
Genome-Wide Mapping of Histone H3 Lysine 4 Trimethylation (H3K4me3) and Its Involvement in Fatty Acid Biosynthesis in Sunflower Developing Seeds

 , ,
, ,  , and
, and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Biological Material and Growth Condition
2.2. Isolation and Immunoprecipitation of Chromatin
2.3. Computational Analysis of Sequencing Data
2.3.1. Quality Analysis and Filtering
2.3.2. Mapping Sequences
2.3.3. Aligned Sequences Evaluation and Peak-Calling
2.3.4. ChIP Peaks Annotation and Functional Enrichment Analysis
2.3.5. Motif Analysis: WRI1 Binding Sequences
2.4. mRNA Preparation and cDNA Synthesis
2.5. Quantitative Real-Time PCR
2.6. Analysis of Fatty Acid Composition
2.7. Statistical Analysis
3. Results and Discussion
3.1. Computational Analysis of Sequencing Data
3.2. Distribution of H3K4m3 Histone Modification in the Sunflower Genome
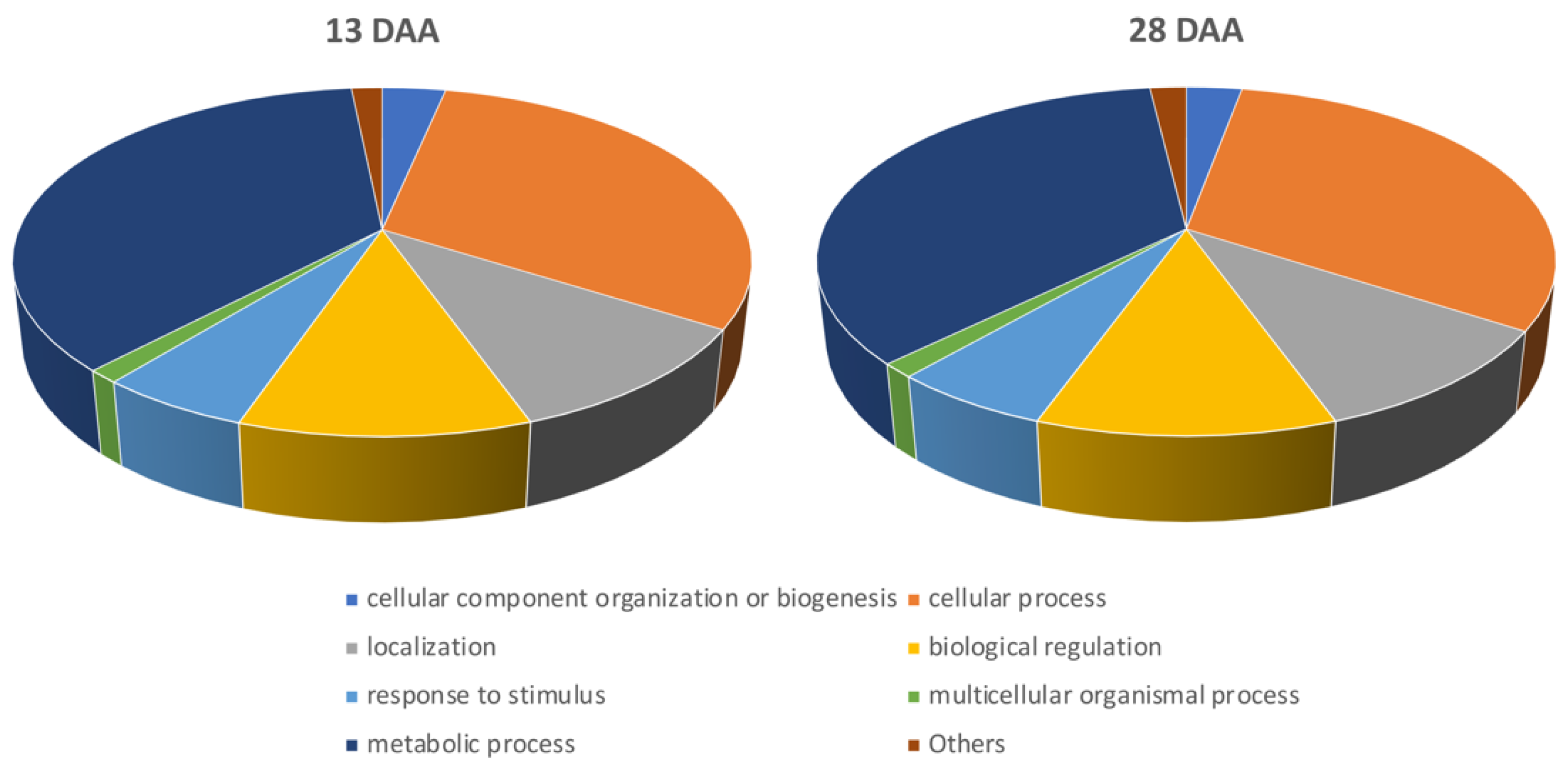
3.3. Functional Enrichment Analysis
3.4. Functional Annotation of Genes Involved in Fatty Acid Biosynthesis
3.5. Fatty Acid Composition in Developing Sunflower Seeds
3.6. WRI1 Gene Expression in Developing Sunflower Seeds
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Berger, S.L. The complex language of chromatin regulation during transcription. Nature 2007, 447, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Pfluger, J.; Wagner, D. Histone modifications and dynamic regulation of genome accessibility in plants. Curr. Opin. Plant Biol. 2007, 10, 645–652. [Google Scholar] [CrossRef]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- Millar, C.B.; Grunstein, M. Genome-wide patterns of histone modifications in yeast. Nat. Rev. Mol. Cell Biol. 2006, 7, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Barrera, L.O.; Ren, B. The transcriptional regulatory code of eukaryotic cells –insights from genome-wide analysis of chromatin organization and transcription factor binding. Curr. Opin. Cell Biol. 2006, 18, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. SnapShot: Histone-modifying enzymes. Cell 2007, 131, 822. [Google Scholar] [CrossRef] [PubMed]
- Schones, D.E.; Zhao, K. Genome-wide approaches to studying chromatin modifications. Nat. Rev. Genet. 2008, 9, 179–191. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zang, C.; Rosenfeld, J.A.; Schones, D.E.; Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Peng, W.; Zhang, M.Q.; et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet. 2008, 40, 897–903. [Google Scholar] [CrossRef]
- Zhou, V.W.; Goren, A.; Bernstein, B.E. Charting histone modifications and the functional organization of mammalian genomes. Nat. Rev. Genet. 2010, 12, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.J.; Tian, L. Roles of dynamic and reversible histone acetylation in plant development and polyploidy. Biochim. Biophys. Acta 2007, 1769, 295–307. [Google Scholar] [CrossRef]
