
Optimizing Protocols for Arabidopsis Shoot and Root Protoplast Cultivation




Abstract
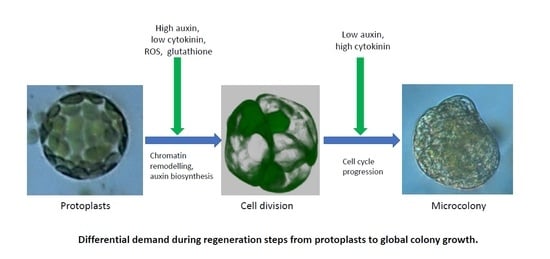
1. Introduction
2. Results and Discussion
2.1. Shoot-Derived Protoplasts
2.1.1. Starting Material
2.1.2. Protoplast Sources
2.1.3. Protoplasts Isolation Procedure
2.1.4. Medium for Protoplast Cultivation
2.1.5. Osmotic Pressure Adjustment for Selecting Cells with High Proliferative Capacity
2.1.6. Organic Acid Supplementation and Protoplast Plating Density
2.1.7. Cell Viability/Activity Assays
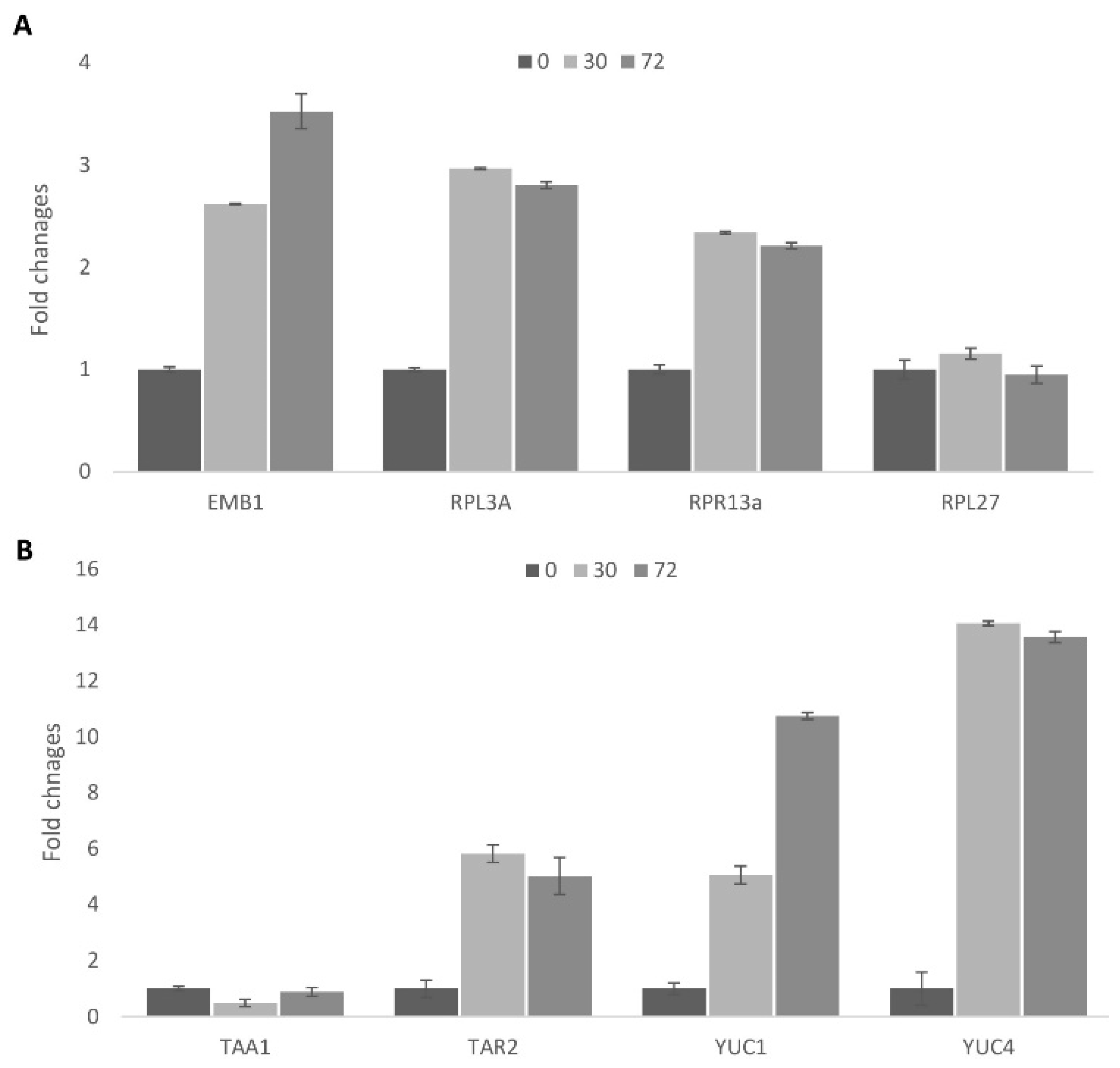
2.1.8. Analysis of Gene Expression during Cell Re-Programming
2.1.9. Activation of Auxin Biosynthesis during Cell Activation
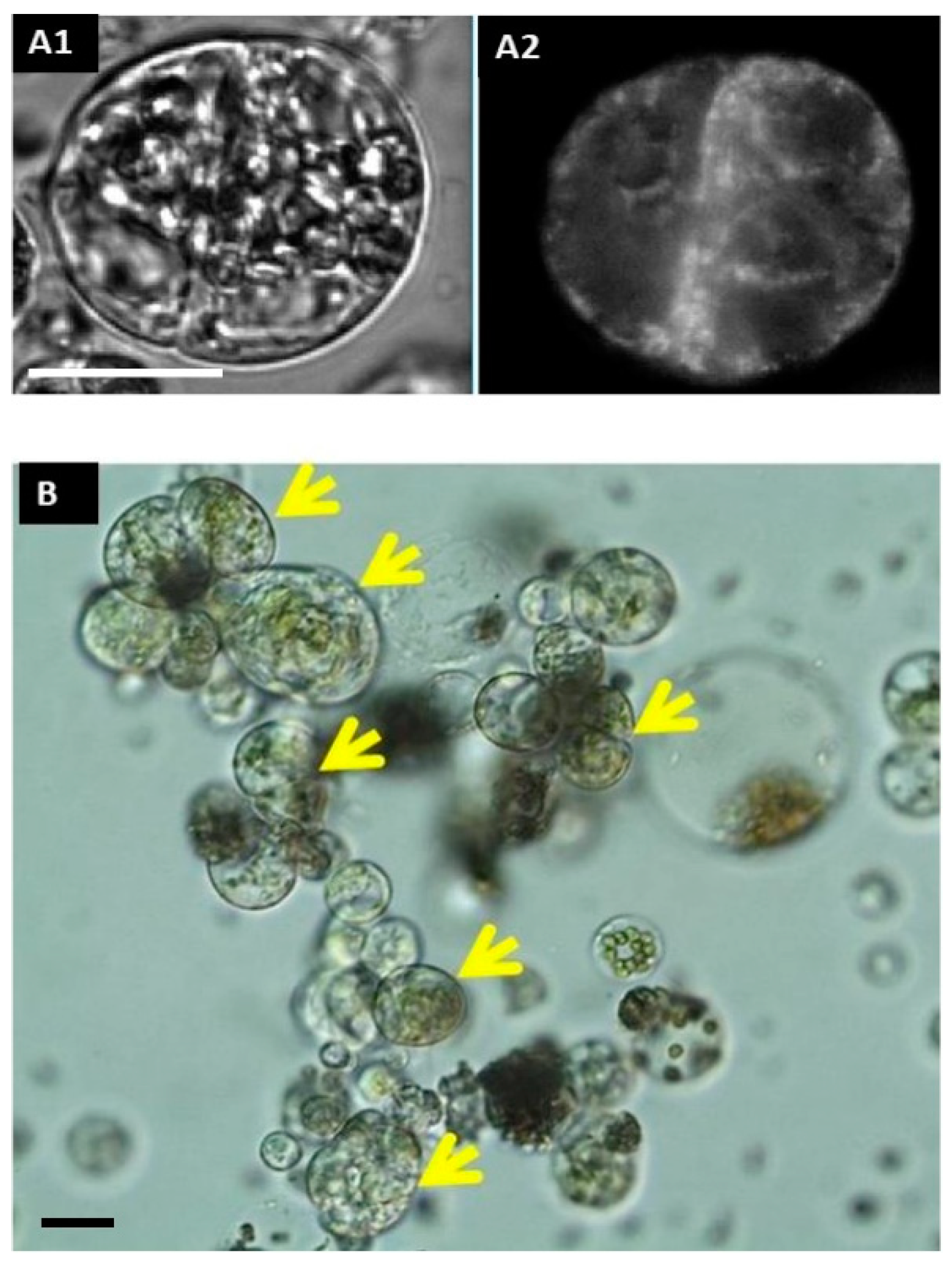
2.1.10. Detection of Protoplast Competence to Undergo the Cell Cycle
2.1.11. Nutrient Adjustment and Induction of Cell Division
2.1.12. Type of Cells and Plant Regeneration
2.2. Application of Shoot-Derived Protoplasts to Investigate the Effect of Different Compounds on Cell Reprogramming
2.3. Root-Derived Protoplasts
3. Materials and Methods
3.1. Plant Material and Growth Conditions
3.2. Protoplasts Isolation and Cultivation
3.2.1. Enzyme Preparation
3.2.2. Protoplasts Isolation
3.2.3. Protoplasts Cultivation
3.2.4. “Creation” of Totipotent Cells
3.3. Cell Viability/activity Determination
3.4. Cell Proliferation Assay
