
Towards a Joint International Database: Alignment of SSR Marker Data for European Collections of Cherry Germplasm

 , , , and
, , , and
Abstract
1. Introduction
2. Results
2.1. Standardisation of the Great Britain (GB) Dataset
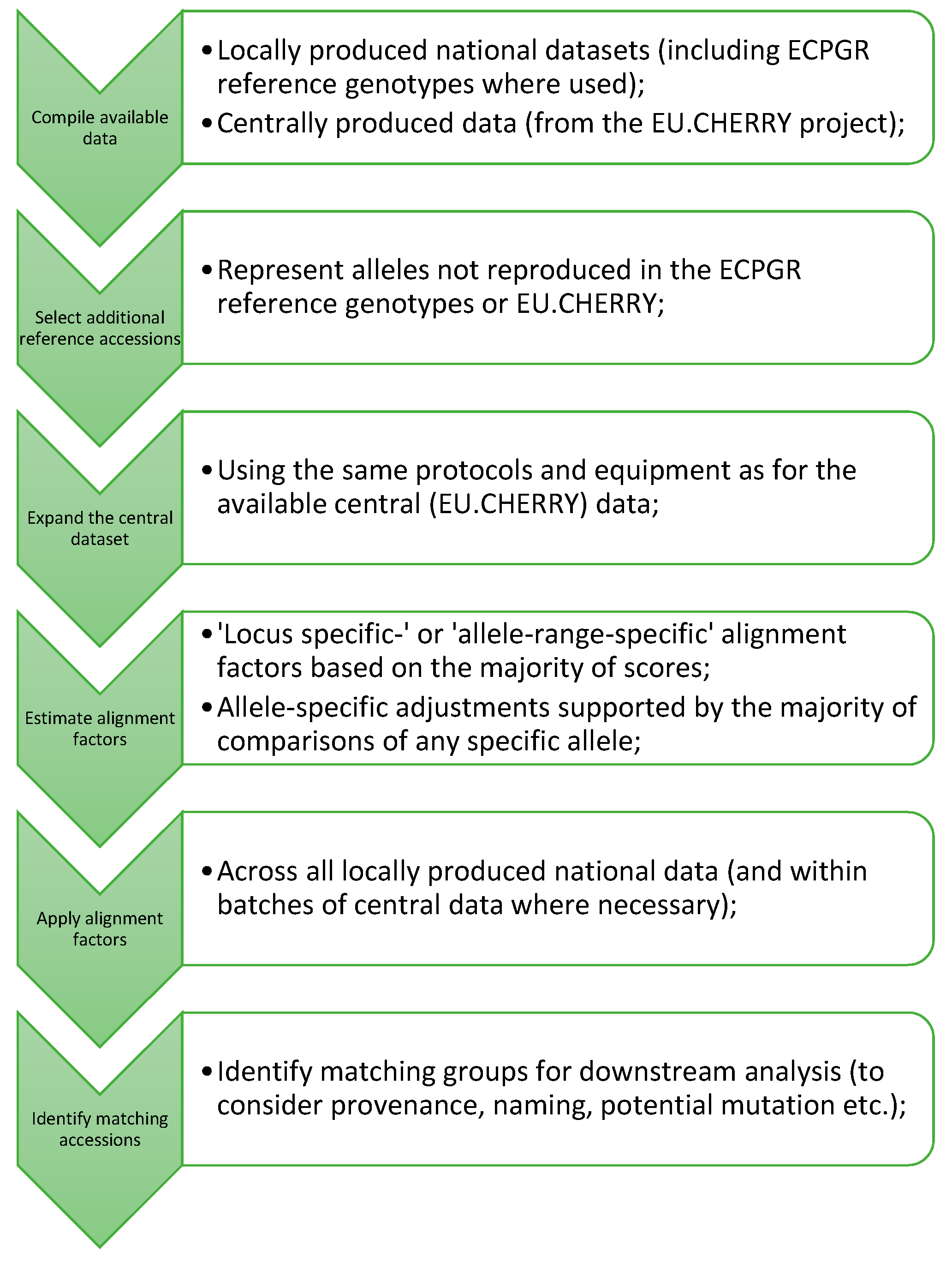
2.2. Production of a Central Dataset
2.3. Alignment of National Datasets to the Central Data
2.4. Diversity Metrics for the Aligned Dataset
2.5. Identification of Matching Accessions
3. Discussion
3.1. Consistency of Alignment Factors
3.2. Selection of Reference Samples
3.3. Identification of Errors Through Data Alignment
3.4. Comparison to Alignment Attempts in Other Species
3.5. Discriminatory Power of the Aligned Dataset
3.6. Genetic Diversity in the Aligned Dataset
3.7. Summary
4. Materials and Methods
4.1. National De Novo Genotyping of GB Samples
4.2. Compilation of National Datasets and Central Data from EU.CHERRY
4.3. Selection of Additional Reference Accessions
4.4. Expansion of the Central SSR Dataset
4.5. Alignment of Data and Estimation of Alignment Factors
4.6. Identification of Matching Accessions
4.7. Generation of Diversity Metrics
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Genesys Global Portal on Plant Genetic Resources. Available online: www.genesys-pgr.org (accessed on 11 February 2021).
- Hardy, O.J.; Vekemans, X. SPAGEDi: A versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol. Ecol. Notes 2002, 2, 618–620. [Google Scholar] [CrossRef]
- Benediková, D.; Giovannini, D. Review on genetic resources in the ECPGR Prunus working group. Acta Hortic. 2013, 981, 43–51. [Google Scholar] [CrossRef]
- Delmas, M.; Höfer, M.; Giovannini, D. Passport and Priority Descriptors for Cherry. 2018. Available online: https://www.ecpgr.cgiar.org/fileadmin/templates/ecpgr.org/upload/WG_UPLOADS_PHASE_IX/PRUNUS/EuCherry_descriptors_GUIDELINESv1_FINAL__corr_28_08_2018.pdf (accessed on 26 May 2021).
- Peakall, R.; Smouse, P.E. GENALEX 6: Genetic analysis in excel. Population genetic software for teaching and research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar] [CrossRef]
- Dirlewanger, E.; Cosson, P.; Tavaud, M.; Aranzana, M.J.; Poizat, C.; Zanetto, A.; Arus, P.; Laigret, F. Development of microsatellite markers in peach Prunus persica (L.) Batsch and their use in genetic diversity analysis in peach and sweet cherry (Prunus avium L.). Theor. Appl. Genet. 2002, 105, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Struss, D.; Ahmad, R.; Southwick, S.M.; Boritzki, M. Analysis of sweet cherry (Prunus avium L.) cultivars using SSR and AFLP markers. J. Am. Soc. Hortic. Sci. 2003, 128, 904–909. [Google Scholar] [CrossRef]