- Luo, M.; Hung, F.-Y.; Yang, S.; Liu, X.; Wu, K. Histone lysine demethylases and their functions in plants. Plant Mol. Biol. Rep. 2013, 32, 558–565. [Google Scholar] [CrossRef]
- Wang, T.; Xing, J.; Liu, X.; Liu, Z.; Yao, Y.; Hu, Z.; Peng, H.; Xin, M.; Zhou, D.X.; Zhang, Y.; et al. Histone acetyltransferase general control non-repressed protein 5 (GCN 5) affects the fatty acid composition of Arabidopsis thaliana seeds by acetylating fatty acid desaturase3 (FAD 3). Plant J. 2016, 88, 794–808. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Deng, S.; Wang, H.; Ye, J.; Wu, H.W.; Sun, H.X.; Chua, N.H. CURLY LEAF regulates gene sets coordinating seed size and lipid biosynthesis. Plant Physiol. 2016, 171, 424–436. [Google Scholar] [CrossRef] [PubMed]
- Thelen, J.J.; Ohlrogge, J.B. Both antisense and sense expression of biotin carboxyl carrier protein isoform 2 inactivates the plastid acetylcoenzyme A carboxylase in Arabidopsis thaliana. Plant J. 2002, 32, 419–431. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, D.H.; Flintham, J.E.; Hills, M.J. Genetic control of storage oil synthesis in seeds of Arabidopsis. Plant Physiol. 2004, 136, 3341–3349. [Google Scholar] [CrossRef]
- Garcés, R.; García, J.M.; Mancha, M. Lipid characterization in seeds of a high oleic acid sunflower mutant. Phytochemistry 1989, 28, 597–2600. [Google Scholar] [CrossRef]
- Osorio, J.; Fernández-Martínez, J.; Mancha, M.; Garcés, R. Mutant sunflowers with high concentration of saturated fatty acids in the oil. Crop Sci. 1995, 35, 739–742. [Google Scholar] [CrossRef]
- Fernández-Martínez, J.M.; Mancha, M.; Osorio, J.; Garcés, R. Sunflower mutant containing high levels of palmitic acid in high oleic background. Euphytica 1997, 97, 113–116. [Google Scholar] [CrossRef]
- Fernández-Moya, V.; Martínez-Force, E.; Garcés, R. Oils from improved high stearic acid sunflower seeds. J. Agric. Food Chem. 2005, 53, 5326–5330. [Google Scholar] [CrossRef]
- Salas, J.J.; Martínez-Force, E.; Harwood, J.L.; Venegas-Calerón, M.; Aznar-Moreno, J.A.; Moreno-Pérez, A.J.; Ruíz-López, N.; Serrano-Vega, M.J.; Graham, I.A.; Mullen, R.T.; et al. Biochemistry of high stearic sunflower, a new source of saturated fats. Prog. Lipid Res. 2014, 55, 30–42. [Google Scholar] [CrossRef]
- Ohlrogge, J.B.; Jaworski, J.G. Regulation of fatty acid synthesis. Annu. Rev. Plant Biol. 1997, 48, 109e136. [Google Scholar] [CrossRef]
- Troncoso-Ponce, M.A.; Kilaru, A.; Cao, X.; Durrett, T.P.; Fan, J.; Jensen, J.K.; Thrower, N.A.; Pauly, M.; Wilkerson, C.; Ohlrogge, J.B. Comparative deep transcriptional profiling of four developing oilseeds. Plant J. 2011, 68, 1014–1027. [Google Scholar] [CrossRef] [PubMed]
- Baud, S.; Santos Mendoza, M.; To, A.; Harscoe, T.E.; Lepiniec, L.; Dubreucq, B. WRINKLED1 specifies the regulatory action of LEAFY COTYLEDON2 towards fatty acid metabolism during seed maturation in Arabidopsis. Plant J. 2007, 50, 825–838. [Google Scholar] [CrossRef] [PubMed]
