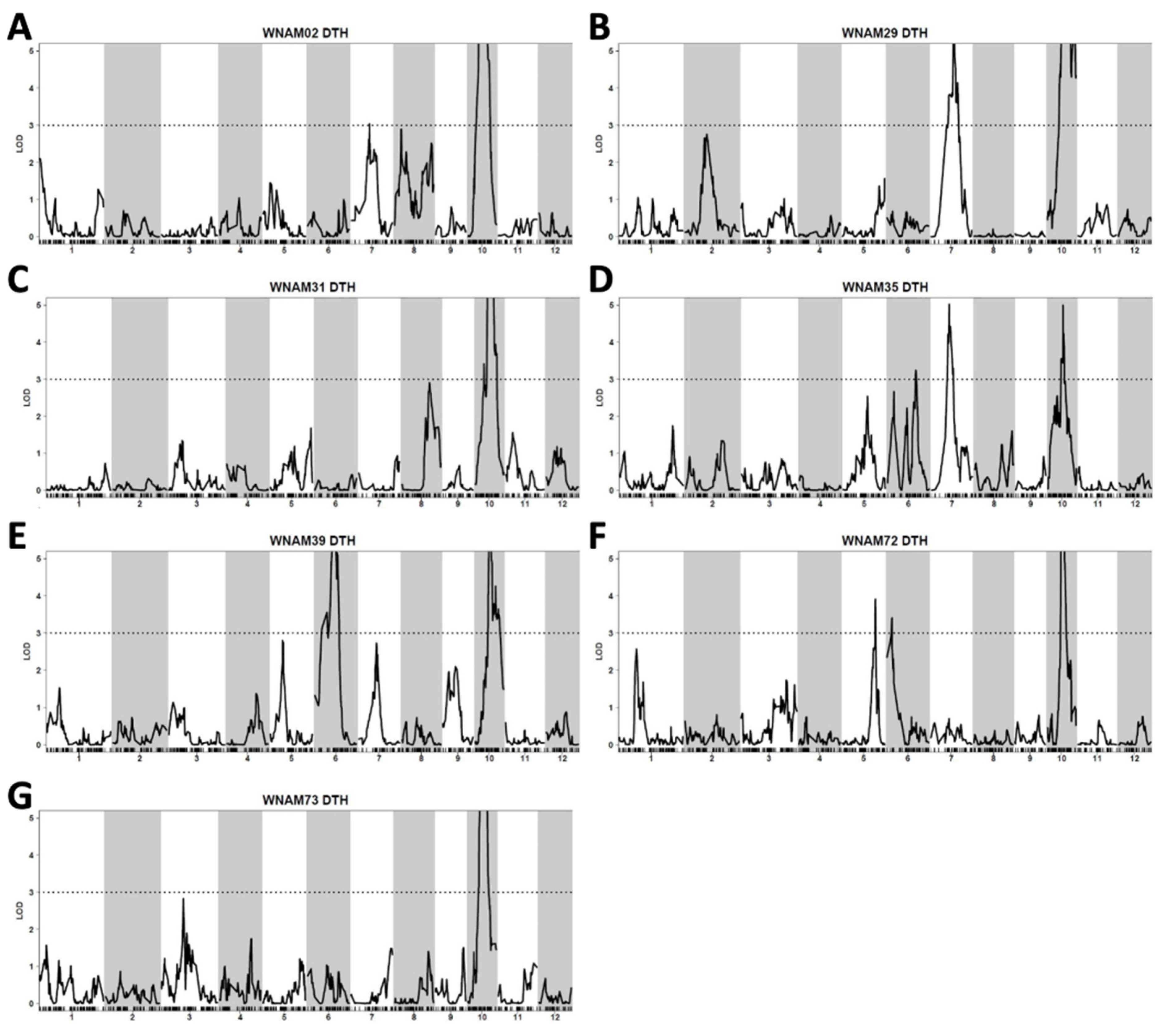
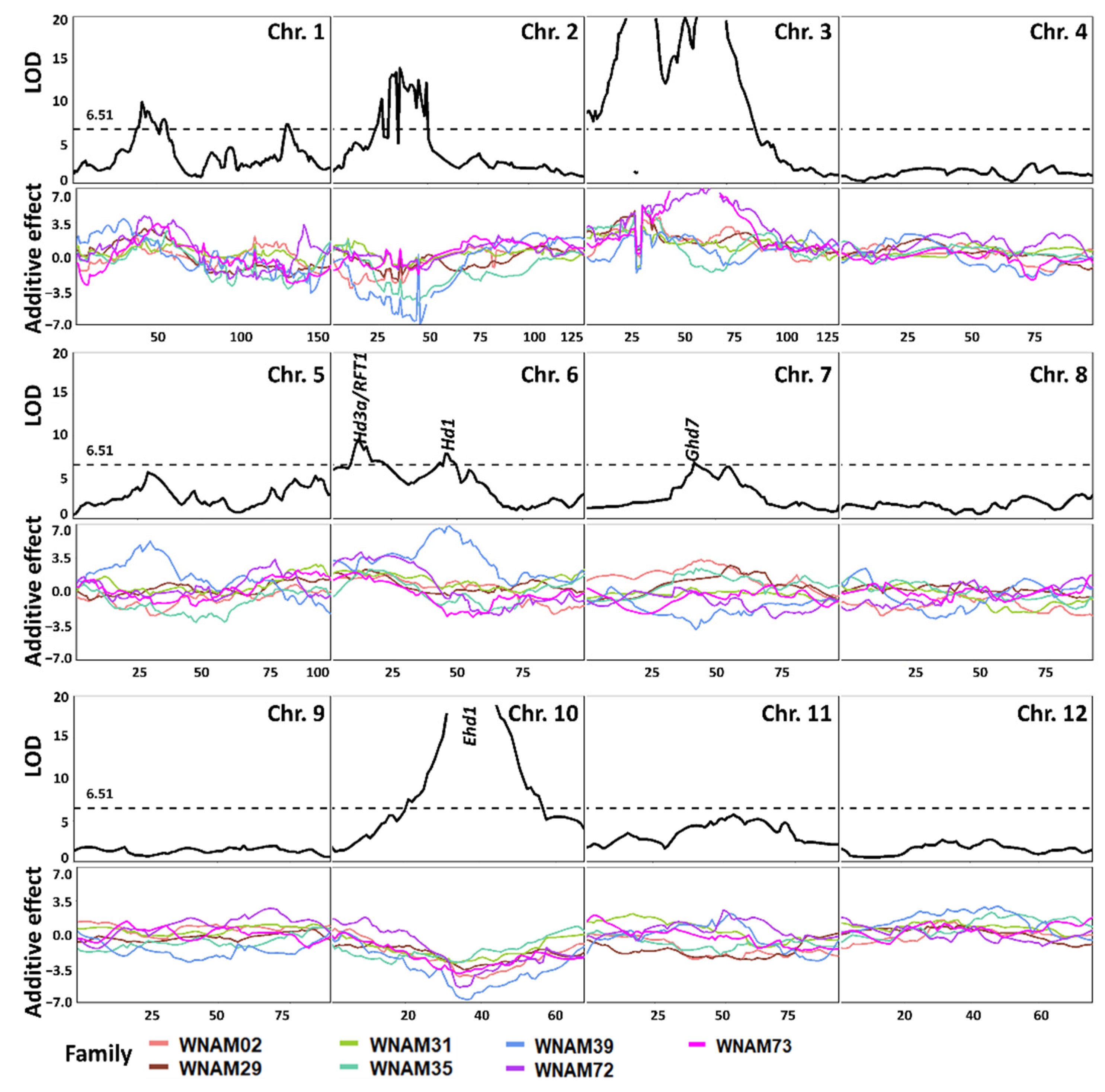
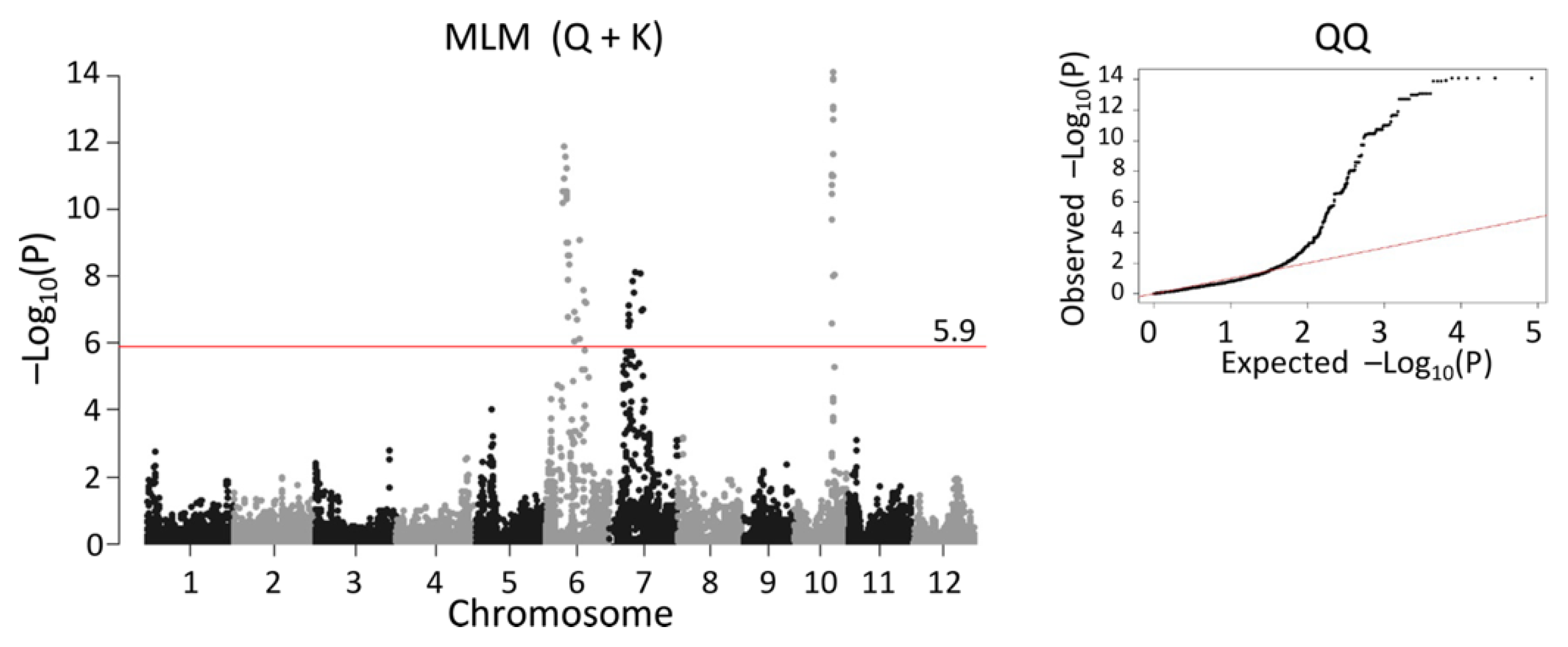
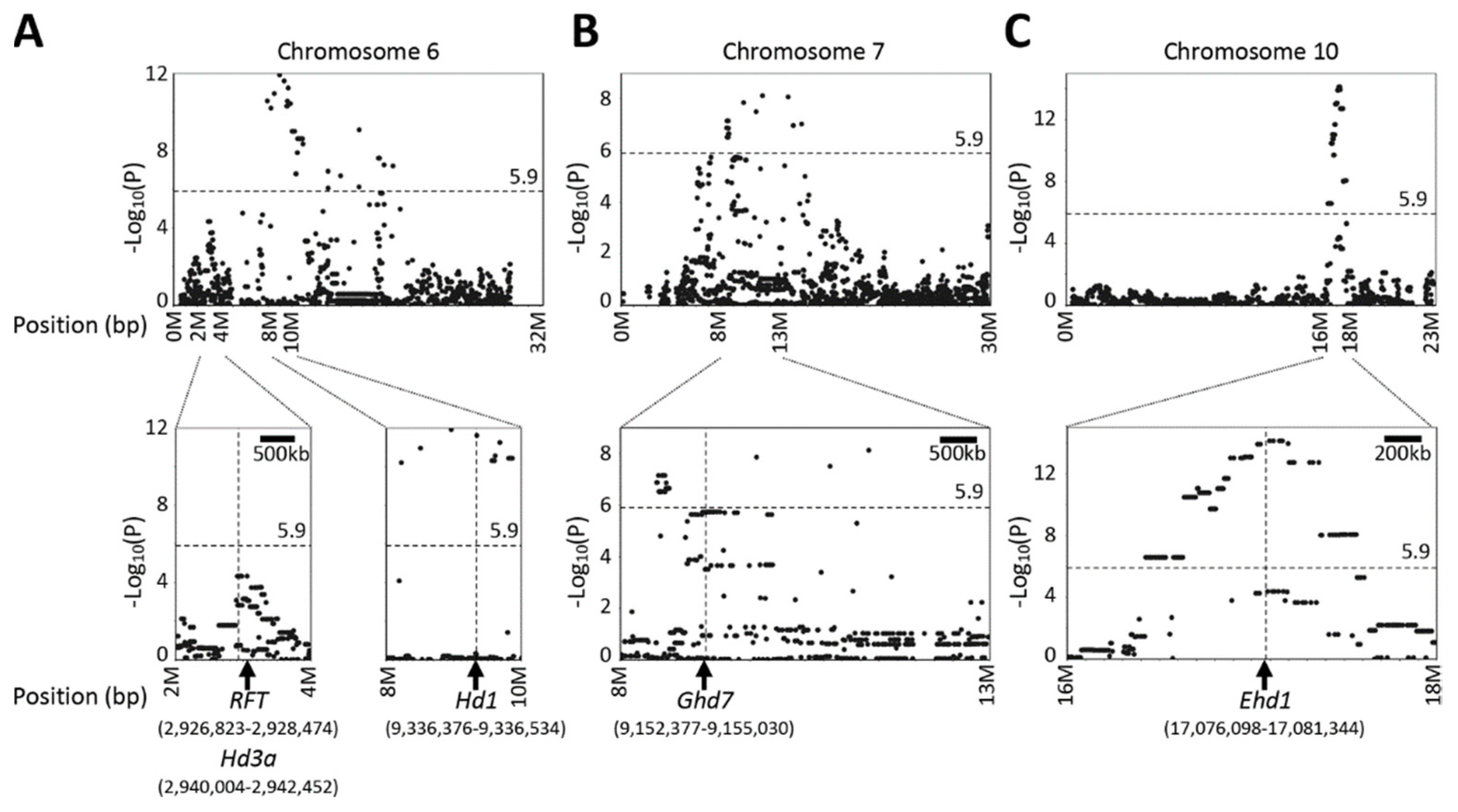
2.5. QTL Detected by Single-Family Analysis, Joint Analysis, and GWAS-Based Methods
2.6. Evaluation of Mapping Accuracy
3. Discussion
3.1. Development of aus-NAM Population
3.2. Genotyping of aus-NAM Population
3.3. Accuracy of QTL Mapping Using aus-NAM Population
3.4. Prospects
4. Materials and Methods
4.1. Plant Materials
4.2. Trait Evaluation and Statistical Analysis
4.3. Genotyping By Sequencing (GBS)
4.4. Whole-Genome Resequencing of T65 and aus Founders
4.5. Projection of Parental Variants and Population Structure Estimation
4.6. Simple QTL Mapping
4.7. Joint QTL Mapping
4.8. Genome-Wide Association Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Reyes, V.P.; Angeles-Shim, R.B.; Mendioro, M.S.; Manuel, M.C.C.; Lapis, R.S.; Shim, J.; Sunohara, H.; Nishiuchi, S.; Kikuta, M.; Makihara, D.; et al. Marker-Assisted Introgression and Stacking of Major QTLs Controlling Grain Number (Gn1a) and Number of Primary Branching (WFP) to NERICA Cultivars. Plants 2021, 10, 844. [Google Scholar] [CrossRef]
- Ikeda, M.; Miura, K.; Aya, K.; Kitano, H.; Matsuoka, M. Genes offering the potential for designing yield-related traits in rice. Curr. Opin. Plant Biol. 2013, 16, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Yano, M.; Sasaki, T. Genetic and molecular dissection of quantitative traits in rice. Plant Mol. Biol. 1997, 35, 145–153. [Google Scholar] [CrossRef]