- Wünsch, A.; Hormaza, J.I. Molecular characterisation of sweet cherry (Prunus avium L.) genotypes using peach [Prunus persica (L.) Batsch] SSR sequences. Heredity 2002, 89, 56–63. [Google Scholar] [CrossRef]
- Clarke, J.B.; Tobutt, K.R. Development and characterization of polymorphic microsatellites from Prunus avium “Napoleon”. Mol. Ecol. Notes 2003, 3, 578–580. [Google Scholar] [CrossRef]
- Wünsch, A.; Hormaza, J.I. Molecular evaluation of genetic diversity and S-allele composition of local Spanish sweet cherry (Prunus avium L.) cultivars. Genet. Resour. Crop. Evol. 2004, 51, 635–641. [Google Scholar] [CrossRef]
- Gulen, H.; Ipek, A.; Ergin, S.; Akcay, E.; Eris, A. Assessment of genetic relationships among 29 introduced and 49 local sweet cherry accessions in Turkey using AFLP and SSR markers. J. Hortic. Sci. Biotechnol. 2010, 85, 427–431. [Google Scholar] [CrossRef]
- Stanys, V.; Baniulis, D.; Morkunaite-Haimi, S.; Siksnianiene, J.B.; Frercks, B.; Gelvonauskiene, D.; Stepulaitiene, I.; Staniene, G.; Siksnianas, T. Characterising the genetic diversity of Lithuanian sweet cherry (Prunus avium L.) cultivars using SSR markers. Sci. Hortic. 2012, 142, 136–142. [Google Scholar] [CrossRef]
- Marchese, A.; Giovannini, D.; Leone, A.; Mafrica, R.; Palasciano, M.; Cantini, C.; Di Vaio, C.; De Salvador, F.R.; Giacalone, G.; Caruso, T.; et al. S-genotype identification, genetic diversity and structure analysis of Italian sweet cherry germplasm. Tree Genet. Genomes 2017, 13, 20. [Google Scholar] [CrossRef]
- Marchese, A.; Tobutt, K.R.; Raimondo, A.; Motisi, A.; Bošković, R.I.; Clarke, J.; Caruso, T. Morphological characteristics, microsatellite fingerprinting and determination of incompatibility genotypes of Sicilian sweet cherry cultivars. J. Hortic. Sci. Biotechnol. 2007, 82, 41–48. [Google Scholar] [CrossRef]
- Ivanovych, Y.; Volkov, R. Genetic relatedness of sweet cherry (Prunus avium L.) cultivars from Ukraine determined by microsatellite markers. J. Hortic. Sci. Biotechnol. 2018, 93, 64–72. [Google Scholar] [CrossRef]
- Lacis, G.; Rashal, I.; Ruisa, S.; Trajkovski, V.; Iezzoni, A.F. Assessment of genetic diversity of Latvian and Swedish sweet cherry (Prunus avium L.) genetic resources collections by using SSR (microsatellite) markers. Sci. Hortic. 2009, 121, 451–457. [Google Scholar] [CrossRef]