3.5. Quantitative Real-Time PCR Assay (qPCR)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Birnbaum, K.D.; Roudier, F. Epigenetic memory and cell fate reprogramming in plants. Regeneration 2017, 4, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Pasternak, T.; Lystvan, K.; Betekhtin, A.; Hasterok, R. From Single Cell to Plants: Mesophyll Protoplasts as a Versatile System for Investigating Plant Cell Reprogramming. Int. J. Mol. Sci. 2020, 21, 4195. [Google Scholar] [CrossRef] [PubMed]
- Negrutiu, I.; Beeftink, F.; Jacobs, M. Arabidopsis thaliana as a model system in somatic cell genetics I. Cell and tissue culture. Plant Sci. Lett. 1975, 5, 293–304. [Google Scholar] [CrossRef]
- Damm, B.; Willmitzer, L. Regeneration of fertile plants from protoplasts of different Arabidopsis thaliana genotypes. Mol. Gen. Genet. 1988, 213, 15–20. [Google Scholar] [CrossRef]
- Karesch, H.; Bilang, R.; Potrykus, I. Arabidopsis thaliana: Protocol for plant regeneration from protoplasts. Plant Cell Rep. 1991, 9, 575–578. [Google Scholar] [CrossRef] [PubMed]
- O′Neill, C.M.; Mathias, R.J. Regeneration of plants from protoplasts of Arabidopsis thaliana L. cv. Columbia (C24), via direct embryogenesis. J. Exp. Bot. 1993, 44, 1579–1585. [Google Scholar] [CrossRef]
- Chupeau, M.C.; Granier, F.; Pichon, O.; Renou, J.P.; Gaudin, V.; Chupeau, Y. Characterization of the early events leading to totipotency in an Arabidopsis protoplast liquid culture by temporal transcript profiling. Plant Cell 2013, 25, 2444–2463. [Google Scholar] [CrossRef]