- Ogura, T.; Busch, W. From phenotypes to causal sequences: Using genome wide association studies to dissect the sequence basis for variation of plant development. Curr. Opin. Plant Biol. 2015, 23, 98–108. [Google Scholar] [CrossRef]
- Mogga, M.; Sibiya, J.; Shimelis, H.; Lamo, J.; Yao, N. Diversity analysis and genome-wide association studies of grain shape and eating quality traits in rice (Oryza sativa L.) using DArT markers. PLoS ONE 2018, 13, e0198012. [Google Scholar] [CrossRef] [Green Version]
- Yano, K.; Yamamoto, E.; Aya, K.; Takeuchi, H.; Lo, P.C.; Hu, L.; Yamasaki, M.; Yoshida, S.; Kitano, H.; Hirano, K.; et al. Genome-wide association study using whole-genome sequencing rapidly identifies new genes influencing agronomic traits in rice. Nat. Genet. 2016, 48, 927–934. [Google Scholar] [CrossRef] [PubMed]
- Norton, G.J.; Travis, A.J.; Douglas, A.; Fairley, S.; Alves, E.P.; Ruang-Areerate, P.; Naredo, M.E.B.; McNally, K.L.; Hossain, M.; Islam, M.R.; et al. Genome wide association mapping of grain and straw biomass traits in the rice Bengal and Assam Aus Panel (BAAP) grown under alternate wetting and drying and permanently flooded irrigation. Front. Plant Sci. 2018, 9, 1223. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Liu, H.; Wu, L.; Warburton, M.; Yan, J. Genome-wide association studies in maize: Praise and stargaze. Mol. Plant 2017, 10, 359–374. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.; Gore, M.; Buckler, E.S.; Yu, J. Status and prospects of association mapping in plants. Plant Genome 2008, 1, 5–20. [Google Scholar] [CrossRef]