- Kong, Q.; Yuan, L.; Ma, W. WRINKLED1, a “Master Regulator” in Transcriptional Control of Plant Oil Biosynthesis. Plants 2019, 8, 238. [Google Scholar] [CrossRef]
- Kong, Q.; Yang, Y.; Guo, L.; Yuan, L.; Ma, W. Molecular basis of plant oil biosynthesis: Insights gained from studying the WRINKLED1 transcription factor. Front. Plant Sci. 2020, 11, 24. [Google Scholar] [CrossRef] [PubMed]
- Fei, W.; Yang, S.; Hu, J.; Yang, F.; Qu, G.; Peng, D.; Zhou, B. Research advances of WRINKLED1 (WRI1) in plants. Funct. Plant Biol. 2020, 47, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Focks, N.; Benning, C. Wrinkled1: A novel, low-seed-oil mutant of Arabidopsis with a deficiency in the seed-specific regulation of carbohydrate metabolism. Plant Physiol. 1998, 118, 91–101. [Google Scholar] [CrossRef]
- Baud, S. Seeds as oil factories. Plant Reprod. 2018, 31, 213–235. [Google Scholar] [CrossRef]
- Braybrook, S.A.; Harada, J.J. LECs go crazy in embryo development. Trends Plant Sci. 2008, 13, 624–630. [Google Scholar] [CrossRef]
- Santos-Mendoza, M.; Dubreucq, B.; Baud, S.; Parcy, F.; Caboche, M.; Lepiniec, L. Deciphering gene regulatory networks that control seed development and maturation in Arabidopsis. Plant J. 2008, 54, 608–620. [Google Scholar] [CrossRef]
- Verdier, J.; Thompson, R.D. Transcriptional regulation of storage protein synthesis during dicotyledon seed filling. Plant Cell Physiol. 2008, 49, 1263–1271. [Google Scholar] [CrossRef] [PubMed]
- Roscoe, T.T.; Guilleminot, J.; Bessoule, J.J.; Berger, F.; Devic, M. Complementation of seed maturation phenotypes by ectopic expression of ABSCISIC ACID INSENSITIVE3, FUSCA3 and LEAFY COTYLEDON2 in Arabidopsis. Plant Cell Physiol. 2015, 56, 1215–1228. [Google Scholar] [CrossRef] [PubMed]
- Müller, K.; Bouyer, D.; Schnittger, A.; Kermode, A.R. Evolutionarily conserved histone methylation dynamics during seed life-cycle transitions. PLoS ONE 2012, 7, e51532. [Google Scholar] [CrossRef]
- Molitor, A.M.; Bu, Z.; Yu, Y.; Shen, W.H. Arabidopsis AL PHD-PRC1 complexes promote seed germination through H3K4me3-to-H3K27me3 chromatin state switch in repression of seed developmental genes. PLoS Genet. 2014, 10, e1004091. [Google Scholar] [CrossRef] [PubMed]
- ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Sequeira-Mendes, J.; Aragüez, I.; Peiró, R.; Mendez-Giraldez, R.; Zhang, X.; Jacobsen, S.E.; Bastolla, U.; Gutierrez, C. The functional topography of the Arabidopsis genome is organized in a reduced number of linear motifs of chromatin states. Plant Cell 2014, 26, 2351–2366. [Google Scholar] [CrossRef]
- Santos-Pereira, J.M.; Gallardo-Fuentes, L.; Neto, A.; Acemel, R.D.; Tena, J.J. Pioneer and repressive functions of p63 during zebrafish embryonic ectoderm specification. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef]
- Hannon, G.J. FASTX-Toolkit. 2010. Available online: http://hannonlab.cshl.edu/fastx_toolkit/ (accessed on 18 December 2019).