- Frei, A.; Szalatnay, D.; Zollinger, T.; Frey, J. Molecular characterisation of the national collection of Swiss cherry cultivars. J. Hortic. Sci. Biotechnol. 2010, 85, 277–282. [Google Scholar] [CrossRef]
- Patzak, J.; Henychova, A.; Paprstein, F.; Sedlak, J. Evaluation of genetic variability within sweet cherry (Prunus avium L.) genetic resources by molecular SSR markers. Acta Sci. Pol. Hortorum Cultus 2019, 18, 157–165. [Google Scholar] [CrossRef]
- Sehic, J.; Nybom, H.; Hjeltnes, S.H.; Gasi, F. DNA marker-assisted identification of Prunus accessions. Acta Hortic. 2015, 1101, 153–158. [Google Scholar] [CrossRef]
- Bühlmann, A.; Gassmann, J.; Ingenfeld, A.; Hunziker, K.; Kellerhals, M.; Frey, J.E. Molecular characterisation of the swiss fruit genetic resources. Erwerbs-Obstbau 2015, 57, 29–34. [Google Scholar] [CrossRef]
- Höfer, M.; Braun-Lüllemann, A.; Schiffler, J.; Schuster, M.; Flachowsky, H. Pomological and Molecular Characterization of Sweet Cherry Cultivars (Prunus avium L.) of the German Fruit Genebank; OpenAgrar Repository: Germany, 2021. [Google Scholar] [CrossRef]
- Liu, C.; Qi, X.; Song, L.; Li, Y.; Li, M. Species identification, genetic diversity and population structure of sweet cherry commercial cultivars assessed by SSRs and the gametophytic self-incompatibility locus. Sci. Hortic. 2018, 237, 28–35. [Google Scholar] [CrossRef]
- Mariette, S.; Tavaud, M.; Arunyawat, U.; Capdeville, G.; Millan, M.; Salin, F. Population structure and genetic bottleneck in sweet cherry estimated with SSRs and the gametophytic self-incompatibility locus. BMC Genet. 2010, 11, 77. [Google Scholar] [CrossRef]
- Clarke, J.B.; Tobutt, K.R. A standard set of accessions, microsatellites and genotypes for harmonising the fingerprinting of cherry collections for the ECPGR. Acta Hortic. 2009, 814, 615–618. [Google Scholar] [CrossRef]
- Delmas, M.; Giovannini, D.; Stanivuković, S.; Paprstein, F.; Kaldmäe, H.; Höfer, M.; Lacis, G.; Ordidge, M.; Fernandez, F.; Lateur, M.; et al. Collaborative Action for Updating, Documenting and Communicating the Cherry Patrimonial Richness in EU (EU.CHERRY). 2019. Available online: https://www.ecpgr.cgiar.org/fileadmin/bioversity/publications/pdfs/EU.CHERRY_Final_Activity_Report_final_web_27_09_2019.pdf (accessed on 26 May 2021).