- Korte, A.; Farlow, A. The advantages and limitations of trait analysis with GWAS: A review. Plant Methods 2013, 9, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Holland, J.B.; McMullen, M.D.; Buckler, E.S. Genetic design and statistical power of nested association mapping in maize. Genetics 2008, 178, 539–551. [Google Scholar] [CrossRef] [Green Version]
- Dell’Acqua, M.; Gatti, D.M.; Pea, G.; Cattonaro, F.; Coppens, F.; Magris, G.; Hlaing, A.L.; Aung, H.H.; Nelissen, H.; Baute, J.; et al. Genetic properties of the MAGIC maize population: A new platform for high definition QTL mapping in Zea mays. Genome Biol. 2015, 16, 167. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Y.; Tong, H.; Yang, X.; Xu, S.; Pan, Q.; Qiao, F.; Raihan, M.S.; Luo, Y.; Liu, H.; Zhang, X.; et al. Genome-wide dissection of the maize ear genetic architecture using multiple populations. New Phytol. 2016, 210, 1095–1106. [Google Scholar] [CrossRef] [Green Version]
- McMullen, M.D.; Kresovich, S.; Villeda, H.S.; Bradbury, P.; Li, H.; Sun, Q. Supporting online material for: Genetic properties of the maize nested association mapping population. Science 2009, 325, 737–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buckler, E.S.; Holland, J.B.; Bradbury, P.J.; Acharya, C.B.; Brown, P.J.; Browne, C.; Ersoz, E.; Flint-Garcia, S.; Garcia, A.; Glaubitz, J.C.; et al. The genetic architecture of maize flowering time. Science 2009, 325, 714–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, F.; Bradbury, P.J.; Brown, P.J.; Hung, H.; Sun, Q.; Flint-Garcia, S.; Rocheford, T.R.; McMullen, M.D.; Holland, J.B.; Buckler, E.S. Genome-wide association study of leaf architecture in the maize nested association mapping population. Nat. Genet. 2011, 43, 159–162. [Google Scholar] [CrossRef] [PubMed]