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Badouin, H.; Gouzy, J.; Grassa, C.J.; Murat, F.; Staton, S.E.; Cottret, L.; Lelandais-Brière, C.; Owens, G.L.; Carrère, S.; Mayjonade, B.; et al. The sunflower genome provides insights into oil metabolism, flowering and Asterid evolution. Nature 2017, 546, 148–152. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Yan, H.; Evans, J.; Kalmbach, M.; Moore, R.; Middha, S.; Luban, S.; Wang, L.; Bhagwate, A.; Li, W.; Sun, Z.; et al. HiChIP: A high-throughput pipeline for integrative analysis of ChIP-Seq data. BMC Bioinform. 2014, 15, 280. [Google Scholar] [CrossRef] [PubMed]
- Marinov, G.K.; Kundaje, A.; Park, P.J.; Wold, B.J. Large-scale quality analysis of published ChIP-seq data. G3 Genes Genomes Genet. 2014, 4, 209–223. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, J.M. Improved peak-calling with MACS2. BioRxiv 2018, 496521. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; He, Q.Y. ChIPseeker: An R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 2015, 31, 2382–2383. [Google Scholar] [CrossRef]
- Thomas, P.D.; Campbell, M.J.; Kejariwal, A.; Mi, H.; Karlak, B.; Daverman, R.; Diemer, K.; Muruganujan, A.; Narechania, A. PANTHER: A library of protein families and subfamilies indexed by function. Genome Res. 2003, 13, 2129–2141. [Google Scholar] [CrossRef]
- Mi, H.; Dong, Q.; Muruganujan, A.; Gaudet, P.; Lewis, S.; Thomas, P.D. PANTHER version 7: Improved phylogenetic trees, orthologs and collaboration with the Gene Ontology Consortium. Nucleic Acids Res. 2010, 38, D204–D210. [Google Scholar] [CrossRef]
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Cheng, J.X.; Murre, C.; Singh, H.; Glass, C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 2010, 38, 576–589. [Google Scholar] [CrossRef]
- Moreno-Pérez, A.J.; Sánchez-García, A.; Salas, J.J.; Garcés, R.; Martínez-Force, E. Acyl-ACP thioesterases from macadamia (Macadamia tetraphylla) nuts: Cloning, characterization and their impact on oil composition. Plant Physiol. Biochem. 2011, 49, 82–87. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Hara, A.; Radin, N.S. Lipid extraction of tissues with a low-toxicity solvent. Anal. Biochem. 1978, 90, 420–425. [Google Scholar] [CrossRef]
- Haque, M.E.; Han, B.; Wang, B.; Wang, Y.; Liu, A. Development of an efficient chromatin immunoprecipitation method to investigate protein-DNA interaction in oleaginous castor bean seeds. PLoS ONE 2018, 13, e0197126. [Google Scholar] [CrossRef]
- Gómez-Díaz, E.; Rivero, A.; Chandre, F.; Corces, V.G. Insights into the epigenomic landscape of the human malaria vector Anopheles gambiae. Front. Genet. 2014, 5, 277. [Google Scholar]
- Landt, S.G.; Marinov, G.K.; Kundaje, A.; Kheradpour, P.; Pauli, F.; Batzoglou, S.; Bernstein, B.E.; Bickel, P.; Brown, J.B.; Cayting, P.; et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 2012, 22, 1813–1831. [Google Scholar] [CrossRef]