- Jones, C.J.; Edwards, K.J.; Castaglione, S.; Winfield, M.O.; Sala, F.; van de Wiel, C.; Bredemeijer, G.; Vosman, B.; Matthes, M.; Daly, A.; et al. Reproducibility testing of RAPD, AFLP and SSR markers in plants by a network of European laboratories. Mol. Breed. 1997, 3, 381–390. [Google Scholar] [CrossRef]
- Bredemeijer, G.; Cooke, R.; Ganal, M.; Peeters, R.; Isaac, P.; Noordijk, Y.; Rendell, S.; Jackson, J.; Röder, M.; Wendehake, K.; et al. Construction and testing of a microsatellite database containing more than 500 tomato varieties. Theor. Appl. Genet. 2002, 105, 1019–1026. [Google Scholar] [CrossRef]
- Röder, M.; Wendehake, K.; Korzun, V.; Bredemeijer, G.; Laborie, D.; Bertrand, L.; Isaac, P.; Rendell, S.; Jackson, J.; Cooke, R.; et al. Construction and analysis of a microsatellite-based database of European wheat varieties. Theor. Appl. Genet. 2002, 106, 67–73. [Google Scholar] [CrossRef]
- George, M.L.C.; Regalado, E.; Li, W.; Cao, M.; Dahlan, M.; Pabendon, M.; Warburton, M.L.; Xianchun, X.; Hoisington, D. Molecular characterization of Asian maize inbred lines by multiple laboratories. Theor. Appl. Genet. 2004, 109, 80–91. [Google Scholar] [CrossRef]
- This, P.; Jung, A.; Boccacci, P.; Borrego, J.; Botta, R.; Costantini, L.; Crespan, M.; Dangl, G.S.; Eisenheld, C.; Ferreira-Monteiro, F.; et al. Development of a standard set of microsatellite reference alleles for identification of grape cultivars. Theor. Appl. Genet. 2004, 109, 1448–1458. [Google Scholar] [CrossRef]
- Cryer, N.C.; Fenn, M.G.E.; Turnbull, C.J.; Wilkinson, M.J. Allelic size standards and reference genotypes to unify international cocoa (Theobroma cacao L.) microsatellite data. Genet. Resour. Crop. Evol. 2006, 53, 1643–1652. [Google Scholar] [CrossRef]
- Baric, S.; Monschein, S.; Hofer, M.; Grill, D.; Dalla Via, J. Comparability of genotyping data obtained by different procedures–an inter-laboratory survey. J. Hortic. Sci. Biotechnol. 2008, 83, 183–190. [Google Scholar] [CrossRef]
- Doveri, S.; Sabino Gil, F.; Díaz, A.; Reale, S.; Busconi, M.; da Câmara Machado, A.; Martín, A.; Fogher, C.; Donini, P.; Lee, D. Standardization of a set of microsatellite markers for use in cultivar identification studies in olive (Olea europaea L.). Sci. Hortic. 2008, 116, 367–373. [Google Scholar] [CrossRef]
- Antonius, K.; Karhu, S.; Kaldmae, H.; Lacis, G.; Rugenius, R.; Baniulis, D.; Sasnauskas, A.; Schulte, E.; Kuras, A.; Korbin, M.; et al. Development of the Northern European Ribes core collection based on a microsatellite (SSR) marker diversity analysis. Plant. Genet. Resour. Charact. Util. 2012, 10, 70–73. [Google Scholar] [CrossRef]
