
DNA Modification Patterns within the Transposable Elements of the Fig (Ficus carica L.) Genome

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Sequence Collection
2.2. Genome-Wide Transposable Elements Distribution Analysis
2.3. Full-Length LTR-REs Modification Analysis
2.4. Distribution Analysis of Methylated LTR-REs According to Their Centromere Proximity
2.5. Analysis of the Proximity of Methylated LTR-REs to Genes
3. Results
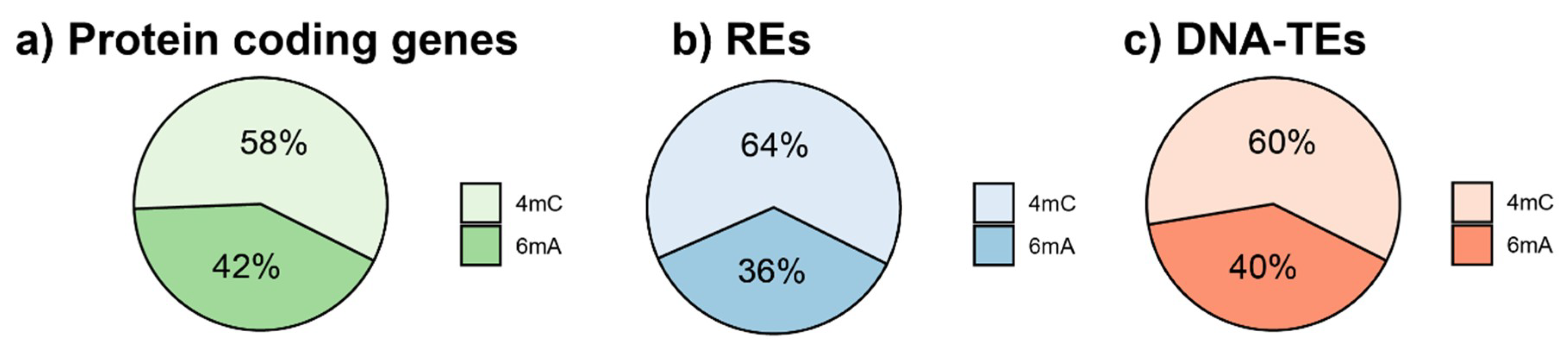
3.1. Genome-Wide DNA Modifications of the Fig Repetitive Component
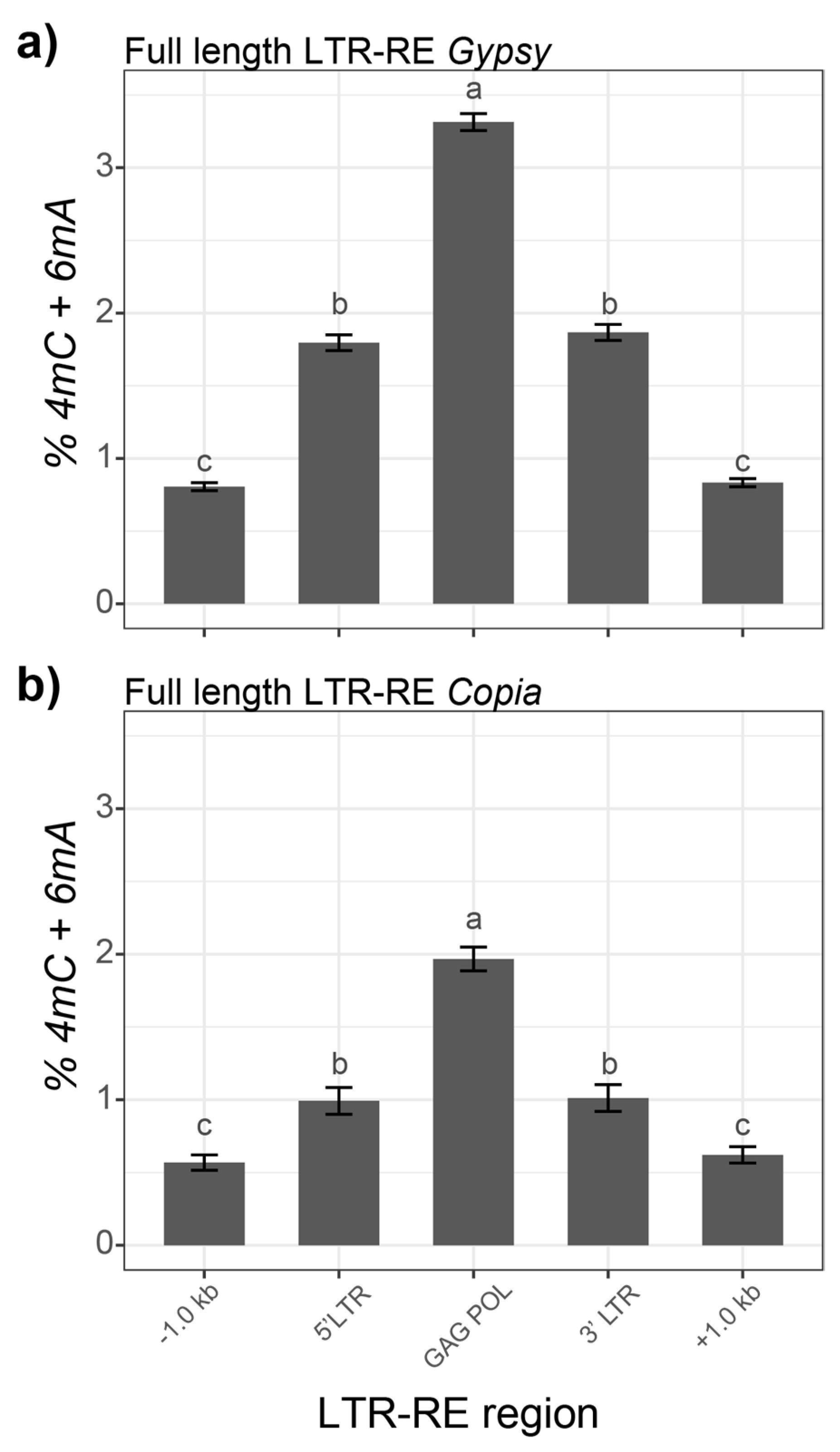
3.2. 4mC and 6mA DNA Modifications in Fig Full-Length LTR-REs
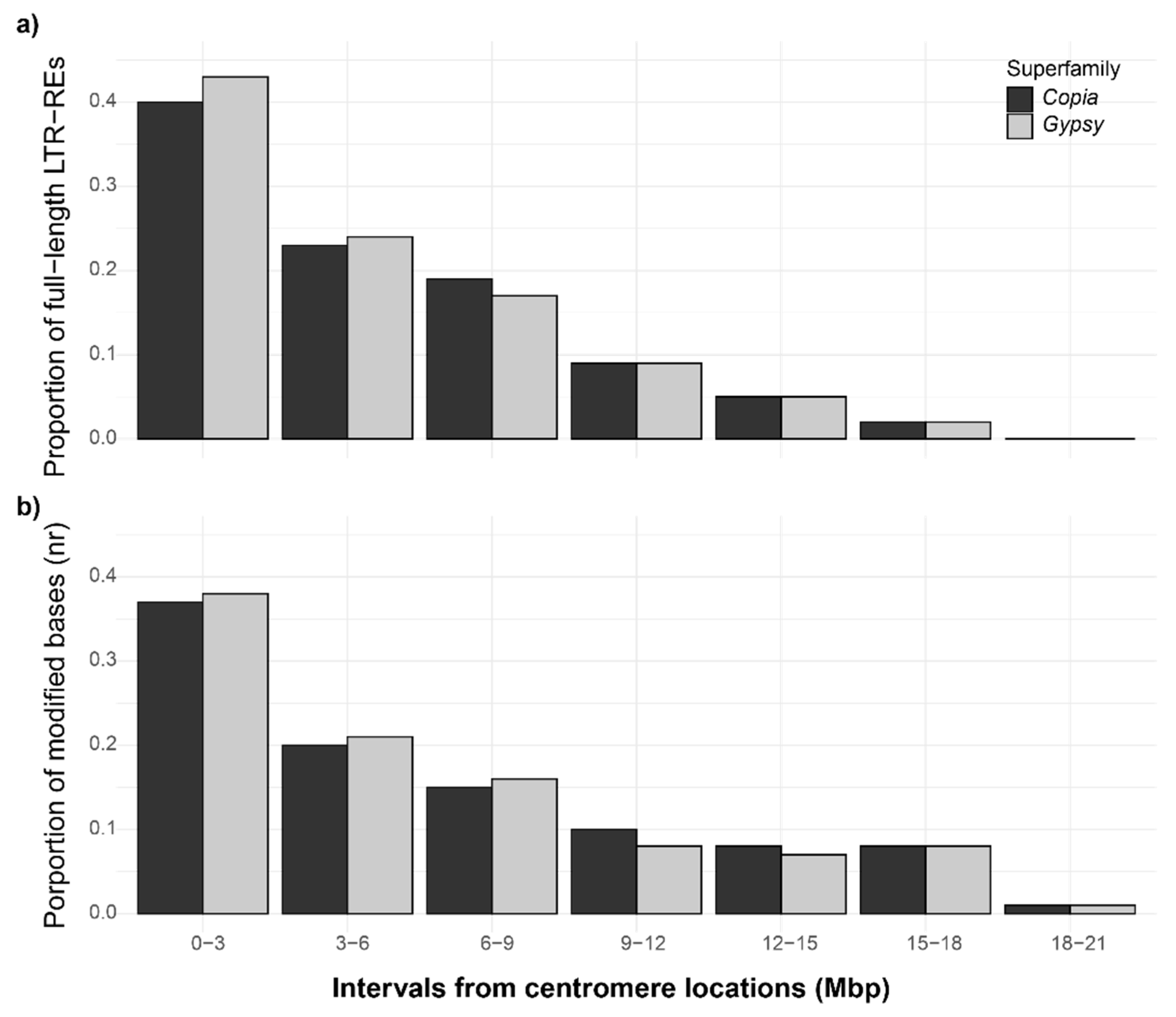
3.3. Unconventional DNA Modifications in Fig Full-Length LTR-Res, and Their Proximity to Centromeric Regions
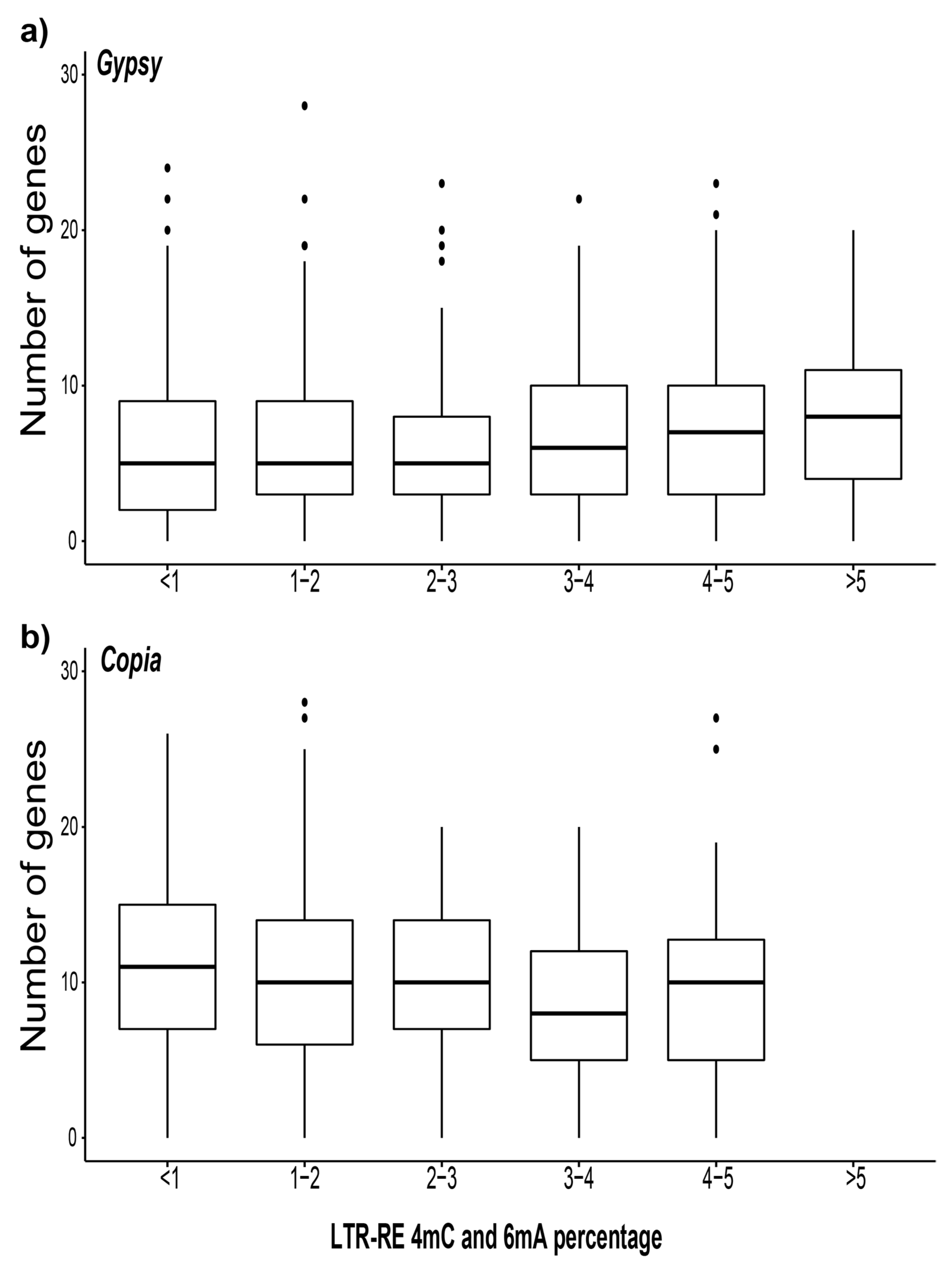
3.4. Analysis of the Gene Proximity to Methylated LTR-REs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| TE | transposon |
| RE | retrotransposon |
| LTR | long terminal repeat |
| 5mC | 5-methyl cytosine |
| SMRT | single-molecule real-time |
| PacBio | Pacific Biosciences |
| 4mC | N4-methylcytosine |
| 6mA | N6-methyladenine |
| GO | Gene Ontology |
References
- Wicker, T.; Sabot, F.; Hua-Van, A.; Bennetzen, J.L.; Capy, P.; Chalhoub, B.; Flavell, A.; Leroy, P.; Morgante, M.; Panaud, O.; et al. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 2007, 8, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Lisch, D. How important are transposons for plant evolution? Nat. Rev. Genet. 