- Peiffer, J.A.; Romay, M.C.; Gore, M.A.; Flint-Garcia, S.A.; Zhang, Z.; Millard, M.J. The genetic architecture of maize height. Genetics 2014, 196, 1337–1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fragoso, C.A.; Moreno, M.; Wang, Z.; Heffelfinger, C.; Arbelaez, L.J.; Aguirre, J.A.; Franco, N.; Romero, L.E.; Labadie, K.; Zhao, H.; et al. Genetic architecture of a rice nested association mapping population. G3 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Bajgain, P.; Rouse, M.N.; Tsilo, T.J.; Macharia, G.K.; Bhavani, S.; Jin, Y.; Anderson, J.A. Nested association mapping of stem rust resistance in wheat using genotyping by sequencing. PLoS ONE 2016, 11, e0155760. [Google Scholar] [CrossRef] [Green Version]
- Maurer, A.; Draba, V.; Jiang, Y.; Schnaithmann, F.; Sharma, R.; Schumann, E.; Kilian, B.; Reif, J.C.; Pillen, K. Modelling the genetic architecture of flowering time control in barley through nested association mapping. BMC Genom. 2015, 16, 290. [Google Scholar] [CrossRef] [Green Version]
- Song, Q.; Yan, L.; Quigley, C.; Jordan, B.D.; Fickus, E.; Schroeder, S.; Song, B.H.; Charles An, Y.Q.; Hyten, D.; Nelson, R.; et al. Genetic characterization of the soybean nested association mapping population. Plant Genome 2017, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouchet, S.; Olatoye, M.O.; Marla, S.R.; Perumal, R.; Tesso, T.; Yu, J.; Tuinstra, M.; Morris, G.P. Increased power to dissect adaptive traits in global sorghum diversity using a nested association mapping population. Genetics 2017, 206, 573–585. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Guo, C.; Wang, B.; Ye, J.; Liu, M.; Wu, Z.; Xiao, Y.; Zhang, Q.; Li, H.; King, G.J.; et al. Genetic properties of a nested association mapping population constructed with semi-winter and spring oilseed rapes. Front. Plant Sci. 2018, 9, 1740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garris, A.J.; Tai, T.H.; Coburn, J.; Kresovich, S.; McCouch, S. Genetic structure and diversity in Oryza sativa L. Genetics 2005, 169, 1631. [Google Scholar] [CrossRef] [Green Version]
- Travis, A.J.; Norton, G.J.; Datta, S.; Sarma, R.; Dasgupta, T.; Savio, F.L.; Macaulay, M.; Hedley, P.E.; McNally, K.L.; Sumon, M.H.; et al. Assessing the genetic diversity of rice originating from Bangladesh, Assam and west Bengal. Rice (New York) 2015, 8, 35. [Google Scholar] [CrossRef] [Green Version]
- Kihupi, A.; Angeles, E.; Khush, G. Genetic analysis of resistance to bacterial blight, Xanthomonas oryzae pv. oryzae, in rice, Oryza sativa L. Euphytica 2001, 117, 39–46. [Google Scholar] [CrossRef]
- Takehisa, H.; Yasuda, M.; Fukuta, Y.; Kobayashi, N.; Hayashi, N.; Nakashita, H.; Abe, T.; Sato, T. Genetic analysis of resistance genes in an Indica-type rice (Oryza sativa L.), Kasalath, using DNA markers. Breed. Sci. 2009, 59, 253–260. [Google Scholar] [CrossRef] [Green Version]
- Kojima, Y.; Ebana, K.; Fukuoka, S.; Nagamine, T.; Kawase, M. Development of an RFLP-based rice diversity research set of germplasm. Breed. Sci. 2005, 55, 431–440. [Google Scholar] [CrossRef] [Green Version]
- Izawa, T.; Oikawa, T.; Sugiyama, N.; Tanisaka, T.; Yano, M.; Shimamoto, K. Phytochrome mediates the external light signal to repress FT orthologs in photoperiodic flowering of rice. Genes Dev. 2002, 16, 2006–2020. [Google Scholar] [CrossRef] [Green Version]