- Stephenson, J.J.; Campbell, M.R.; Hess, J.E.; Kozfkay, C.; Matala, A.P.; McPhee, M.V.; Moran, P.; Narum, S.R.; Paquin, M.M.; Schlei, O.; et al. A centralized model for creating shared, standardized, microsatellite data that simplifies inter-laboratory collaboration. Conserv. Genet. 2009, 10, 1145–1149. [Google Scholar] [CrossRef]
- Ellis, J.S.; Gilbey, J.; Armstrong, A.; Balstad, T.; Cauwelier, E.; Cherbonnel, C.; Consuegra, S.; Coughlan, J.; Cross, T.F.; Crozier, W.; et al. Microsatellite standardization and evaluation of genotyping error in a large multi-partner research programme for conservation of Atlantic salmon (Salmo salar L.). Genetica 2011, 139, 353–367. [Google Scholar] [CrossRef]
- Urrestarazu, J.; Denance, C.; Ravon, E.; Guyader, A.; Guisnel, R.; Feugey, L.; Poncet, C.; Lateur, M.; Houben, P.; Ordidge, M.; et al. Analysis of the genetic diversity and structure across a wide range of germplasm reveals prominent gene flow in apple at the European level. BMC Plant. Biol. 2016, 16. [Google Scholar] [CrossRef]
- Denancé, C.; Muranty, H.; Durel, C.-E. MUNQ-Malus UNiQue Genotype Code for Grouping Apple Accessions Corresponding to a Unique Genotypic Profile, V1 ed.; Portail Data INRAE: France, 2020. [Google Scholar] [CrossRef]
- Muranty, H.; Denancé, C.; Feugey, L.; Crépin, J.-L.; Barbier, Y.; Tartarini, S.; Ordidge, M.; Troggio, M.; Lateur, M.; Nybom, H.; et al. Using whole-genome SNP data to reconstruct a large multi-generation pedigree in apple germplasm. BMC Plant. Biol. 2020, 20, 2. [Google Scholar] [CrossRef] [PubMed]
- Deemer, D.L.; Nelson, C.D. Standardized SSR allele naming and binning among projects. Biotechniques 2010, 49, 835–836. [Google Scholar] [CrossRef] [PubMed]
- Sutton, J.T.; Robertson, B.C.; Jamieson, I.G. Dye shift: A neglected source of genotyping error in molecular ecology. Mol. Ecol. Resour. 2011, 11, 514–520. [Google Scholar] [CrossRef] [PubMed]
- Bruland, O.; Almqvist, E.W.; Goldberg, Y.P.; Boman, H.; Hayden, M.R.; Knappskog, P.M. Accurate determination of the number of CAG repeats in the Huntington disease gene using a sequence-specific internal DNA standard. Clin. Genet. 1999, 55, 198–202. [Google Scholar] [CrossRef]
- Leclair, B.; Frégeau, C.J.; Bowen, K.L.; Fourney, R.M. Precision and accuracy in fluorescent short tandem repeat DNA typing: Assessment of benefits imparted by the use of allelic ladders with the AmpFlSTR®Profiler Plus™ kit. Electrophoresis 2004, 25, 790–796. [Google Scholar] [CrossRef]
- Fernandez, I.; Marti, A.; Athanson, B.; Koepke, T.; Font, I.; Forcada, C.; Dhingra, A.; Oraguzie, N. Genetic diversity and relatedness of sweet cherry (Prunus avium L.) cultivars based on single nucleotide polymorphic markers. Front. Plant Sci. 2012, 3, 116. [Google Scholar] [CrossRef]