2013, 14, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Vitte, C.; Fustier, M.A.; Alix, K.; Tenaillon, M.I. The bright side of transposons in crop evolution. Brief. Funct. Genom. 2014, 13, 276–295. [Google Scholar] [CrossRef] [PubMed]
- Wicker, T.; Keller, B. Genome-wide comparative analysis of copia retrotransposons in Triticeae, rice, and Arabidopsis reveals conserved ancient evolutionary lineages and distinct dynamics of individual copia families. Genome Res. 2007, 17, 1072–1081. [Google Scholar] [CrossRef] [PubMed]
- Llorens, C.; Futami, R.; Covelli, L.; Domínguez-Escribá, L.; Viu, J.M.; Tamarit, D.; Aguilar-Rodríguez, J.; Vicente-Ripolles, M.; Fuster, G.; Bernet, G.P.; et al. The Gypsy Database (GyDB) of mobile genetic elements: Release 2.0. Nucleic Acids Res. 2011, 39, 70–74. [Google Scholar] [CrossRef]
- Buti, M.; Moretto, M.; Barghini, E.; Mascagni, F.; Natali, L.; Brilli, M.; Lomsadze, A.; Sonego, P.; Giongo, L.; Alonge, M.; et al. The genome sequence and transcriptome of Potentilla Micrantha and their comparison to Fragaria Vesca (the Woodland Strawberry). Gigascience 2018, 7, giy010. [Google Scholar] [CrossRef]
- Neumann, P.; Novák, P.; Hoštáková, N.; MacAs, J. Systematic survey of plant LTR-retrotransposons elucidates phylogenetic relationships of their polyprotein domains and provides a reference for element classification. Mob. DNA 2019, 10, 1. [Google Scholar] [CrossRef]