- Du, Z.; Li, H.; Wei, Q.; Zhao, X.; Wang, C.; Zhu, Q.; Yi, X.; Xu, W.; Liu, X.S.; Jin, W.; et al. Genome-wide analysis of histone modifications: H3K4me2, H3K4me3, H3K9ac, and H3K27ac in Oryza sativa L. Japonica. Mol. Plant. 2013, 6, 1463–1472. [Google Scholar] [CrossRef]
- Zhang, X.; Bernatavichute, Y.V.; Cokus, S.; Pellegrini, M.; Jacobsen, S.E. Genome-wide analysis of mono-, di-and trimethylation of histone H3 lysine 4 in Arabidopsis thaliana. Genome Biol. 2009, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Ha, M.; Ng, D.W.; Li, W.H.; Chen, Z.J. Coordinated histone modifications are associated with gene expression variation within and between species. Genome Res. 2011, 21, 590–598. [Google Scholar] [CrossRef]
- Van Dijk, K.; Ding, Y.; Malkaram, S.; Riethoven, J.J.M.; Liu, R.; Yang, J.; Laczko, P.; Chen, H.; Xia, Y.; Ladunga, I.; et al. Dynamic changes in genome-wide histone H3 lysine 4 methylation patterns in response to dehydration stress in Arabidopsis thaliana. BMC Plant Biol. 2010, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, X.; He, K.; Ma, Y.; Su, N.; He, H.; Stolc, V.; Tongprasit, W.; Jin, W.; Jiang, J.; et al. High-resolution mapping of epigenetic modifications of the rice genome uncovers interplay between DNA methylation, histone methylation, and gene expression. Plant Cell 2008, 20, 259–276. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; David, A.P.; Dolinkski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Jones, A.; Davies, H.M.; Voelker, T.A. Palmitoyl-acyl carrier protein (ACP) thioesterase and the evolutionary origin of plant Acyl-ACP thio-esterases. Plant Cell 1995, 7, 359e371. [Google Scholar]
- Cagliari, A.; Turchetto-Zolet, A.C.; Korbes, A.P.; dos Santos Maraschin, F.; Margis, R.; Margis-Pinheiro, M. New insights on the evolution of Leafy cotyledon1 (LEC1) type genes in vascular plants. Genomics 2014, 103, 380–387. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Perkins, M.; Skori, L.; Hickerson, N.M.; Jamshed, M.; Samuel, M.A. Genetic manipulation of ABI3 confers frost-tolerant seed degreening in canola. Plant Biotechnol. J. 2020, 18, 602. [Google Scholar] [CrossRef]
- Aziz, U.; Tang, T.; Saleem, N.; Yang, Z.; Zhang, M. New insights revealed the evolution of the AFL subfamily of B3 transcription factors from chlorophyta and its requisite in land plants. Pak. J. Agric. Sci. 2020, 57, 129–1235. [Google Scholar]
- Fatihi, A.; Boulard, C.; Bouyer, D.; Baud, S.; Dubreucq, B.; Lepiniec. Deciphering and modifying LAFL transcriptional regulatory network in seed for improving yield and quality of storage compounds. Plant Sci. 2016, 250, 198–204. [Google Scholar] [CrossRef]
- Lepiniec, L.; Devic, M.; Roscoe, T.J.; Bouyer, D.; Zhou, D.X.; Boulard, C.; Baud, S.; Dubreucq, B. Molecular and epigenetic regulations and functions of the LAFL transcriptional regulators that control seed development. Plant Reprod. 2018, 31, 291–307. [Google Scholar] [CrossRef]