- Kojima, S.; Takahashi, Y.; Kobayashi, Y.; Monna, L.; Sasaki, T.; Araki, T.; Yano, M. Hd3a, a rice ortholog of the Arabidopsis FT gene, promotes transition to flowering downstream of Hd1 under short-day conditions. Plant Cell Physiol. 2002, 43, 1096–1105. [Google Scholar] [CrossRef] [Green Version]
- Yano, M.; Katayose, Y.; Ashikari, M.; Yamanouchi, U.; Monna, L.; Fuse, T.; Baba, T.; Yamamoto, K.; Umehara, Y.; Nagamura, Y.; et al. Hd1, a major photoperiod sensitivity quantitative trait locus in rice, is closely related to the arabidopsis flowering time gene CONSTANS. Plant Cell 2000, 12, 2473–2483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, W.; Xing, Y.; Weng, X.; Zhao, Y.; Tang, W.; Wang, L.; Zhou, H.; Yu, S.; Xu, C.; Li, X.; et al. Natural variation in Ghd7 is an important regulator of heading date and yield potential in rice. Nat. Genet. 2008, 40, 761–767. [Google Scholar] [CrossRef]
- Doi, K.; Izawa, T.; Fuse, T.; Yamanouchi, U.; Kubo, T.; Shimatani, Z.; Yano, M.; Yoshimura, A. Ehd1, a B-type response regulator in rice, confers short-day promotion of flowering and controls FT-like gene expression independently of Hd1. Genes Dev. 2004, 18, 926–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, E.; Yonemaru, J.-i.; Yamamoto, T.; Yano, M. OGRO: The overview of functionally characterized genes in rice online database. Rice 2012, 5, 26. [Google Scholar] [CrossRef] [Green Version]
- Poland, J.A.; Brown, P.J.; Sorrells, M.E.; Jannink, J.-L. Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by- sequencing approach. PLoS ONE 2012, 7, e032253. [Google Scholar] [CrossRef] [Green Version]