- Vangelisti, A.; Mascagni, F.; Giordani, T.; Sbrana, C.; Turrini, A.; Cavallini, A.; Giovannetti, M.; Natali, L. Arbuscular mycorrhizal fungi induce the expression of specific retrotransposons in roots of sunflower (Helianthus annuus L.). PLoS ONE 2019, 14, e0212371. [Google Scholar] [CrossRef]
- McClintock, B. The origin and behavior of mutable loci in maize. Proc. Natl. Acad. Sci. USA 1950, 36, 344–355. [Google Scholar] [CrossRef] [PubMed]
- Shang, Y.; Yang, F.; Schulman, A.H.; Zhu, J.; Jia, Y.; Wang, J.; Zhang, X.Q.; Jia, Q.; Hua, W.; Yang, J.; et al. Gene deletion in barley mediated by LTR-retrotransposon BARE. Sci. Rep. 2017, 7, 43766. [Google Scholar] [CrossRef]
- Thind, A.K.; Wicker, T.; Müller, T.; Ackermann, P.M.; Steuernagel, B.; Wulff, B.B.; Spannagl, M.; Twardziok, S.O.; Felder, M.; Lux, T.; et al. Chromosome-scale comparative sequence analysis unravels molecular mechanisms of genome dynamics between two wheat cultivars. Genome Biol. 2018, 19, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Bird, A. Perceptions of epigenetics. Nature 2007, 447, 396. [Google Scholar] [CrossRef]
- Law, J.A.; Jacobsen, S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010, 11, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Reinders, J.; Wulff, B.B.H.; Mirouze, M.; Marí-Ordóñez, A.; Dapp, M.; Rozhon, W.; Bucher, E.; Theiler, G.; Paszkowski, J. Compromised stability of DNA methylation and transposon immobilization in mosaic Arabidopsis epigenomes. Genes Dev. 2009, 23, 939–950. [Google Scholar] [CrossRef]