- Zheng, Y.; Ren, N.; Wang, H.; Stromberg, A.J.; Perry, S.E. Global identification of targets of the Arabidopsis MADS domain protein AGAMOUS-Like15. Plant Cell 2009, 21, 2563–2577. [Google Scholar] [CrossRef] [PubMed]
- Barthole, G.; To, A.; Marchive, C.; Brunaud, V.; Soubigou-Taconnat, L.; Berger, N.; Dubreucq, B.; Lepiniec, L.; Baud, S. MYB118 represses endosperm maturation in seeds of Arabidopsis. Plant Cell 2014, 26, 3519–3537. [Google Scholar] [CrossRef]
- Marchive, C.; Nikovics, K.; To, A.; Lepiniec, L.; Baud, S. Transcriptional regulation of fatty acid production in higher plants: Molecular bases and biotechnological outcomes. Eur. J. Lipid Sci. Technol. 2014, 116, 1332–1343. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 1–9. [Google Scholar] [CrossRef]
- Martínez-Force, E.; Cantisán, S.; Serrano-Vega, M.J.; Garcés, R. Acyl-acyl carrier protein thioesterase activity from sunflower (Helianthus annuus L.) seeds. Planta 2000, 211, 673–678. [Google Scholar] [CrossRef]
- Martínez-Force, E.; Álvarez-Ortega, R.; Garcés, R. Enzymatic characterisation of high-palmitic acid sunflower (Helianthus annuus L.) mutants. Planta 1999, 207, 533–538. [Google Scholar] [CrossRef]
- Garcés, R.; Sarmiento, C.; Mancha, M. Temperature regulation of oleate desaturase in sunflower (Helianthus annuus L.) seeds. Planta 1992, 186, 461–465. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Hua, W.; Zhan, G.; Wei, F.; Wang, X.; Liu, G.; Wang, H. Increasing seed mass and oil content in transgenic Arabidopsis by the overexpression of wri1- like gene from Brassica napus. Plant Physiol. Biochem. 2010, 48, 9–15. [Google Scholar] [CrossRef]
- An, D.; Kim, H.; Ju, S.; Go, Y.S.; Kim, H.U.; Suh, M.C. Expression of Camelina WRINKLED1 isoforms rescue the seed phenotype of the Arabidopsis wri1 mutant and increase the triacylglycerol content in tobacco leaves. Front. Plant Sci. 2017, 8, 34. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Dong, X.S.; Wu, B.; Xu, Z.N.; Zhou, Z.X.; Lin, X.G.; Wang, J.; Zang, X.; Ma, W.H. Physiological, histological and molecular analysis of avocado mesocarp fatty acids during fruit development. J. Agric. Sci. 2019, 11, 95–104. [Google Scholar]
- Ji, X.J.; Mao, X.; Hao, Q.T.; Liu, B.L.; Xue, J.A.; Li, R.Z. Splice variants of the castor WRI1 gene upregulate fatty acid and oil biosynthesis when expressed in tobacco leaves. Int. J. Mol. Sci. 2018, 19, 146. [Google Scholar]
- Kim, M.J.; Yang, S.W.; Mao, H.Z.; Veena, S.P.; Yin, J.L.; Chua, N.H. Gene silencing of Sugar-dependent 1 (JcSDP1), encoding a patatin-domain triacylglycerol lipase, enhances seed oil accumulation in Jatropha curcas. Biotechnol. Biofuels 2014, 7, 1–16. [Google Scholar] [CrossRef]