- Cabrera, A.; Rosyara, U.R.; De Franceschi, P.; Sebolt, A.; Sooriyapathirana, S.S.; Dirlewanger, E.; Quero-Garcia, J.; Schuster, M.; Iezzoni, A.F.; van der Knaap, E. Rosaceae conserved orthologous sequences marker polymorphism in sweet cherry germplasm and construction of a SNP-based map. Tree Genet. Genomes 2012, 8, 237–247. [Google Scholar] [CrossRef]
- Ganopoulos, I.; Tsaballa, A.; Xanthopoulou, A.; Madesis, P.; Tsaftaris, A. Sweet cherry cultivar identification by High-Resolution-Melting (HRM) analysis using gene-based SNP markers. Plant. Mol. Biol. Report. 2013, 31, 763–768. [Google Scholar] [CrossRef]
- Moran, P.; Teel, D.J.; LaHood, E.S.; Drake, J.; Kalinowski, S. Standardising multi-laboratory microsatellite data in Pacific salmon: An historical view of the future. Ecol. Freshw. Fish. 2006, 15, 597–605. [Google Scholar] [CrossRef]
- Rosyara, U.R.; Bink, M.C.A.M.; van de Weg, E.; Zhang, G.; Wang, D.; Sebolt, A.; Dirlewanger, E.; Quero-Garcia, J.; Schuster, M.; Iezzoni, A.F. Fruit size QTL identification and the prediction of parental QTL genotypes and breeding values in multiple pedigreed populations of sweet cherry. Mol. Breed. 2013, 32, 875–887. [Google Scholar] [CrossRef]
- Sandefur, P.; Oraguzie, N.; Peace, C. A DNA test for routine prediction in breeding of sweet cherry fruit color, Pav-Rf-SSR. Mol. Breed. 2016, 36, 33. [Google Scholar] [CrossRef]
- Höfer, M.; Peil, A. Phenotypic and genotypic characterization in the collection of sour and duke cherries (Prunus cerasus and ×P. ×gondouini) of the Fruit Genebank in Dresden-Pillnitz, Germany. Genet. Resour. Crop. Evol. 2015, 62, 551–566. [Google Scholar] [CrossRef]
- Xuan, H.; Wang, R.; Büchele, M.; Hartmann, W.; Möller, O. Microsatellite markers (SSR) as a tool to assist in identification of sweet (Prunus avium) and sour cherry (Prunus cerasus). Acta Hortic. 2009, 839, 507–514. [Google Scholar] [CrossRef]
- Edge-Garza, D.A.; Rowland, T.V.; Haendiges, S.; Peace, C. A high-throughput and cost-efficient DNA extraction protocol for the tree fruit crops of apple, sweet cherry, and peach relying on silica beads during tissue sampling. Mol. Breed. 2014, 34, 2225–2228. [Google Scholar] [CrossRef]


{kind=link}
| Locus | Allele Calls Compared | Alignment Factor (bp) | Allele Calls in Agreement with Alignment Factor (%) | Null Alleles (%) | Range of Error from Alignment Factor (bp) |
|---|---|---|---|---|---|
| CPPCT022 | 75 | 0 | 81% | 13% | 1 |
| CPPCT006 | 84 | 0 | 99% | 1% | n/a 2 |
| EMPaS02 | 78 | −3 or −4 1 | 90% | 8% | −1 |
| BPPCT037 | 75 | 1 | 77% | 3% | −1 |
| EMPaS06 | 72 | 0 | 89% | 4% | −1 to 1 |