- Feng, S.; Cokus, S.J.; Zhang, X.; Chen, P.Y.; Bostick, M.; Goll, M.G.; Hetzel, J.; Jain, J.; Strauss, S.H.; Halpern, M.E.; et al. Conservation and divergence of methylation patterning in plants and animals. Proc. Natl. Acad. Sci. USA 2010, 107, 8689–8694. [Google Scholar] [CrossRef] [PubMed]
- Niederhuth, C.E.; Bewick, A.J.; Ji, L.; Alabady, M.S.; Do Kim, K.; Li, Q.; Rohr, N.A.; Rambani, A.; Burke, J.M.; Udall, J.A.; et al. Widespread natural variation of DNA methylation within angiosperms. Genome Biol. 2016, 17, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Ong-Abdullah, M.; Ordway, J.M.; Jiang, N.; Ooi, S.E.; Kok, S.Y.; Sarpan, N.; Azimi, N.; Hashim, A.T.; Ishak, Z.; Rosli, S.K.; et al. Loss of Karma transposon methylation underlies the mantled somaclonal variant of oil palm. Nature 2015, 525, 533–537. [Google Scholar] [CrossRef]
- Hollister, J.D.; Gaut, B.S. Epigenetic silencing of transposable elements: A trade-off between reduced transposition and deleterious effects on neighboring gene expression. Genome Res. 2009, 19, 1419–1428. [Google Scholar] [CrossRef] [PubMed]
- Eichten, S.R.; Ellis, N.A.; Makarevitch, I.; Yeh, C.T.; Gent, J.I.; Guo, L.; McGinnis, K.M.; Zhang, X.; Schnable, P.S.; Vaughn, M.W.; et al. Spreading of heterochromatin is limited to specific families of maize retrotransposons. PLoS Genet. 2012, 8, e1003127. [Google Scholar] [CrossRef]