- Maeo, K.; Tokuda, T.; Ayame, A.; Mitsui, N.; Kawai, T.; Tsukagoshi, H.; Ishiguro, S.; Nakamura, K. An AP2-type transcription factor, WRINKLED1, of Arabidopsis thaliana binds to the AW-box sequence conserved among proximal upstream regions of genes involved in fatty acid synthesis. Plant J. 2009, 60, 476–487. [Google Scholar] [CrossRef]
- Carmona-Rojas, L.; Urrea-Trujillo, A.; Gil-Arrendondo, D.; Atehortúa-Garcés, L.; Pabón-Mora, N. Expression of storage lipid biosynthesis transcription factors and enzymes in Jatropha curcas L. cell suspension cultures and seeds. In Vitro Cell. Dev. Biol. Plant 2020, 57, 1–14. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, Y.; Huang, Y.; Cui, Y.; Hua, J. Gene network of oil accumulation reveals expression profiles in developing embryos and fatty acid composition in Upland cotton. J. Plant Physiol. 2018, 228, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Baud, S.; Lepiniec, L. Regulation of de novo fatty acid synthesis in maturing oilseeds of Arabidopsis. Plant Physiol. Biochem. 2009, 47, 448–455. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Arabidopsis | Sunflower | Fold Enrichment * | |||
|---|---|---|---|---|---|
| Gene ID | Gene Name | Gene ID | Gene Name | 13 DAF | 28 DAF |
| AT3G24650 | ABI3 | HanXRQChr15g0468411 | VIV1 | 6.37 | 5.39 |
| AT3g26790 | FUS3 | HanXRQChr15g0475761 | FUS3 | 5.9 | 6.32 |
| ChIP-Seq Annotation | ||||
|---|---|---|---|---|
| Enzyme Activity | Arabidopsis Gene | Sunflower Homologous Gene | 13 DAA | 28 DAA |
| LS (Lipoyl Synthase) | AT5G08415.1 | HanXRQChr05g0142151 | Yes | Yes |
| HanXRQChr12g0358451 | Yes | Yes | ||
| HanXRQChr17g0559041 | Yes | Yes | ||
| LT (Lipoyl Transferase) | AT4G31050.1 | HanXRQChr09g0268371 | Yes | Yes |
| HanXRQChr05g0141551 | No | Yes | ||
| HanXRQChr02g0056731 | No | Yes | ||
| MCMT (Malonyl-CoA: ACP Malonyltransferase) | AT2G30200.1 | HanXRQChr01g0012161 | No | Yes |
| HanXRQChr01g0012131 | Yes | Yes | ||
| KASI (Ketoacyl-ACP Synthase I) | At5g46290 | HanXRQChr17g0564321 | Yes | Yes |
| HanXRQChr04g0118391 | Yes | Yes | ||
| HanXRQChr13g0415471 | Yes | Yes | ||
| HanXRQChr17g0535001 | No | Yes | ||
| HanXRQChr01g0020441 | No | Yes | ||
| HanXRQChr01g0019841 | No | Yes | ||
| KASII (Ketoacyl-ACP Synthase II) | At1g74960 | HanXRQChr09g0237851 | Yes | Yes |
| HanXRQChr15g0497191 | Yes | Yes | ||
| HanXRQChr06g0178221 | No | No | ||
| HanXRQChr16g0508321 | Yes | Yes | ||
| KASIII (Ketoacyl-ACP Synthase III) | At1g62640 | HanXRQChr02g0049441 | Yes | Yes |
| HanXRQChr05g0148701 | Yes | Yes | ||
| HanXRQChr17g0550351 | Yes | Yes | ||
| KAR (Ketoacyl-ACP Reductase) | At1g24360 | |||
| At1g62610 | ||||
| At3g46170 | HanXRQChr17g0536131 | Yes | Yes | |
| At3g55290 | HanXRQChr10g0318431 | Yes | Yes | |
| At3g55310 | ||||
| HAD (Hydroxyacyl-ACP Dehydrase) | At2g22230 | HanXRQChr03g0066951 | Yes | Yes |
| At5g10160 | ||||
| ER (Enoyl-ACP Reductase) | At2g05990 | HanXRQChr02g0056711 | No | Yes |
| HanXRQChr17g0564011 | Yes | Yes | ||