- Furuta, T.; Ashikari, M.; Jena, K.K.; Doi, K.; Reuscher, S. Adapting genotyping-by-sequencing for rice F2 populations. G3 (Bethesda) 2017, 7, 881–893. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Wang, X.; Ding, X.; Zheng, X.; Yang, Z.; Xu, C.; Hu, Z. Genomic selection of agronomic traits in hybrid rice using an NCII population. Rice 2018, 11, 32. [Google Scholar] [CrossRef] [Green Version]
- Fujino, K.; Yamanouchi, U.; Nonoue, Y.; Obara, M.; Yano, M. Switching genetic effects of the flowering time gene Hd1 in LD conditions by Ghd7 and OsPRR37 in rice. Breed Sci. 2019, 69, 127–132. [Google Scholar] [CrossRef] [Green Version]
- Valluru, R.; Gazave, E.E.; Fernandes, S.B.; Ferguson, J.N.; Lozano, R.; Hirannaiah, P.; Zuo, T.; Brown, P.J.; Leakey, A.D.B.; Gore, M.A.; et al. Deleterious Mutation Burden and Its Association with Complex Traits in Sorghum (Sorghum bicolor). Genetics 2019, 211, 1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kremling, K.A.G.; Chen, S.Y.; Su, M.H.; Lepak, N.K.; Romay, M.C.; Swarts, K.L.; Lu, F.; Lorant, A.; Bradbury, P.J.; Buckler, E.S. Dysregulation of expression correlates with rare-allele burden and fitness loss in maize. Nature 2018, 555, 520–523. [Google Scholar] [CrossRef]
- Gage, J.L.; Monier, B.; Giri, A.; Buckler, E.S. Ten Years of the Maize Nested Association Mapping Population: Impact, Limitations, and Future Directions. Plant Cell 2020, 32, 2083–2093. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020; Available online: https://www.R-project.org/ (accessed on 1 March 2021).