- Quadrana, L.; Silveira, A.B.; Mayhew, G.F.; LeBlanc, C.; Martienssen, R.A.; Jeddeloh, J.A.; Colot, V. The Arabidopsis thaliana mobilome and its impact at the species level. eLife 2016, 5, e15716. [Google Scholar] [CrossRef]
- Choi, K.; Zhao, X.; Tock, A.J.; Lambing, C.; Underwood, C.J.; Hardcastle, T.J.; Serra, H.; Kim, J.; Cho, H.S.; Kim, J.; et al. Nucleosomes and DNA methylation shape meiotic DSB frequency in Arabidopsis thaliana transposons and gene regulatory regions. Genome Res. 2018, 28, 532–546. [Google Scholar] [CrossRef]
- Weigel, D.; Colot, V. Epialleles in plant evolution. Genome Biol. 2012, 13, 1–6. [Google Scholar] [CrossRef]
- Diez, C.M.; Roessler, K.; Gaut, B.S. Epigenetics and plant genome evolution. Curr. Opin. Plant Biol. 2014, 18, 1–8. [Google Scholar] [CrossRef]
- Rhoads, A.; Au, K.F. PacBio sequencing and its applications. Genom. Proteom. Bioinform. 2015, 13, 278–289. [Google Scholar] [CrossRef]
- Ye, G.; Zhang, H.; Chen, B.; Nie, S.; Liu, H.; Gao, W.; Wang, H.; Gao, Y.; Gu, L. De novo genome assembly of the stress tolerant forest species Casuarina equisetifolia provides insight into secondary growth. Plant J. 2019, 97, 779–794. [Google Scholar] [CrossRef] [PubMed]
- Usai, G.; Mascagni, F.; Giordani, T.; Vangelisti, A.; Bosi, E.; Zuccolo, A.; Ceccarelli, M.; King, R.; Hassani-Pak, K.; Zambrano, L.S.; et al. Epigenetic patterns within the haplotype phased fig (Ficus carica L.) genome. Plant J. 2020, 102, 600–614. [Google Scholar] [CrossRef] [PubMed]