| HACPS (Holo-ACP Synthase) | At3g11470 | HanXRQChr08g0212801 | Yes | Yes |
| HanXRQChr07g0194021 | No | Yes | ||
| SAD (Stearoyl-ACP Desaturase) | At1g43800 | HanXRQChr01g0023271 | ||
| At2g43710 | ||||
| At3g02610 | ||||
| At3g02620 | Yes | Yes | ||
| At3g02630 | HanXRQChr11g0343101 | Yes | Yes | |
| At5g16230 | ||||
| At5g16240 | ||||
| FATA (Acyl-ACP Thioesterase A) | At3g25110 | HanXRQChr01g0015251 | Yes | Yes |
| FATB (Acyl-ACP Thioesterase B) | At1g08510 | HanXRQChr09g0240511 | Yes | Yes |
| HanXRQChr06g0180041 | Yes | Yes | ||
| HanXRQChr05g0138201 | No | Yes | ||
| HanXRQChr10g0311291 | Yes | Yes | ||
| HanXRQChr17g0553141 | No | Yes | ||
| HanXRQChr10g0311351 | No | No | ||
| HanXRQChr10g0311341 | No | No | ||
| HanXRQChr10g0311311 | No | No | ||
| HanXRQChr10g0311301 | No | No | ||
| HanXRQChr01g0013271 | Yes | Yes | ||
| HanXRQChr03g0062101 | No | Yes | ||
| HanXRQChr07g0197741 | No | No | ||
| 13 DAA | 28 DAA | |
|---|---|---|
| 16:0 | 12.18 ± 2.64 | 4.74 ± 0.25 * |
| 18:0 | 7.57 ± 0.56 | 2.83 ± 0.53 * |
| 18:1Δ9 | 53.65 ± 13.23 | 71.92 ± 1.27 |
| 18:2Δ9Δ12 | 25.72 ± 10.01 | 20.13 ± 1.31 |
| 20:0 | 0.55 ± 0.08 | 0.13 ± 0.01 * |
| mg fatty acids/mg seed | 0.020 ± 0.003 | 0.125 ± 0.007 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moreno-Pérez, A.J.; Santos-Pereira, J.M.; Martins-Noguerol, R.; DeAndrés-Gil, C.; Troncoso-Ponce, M.A.; Venegas-Calerón, M.; Sánchez, R.; Garcés, R.; Salas, J.J.; Tena, J.J.; et al. Genome-Wide Mapping of Histone H3 Lysine 4 Trimethylation (H3K4me3) and Its Involvement in Fatty Acid Biosynthesis in Sunflower Developing Seeds. Plants 2021, 10, 706. https://doi.org/10.3390/plants10040706
Moreno-Pérez AJ, Santos-Pereira JM, Martins-Noguerol R, DeAndrés-Gil C, Troncoso-Ponce MA, Venegas-Calerón M, Sánchez R, Garcés R, Salas JJ, Tena JJ, et al. Genome-Wide Mapping of Histone H3 Lysine 4 Trimethylation (H3K4me3) and Its Involvement in Fatty Acid Biosynthesis in Sunflower Developing Seeds. Plants. 2021; 10(4):706. https://doi.org/10.3390/plants10040706
Chicago/Turabian StyleMoreno-Pérez, Antonio J., José M. Santos-Pereira, Raquel Martins-Noguerol, Cristina DeAndrés-Gil, M. Adrián Troncoso-Ponce, Mónica Venegas-Calerón, Rosario Sánchez, Rafael Garcés, Joaquín J. Salas, Juan J. Tena, and et al. 2021. "Genome-Wide Mapping of Histone H3 Lysine 4 Trimethylation (H3K4me3) and Its Involvement in Fatty Acid Biosynthesis in Sunflower Developing Seeds" Plants 10, no. 4: 706. https://doi.org/10.3390/plants10040706
APA StyleMoreno-Pérez, A. J., Santos-Pereira, J. M., Martins-Noguerol, R., DeAndrés-Gil, C., Troncoso-Ponce, M. A., Venegas-Calerón, M., Sánchez, R., Garcés, R., Salas, J. J., Tena, J. J., & Martínez-Force, E. (2021). Genome-Wide Mapping of Histone H3 Lysine 4 Trimethylation (H3K4me3) and Its Involvement in Fatty Acid Biosynthesis in Sunflower Developing Seeds. Plants, 10(4), 706. https://doi.org/10.3390/plants10040706