- Dellaporta, S.L.; Wood, J.; Hicks, J.B. A plant DNA minipreparation: Version II. Plant Mol. Biol. Rep. 1983, 1, 19–21. [Google Scholar] [CrossRef]
- Glaubitz, J.C.; Casstevens, T.M.; Lu, F.; Harriman, J.; Elshire, R.J.; Sun, Q.; Buckler, E.S. TASSEL-GBS: A high capacity genotyping by sequencing analysis pipeline. PLoS ONE 2014, 9, e090346. [Google Scholar] [CrossRef]
- Kawahara, Y.; de la Bastide, M.; Hamilton, J.P.; Kanamori, H.; McCombie, W.R.; Ouyang, S.; Schwartz, D.C.; Tanaka, T.; Wu, J.; Zhou, S.; et al. Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice 2013, 6, 4. [Google Scholar] [CrossRef] [Green Version]
- Swarts, K.; Li, H.; Romero Navarro, J.A.; An, D.; Romay, M.C.; Hearne, S.; Acharya, C.; Glaubitz, J.C.; Mitchell, S.; Elshire, R.J.; et al. Novel methods to optimize genotypic imputation for low-coverage, next-generation sequence data in crop plants. Plant Genome 2014, 7. [Google Scholar] [CrossRef] [Green Version]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef]
- Stacklies, W.; Redestig, H.; Scholz, M.; Walther, D.; Selbig, J. PCAmethods—A bioconductor package providing PCA methods for incomplete data. Bioinformatics 2007, 23, 1164–1167. [Google Scholar] [CrossRef] [PubMed]
- Arends, D.; Prins, P.; Jansen, R.C.; Broman, K.W. R/qtl: High-throughput multiple QTL mapping. Bioinformatics 2010, 26, 2990–2992. [Google Scholar] [CrossRef] [Green Version]
- Churchill, G.A.; Doerge, R.W. Empirical threshold values for quantitative trait mapping. Genetics 1994, 963–971. [Google Scholar] [CrossRef]
- Li, H.; Bradbury, P.; Ersoz, E.; Buckler, E.S.; Wang, J. Joint QTL linkage mapping for multiple-cross mating design sharing one common parent. PLoS ONE 2011, 6, e17573. [Google Scholar] [CrossRef] [Green Version]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007. [Google Scholar] [CrossRef] [PubMed]
- Haynes, W. Bonferroni Correction. In Encyclopedia of Systems Biology; Dubitzky, W., Wolkenhauer, O., Cho, K.-H., Yokota, H., Eds.; Springer: New York, NY, USA, 2013; p. 154. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family Name | Founder’s Name | WRC No. 1 | F2 | F5 | Residual Rate |
|---|---|---|---|---|---|
| WNAM02 | Kasalath | WRC02 | 233 | 109 | 46.78% |
| WNAM29 | Kalo Dhan | WRC29 | 219 | 163 | 74.43% |
| WNAM31 | Shoni | WRC31 | 174 | 121 | 69.54% |
| WNAM35 | ARC5955 | WRC35 | 229 | 137 | 59.83% |
| WNAM39 | Badari Dhan | WRC39 | 213 | 126 | 59.15% |
| WNAM72 | DV85 | - | - | 107 | - |
| WNAM73 | ARC10313 | - | - | 132 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kitony, J.K.; Sunohara, H.; Tasaki, M.; Mori, J.-I.; Shimazu, A.; Reyes, V.P.; Yasui, H.; Yamagata, Y.; Yoshimura, A.; Yamasaki, M.; et al. Development of an Aus-Derived Nested Association Mapping (Aus-NAM) Population in Rice. Plants 2021, 10, 1255. https://doi.org/10.3390/plants10061255
Kitony JK, Sunohara H, Tasaki M, Mori J-I, Shimazu A, Reyes VP, Yasui H, Yamagata Y, Yoshimura A, Yamasaki M, et al. Development of an Aus-Derived Nested Association Mapping (Aus-NAM) Population in Rice. Plants. 2021; 10(6):1255. https://doi.org/10.3390/plants10061255
Chicago/Turabian StyleKitony, Justine K., Hidehiko Sunohara, Mikako Tasaki, Jun-Ichi Mori, Akihisa Shimazu, Vincent P. Reyes, Hideshi Yasui, Yoshiyuki Yamagata, Atsushi Yoshimura, Masanori Yamasaki, and et al. 2021. "Development of an Aus-Derived Nested Association Mapping (Aus-NAM) Population in Rice" Plants 10, no. 6: 1255. https://doi.org/10.3390/plants10061255
APA StyleKitony, J. K., Sunohara, H., Tasaki, M., Mori, J.-I., Shimazu, A., Reyes, V. P., Yasui, H., Yamagata, Y., Yoshimura, A., Yamasaki, M., Nishiuchi, S., & Doi, K. (2021). Development of an Aus-Derived Nested Association Mapping (Aus-NAM) Population in Rice. Plants, 10(6), 1255. https://doi.org/10.3390/plants10061255