- Murray, I.A.; Clark, T.A.; Morgan, R.D.; Boitano, M.; Anton, B.P.; Luong, K.; Fomenkov, A.; Turner, S.W.; Korlach, J.; Roberts, R.J. The methylomes of six bacteria. Nucleic Acids Res. 2012, 40, 11450–11462. [Google Scholar] [CrossRef]
- Sánchez-Romero, M.A.; Cota, I.; Casadesús, J. DNA methylation in bacteria: From the methyl group to the methylome. Curr. Opin. Microbiol. 2015, 25, 9–16. [Google Scholar] [CrossRef]
- Beaulaurier, J.; Schadt, E.E.; Fang, G. Deciphering bacterial epigenomes using modern sequencing technologies. Nat. Rev. Genet. 2019, 20, 157–172. [Google Scholar] [CrossRef]
- Liang, Z.; Shen, L.; Cui, X.; Bao, S.; Geng, Y.; Yu, G.; Liang, F.; Xie, S.; Lu, T.; Gu, X.; et al. DNA N6-adenine methylation in Arabidopsis thaliana. Dev. Cell 2018, 45, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Wang, C.; Liu, H.; Zhou, Q.; Liu, Q.; Guo, Y.; Peng, T.; Song, J.; Zhang, J.; Chen, L.; et al. Identification and analysis of adenine N 6-methylation sites in the rice genome. Nat. Plants 2018, 4, 554–563. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Smit, A.F.A.; Hubley, R.; Green, P. RepeatMasker Open-4.0 2013–2015. Available online: http://www.repeatmasker.org (accessed on 1 September 2020).
- de Mendiburu, F.; de Mendiburu, M.F. Package ‘Agricolae’. R Package Version 2020, 1–2. Available online: ftp://mirror.csclub.uwaterloo.ca/CRAN/web/packages/agricolae/agricolae.pdf (accessed on 1 September 2020).