| EMPa004 | 83 | 0 | 90% | 10% | n/a |
| EMPa017 | 68 | 0 | 82% | 9% | 1 |
| EMPa018 | 72 | 0 | 94% | 6% | n/a |
| EMPaS12 | 74 | -8 | 95% | 3% | 1 or −7 |
| EMPaS14 | 80 | 0 | 83% | 16% | 1 |
| Country | Allele Calls 1 | EMPa002 | CPSCT038 | CPPCT022 | CPPCT006 | BPPCT034 | EMPaS02 | PAV-Rf-SSR | BPPCT037 | EMPaS06 | EMPaS12 | EMPaS14 | EMPa004 | EMPa018 | EMPa017 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| France | Compared | 41 | n/a | n/a | n/a | 57 | 55 | n/a | n/a | 49 | 51 | 50 | 53 | 50 | 44 |
| In agreement (%) | 90% | n/a | n/a | n/a | 79% | 95% | n/a | n/a | 90% | 78% | 88% | 94% | 94% | 89% | |
| Null (%) | 10% | n/a | n/a | n/a | 21% | 5% | n/a | n/a | 6% | 18% | 6% | 6% | 6% | 7% | |
| Total | 100% | 100% | 100% | 96% | 96% | 94% | 100% | 100% | 95% | ||||||
| Germany | Compared | 72 | n/a | 78 | 82 | n/a | 73 | n/a | 83 | 79 | 77 | 76 | n/a | n/a | 57 |
| In agreement (%) | 99% | n/a | 97% | 99% | n/a | 100% | n/a | 94% | 95% | 97% | 99% | n/a | n/a | 84% | |
| Null (%) | 1% | n/a | 0% | 1% | n/a | 0% | n/a | 1% | 0% | 1% | 0% | n/a | n/a | 2% | |
| Total | 100% | 97% | 100% | 100% | 95% | 95% | 99% | 99% | 86% | ||||||
| Great Britain | Compared | 65 | 64 | 77 | 81 | 78 | 83 | 61 | 80 | 81 | 80 | 78 | 79 | 76 | 67 |
| In agreement (%) | 80% | 61% | 86% | 91% | 74% | 85% | 46% | 85% | 96% | 97% | 95% | 96% | 97% | 72% | |
| Null (%) | 18% | 36% | 4% | 3% | 24% | 5% | 52% | 3% | 0% | 1% | 1% | 4% | 3% | 6% | |
| Total | 98% | 97% | 89% | 94% | 97% | 90% | 98% | 87% | 96% | 99% | 96% | 100% | 100% | 78% | |
| Italy | Compared | 39 | n/a | 44 | 50 | n/a | 53 | n/a | 54 | 55 | 60 | n/a | n/a | n/a | 48 |
| In agreement (%) | 97% | n/a | 84% | 78% | n/a | 87% | n/a | 69% | 78% | 82% | n/a | n/a | n/a | 77% | |
| Null (%) | 0% | n/a | 5% | 12% | n/a | 6% | n/a | 11% | 5% | 8% | n/a | n/a | n/a | 2% | |
| Total | 97% | 89% | 90% | 92% | 80% | 84% | 90% | 79% | |||||||
| Sweden | Compared | 41 | n/a | 34 | 39 | n/a | n/a | n/a | 43 | n/a | 39 | n/a | n/a | n/a | n/a |
| In agreement (%) | 78% | n/a | 91% | 97% | n/a | n/a | n/a | 65% | n/a | 62% | n/a | n/a | n/a | n/a | |
| Null (%) | 12% | n/a | 9% | 3% | n/a | n/a | n/a | 16% | n/a | 8% | n/a | n/a | n/a | n/a | |
| Total | 90% | 100% | 100% | 81% | 69% | ||||||||||
| Switzerland | Compared | n/a | n/a | 15 | 12 | n/a | 15 | n/a | 14 | 14 | 14 | 13 | n/a | n/a | n/a |
| In agreement (%) | n/a | n/a | 67% | 83% | n/a | 100% | n/a | 93% | 86% | 93% | 69% | n/a | n/a | n/a | |
| Null (%) | n/a | n/a | 0% | 0% | n/a | 0% | n/a | 7% | 0% | 0% | 0% | n/a | n/a | n/a | |
| Total | 67% | 83% | 100% | 100% | 86% | 93% | 69% |
| Locus | Allele no. | N | HO | HE | PIC | NE-I | NE-SI |