- Studer, A.; Zhao, Q.; Ross-Ibarra, J.; Doebly, J. Identification of a functional transposon insertion in the maize domestication gene tb1. Nat. Genet. 2011, 43, 1160–1164. [Google Scholar] [CrossRef] [PubMed]
- Mori, K.; Shirasawa, K.; Nogata, H.; Hirata, C.; Tashiro, K.; Habu, T.; Kim, S.; Himeno, S.; Kuhara, S.; Ikegami, H. Identification of RAN1 orthologue associated with sex determination through whole genome sequencing analysis in fig (Ficus carica L.). Sci. Rep. 2017, 7, 41124. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Luo, G.Z.; Chen, K.; Deng, X.; Yu, M.; Han, D.; Hao, Z.; Liu, J.; Lu, X.; Doré, L.C.; et al. N6-methyldeoxyadenosine marks active transcription start sites in Chlamydomonas. Cell 2015, 161, 879–892. [Google Scholar] [CrossRef]
- Zhang, W.; Spector, T.D.; Deloukas, P.; Bell, J.T.; Engelhardt, B.E. Predicting genome-wide DNA methylation using methylation marks, genomic position, and DNA regulatory elements. Genome Biol. 2015, 16, 14. [Google Scholar] [CrossRef]
- Koziol, M.J.; Bradshaw, C.R.; Allen, G.E.; Costa, A.S.; Frezza, C.; Gurdon, J.B. Identification of methylated deoxyadenosines in vertebrates reveals diversity in DNA modifications. Nat. Struct. Mol. Biol. 2016, 23, 24. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Riaz, A.; Chachar, S.; Ding, Y.; Du, H.; Gu, X. Epigenetic Modifications of mRNA and DNA in Plants. Mol. Plant 2020, 13, 14–30. [Google Scholar] [CrossRef]
- Cokus, S.J.; Feng, S.; Zhang, X.; Chen, Z.; Merriman, B.; Haudenschild, C.D.; Pradhan, S.; Nelson, S.F.; Pellegrini, M.; Jacobsen, S.E. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 2008, 452, 215–219. [Google Scholar] [CrossRef]
- Lisch, D. Epigenetic regulation of transposable elements in plants. Annu. Rev. Plant Biol. 2009, 60, 43–66. [Google Scholar] [CrossRef] [PubMed]
- Zemach, A.; McDaniel, I.E.; Silva, P.; Zilberman, D. Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science 2010, 328, 916–919. [Google Scholar] [CrossRef] [PubMed]
- Zakrzewski, F.; Schmidt, M.; Van Lijsebettens, M.; Schmidt, T. DNA methylation of retrotransposons, DNA transposons and genes in sugar beet (Beta vulgaris L.). Plant J. 2017, 90, 1156–1175. [Google Scholar] [CrossRef]
- Choi, J.Y.; Purugganan, M.D. Evolutionary epigenomics of retrotransposon-mediated methylation spreading in rice. Mol. Biol. Evol. 2018, 35, 365–382. [Google Scholar] [CrossRef] [PubMed]
- Rabinowicz, P.D.; Palmer, L.E.; May, B.P.; Hemann, M.T.; Lowe, S.W.; McCombie, W.R.; Martienssen, R.A. Genes and transposons are differentially methylated in plants, but not in mammals. Genome Res. 2003, 13, 2658–2664. [Google Scholar] [CrossRef]
- Sharma, A.; Presting, G.G. Centromeric retrotransposon lineages predate the maize/rice divergence and differ in abundance and activity. Mol. Genet. Genom. 2008, 279, 133–147. [Google Scholar] [CrossRef]
- Gong, Z.Y.; Wu, Y.F.; Koblizkova, A.; Torres, G.A.; Wang, K.; Iovene, M.; Neumann, P.; Zhang, W.; Novák, P.; Buell, C.R.; et al. Repeatless and repeat-based centromeres in potato: Implications for centromere evolution. Plant Cell 2012, 24, 3559–3574. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Liu, Y.; Liu, C.; Shi, Q.; Huang, Y.; Han, F. Centromere satellite repeats have undergone rapid changes in polyploid wheat subgenomes. Plant Cell 2019, 31, 2035–2051. [Google Scholar] [CrossRef] [PubMed]
- Kirov, I.; Odintsov, S.; Omarov, M.; Gvaramiya, S.; Merkulov, P.; Dudnikov, M.; Ermolaev, A.; Van Laere, K.; Soloviev, A.; Khrustaleva, L. Functional Allium fistulosum centromeres comprise arrays of a long satellite repeat, insertions of retrotransposons and chloroplast DNA. Front. Plant Sci. 2020, 11, 562001. [Google Scholar] [CrossRef]
- Mascagni, F.; Vangelisti, A.; Giordani, T.; Cavallini, A.; Natali, L. A computational comparative study of the repetitive DNA in the genus Quercus L. Tree Genet. Genomes 2020, 16, 11. [Google Scholar] [CrossRef]
- Makarevitch, I.; Waters, A.J.; West, P.T.; Stitzer, M.; Hirsch, C.N.; Ross-Ibarra, J.; Springer, N.M. Transposable elements contribute to activation of maize genes in response to abiotic stress. PLoS Genet. 2015, 11, e1004915. [Google Scholar]
- Wang, G.; Jiang, H.; de León, G.D.T.; Martinez, G.; Köhler, C. Sequestration of a transposon-derived siRNA by a target mimic imprinted gene induces postzygotic reproductive isolation in Arabidopsis. Dev. Cell 2018, 46, 696–705. [Google Scholar] [CrossRef] [PubMed]
- Arnaud, P.; Goubely, C.; Pelissier, T.; Deragon, J.M. SINE retroposons can be used in vivo as nucleation centers for de novo methylation. Mol. Cell. Biol. 2000, 20, 3434–3441. [Google Scholar] [CrossRef] [PubMed]
- Sigman, M.J.; Slotkin, R.K. The first rule of plant transposable element silencing: Location, location, location. Plant Cell 2016, 28, 304–313. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Order | Superfamily | Lineage | 4mC/6mA | GC% |
|---|---|---|---|---|
| Class I | ||||
| LTR | Gypsy | Athila | 1.73 | 36.00 |
| Chromovirus | 1.83 | 35.35 | ||
| Ogre-Tat | 2.03 | 33.17 | ||
| Copia | Ale | 1.61 | 39.12 | |
| Angela | 1.47 | 34.76 | ||
| Bianca | 1.69 | 37.22 | ||
| Ikeros | 1.61 | 32.59 | ||
| Ivana | 1.78 | 35.24 | ||
| SIRE | 1.84 | 36.31 | ||
| TAR | 1.87 | 32.70 | ||
| Tork | 1.27 | 36.12 | ||
| ND | 1.71 | 35.93 | ||
| LINE | 1.63 | 33.88 | ||
| SINE | 0.96 | 36.79 | ||
| Class II | ||||
| Sub-classI | TIR | 1.74 | 34.99 | |
| Sub-classII | Helitron | 1.14 | 37.54 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Usai, G.; Vangelisti, A.; Simoni, S.; Giordani, T.; Natali, L.; Cavallini, A.; Mascagni, F. DNA Modification Patterns within the Transposable Elements of the Fig (Ficus carica L.) Genome. Plants 2021, 10, 451. https://doi.org/10.3390/plants10030451
Usai G, Vangelisti A, Simoni S, Giordani T, Natali L, Cavallini A, Mascagni F. DNA Modification Patterns within the Transposable Elements of the Fig (Ficus carica L.) Genome. Plants. 2021; 10(3):451. https://doi.org/10.3390/plants10030451
Chicago/Turabian StyleUsai, Gabriele, Alberto Vangelisti, Samuel Simoni, Tommaso Giordani, Lucia Natali, Andrea Cavallini, and Flavia Mascagni. 2021. "DNA Modification Patterns within the Transposable Elements of the Fig (Ficus carica L.) Genome" Plants 10, no. 3: 451. https://doi.org/10.3390/plants10030451
APA StyleUsai, G., Vangelisti, A., Simoni, S., Giordani, T., Natali, L., Cavallini, A., & Mascagni, F. (2021). DNA Modification Patterns within the Transposable Elements of the Fig (Ficus carica L.) Genome. Plants, 10(3), 451. https://doi.org/10.3390/plants10030451