|---|---|---|---|---|---|---|---|
| EMPa002 | 15 | 844 | 0.47 | 0.46 | 0.37 | 0.38 | 0.62 |
| CPSCT038 | 6 | 209 | 0.52 | 0.54 | 0.49 | 0.26 | 0.54 |
| CPPCT022 | 17 | 1111 | 0.66 | 0.68 | 0.62 | 0.16 | 0.45 |
| CPPCT006 | 22 | 1122 | 0.73 | 0.75 | 0.70 | 0.11 | 0.40 |
| BPPCT034 | 19 | 376 | 0.76 | 0.74 | 0.70 | 0.10 | 0.41 |
| EMPaS02 | 17 | 1252 | 0.76 | 0.80 | 0.77 | 0.07 | 0.37 |
| PAV-Rf-SSR | 7 | 120 | 0.74 | 0.73 | 0.70 | 0.11 | 0.41 |
| BPPCT037 | 22 | 1125 | 0.80 | 0.80 | 0.78 | 0.07 | 0.37 |
| EMPaS06 | 16 | 1248 | 0.83 | 0.84 | 0.82 | 0.05 | 0.34 |
| EMPaS12 | 17 | 1275 | 0.78 | 0.78 | 0.74 | 0.09 | 0.38 |
| EMPaS14 | 13 | 1107 | 0.63 | 0.58 | 0.50 | 0.25 | 0.52 |
| EMPa004 | 10 | 423 | 0.78 | 0.71 | 0.66 | 0.14 | 0.43 |
| EMPa018 | 11 | 425 | 0.60 | 0.65 | 0.61 | 0.16 | 0.47 |
| EMPa017 | 11 | 855 | 0.35 | 0.37 | 0.35 | 0.42 | 0.67 |
| Mean | 14.5 | 821 | 0.67 | 0.67 | 0.63 | 0.17 | 0.46 |
| Locus | Allele No. |
|---|---|
| EMPa002 | 30 |
| CPSCT038 | 12 |
| CPPCT022 | 29 |
| CPPCT006 | 27 |
| BPPCT034 | 32 |
| EMPaS02 | 23 |
| PAV-Rf-SSR | 7 |
| BPPCT037 | 30 |
| EMPaS06 | 22 |
| EMPaS12 | 28 |
| EMPaS14 | 27 |
| EMPa004 | 15 |
| EMPa018 | 13 |
| EMPa017 | 17 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ordidge, M.; Litthauer, S.; Venison, E.; Blouin-Delmas, M.; Fernandez-Fernandez, F.; Höfer, M.; Kägi, C.; Kellerhals, M.; Marchese, A.; Mariette, S.; et al. Towards a Joint International Database: Alignment of SSR Marker Data for European Collections of Cherry Germplasm. Plants 2021, 10, 1243. https://doi.org/10.3390/plants10061243
Ordidge M, Litthauer S, Venison E, Blouin-Delmas M, Fernandez-Fernandez F, Höfer M, Kägi C, Kellerhals M, Marchese A, Mariette S, et al. Towards a Joint International Database: Alignment of SSR Marker Data for European Collections of Cherry Germplasm. Plants. 2021; 10(6):1243. https://doi.org/10.3390/plants10061243
Chicago/Turabian StyleOrdidge, Matthew, Suzanne Litthauer, Edward Venison, Marine Blouin-Delmas, Felicidad Fernandez-Fernandez, Monika Höfer, Christina Kägi, Markus Kellerhals, Annalisa Marchese, Stephanie Mariette, and et al. 2021. "Towards a Joint International Database: Alignment of SSR Marker Data for European Collections of Cherry Germplasm" Plants 10, no. 6: 1243. https://doi.org/10.3390/plants10061243
APA StyleOrdidge, M., Litthauer, S., Venison, E., Blouin-Delmas, M., Fernandez-Fernandez, F., Höfer, M., Kägi, C., Kellerhals, M., Marchese, A., Mariette, S., Nybom, H., & Giovannini, D. (2021). Towards a Joint International Database: Alignment of SSR Marker Data for European Collections of Cherry Germplasm. Plants, 10(6), 1243. https://doi.org/10.3390/plants10061243