- Mathur, J.; Koncz, C.; Szabados, L. A simple method for isolation, liquid culture, transformation and regeneration of Arabidopsis thaliana protoplasts. Plant Cell Rep. 1995, 14, 221–226. [Google Scholar] [CrossRef]
- Pasternak, T.; Ruperti, B.; Palme, K. A simple high efficiency and low cost in vitro growth system for phenotypic characterization and seed propagation of Arabidopsis thaliana. Biorxiv 2020. [Google Scholar] [CrossRef]
- Masson, J.; Paszkowski, J. The culture response of Arabidopsis thaliana protoplasts is determined by the growth conditions of donor plants. Plant J. 1992, 2, 829–833. [Google Scholar] [CrossRef]
- Donnelly, P.M.; Bonetta, D.; Tsukaya, H.; Dengler, R.E.; Dengler, N.G. Cell cycling and cell enlargement in developing leaves of Arabidopsis. Dev. Biol. 1999, 215, 407–419. [Google Scholar] [CrossRef]
- Moreno, S.; Canales, J.; Hong, L.; Robinson, D.; Roeder, A.H.; Gutiérrez, R.A. Nitrate Defines Shoot Size through Compensatory Roles for Endoreplication and Cell Division in Arabidopsis thaliana. Curr. Biol. 2020, 30, 1988–2000. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, E.J. The essential nutrient elements: Requirements and interactions in plants. In Plant Physiology; Editor Steward FC Academic Press, Inc.: New York, NY, USA, 1963; Volume 3, pp. 137–360. [Google Scholar]
- Hewitt, E.J. Sand and Water Culture Methods Used in the Study of Plant Nutrition; Farnham Royal, Bucks, England Commonwealth Agricultural Bureaux; Cambridge University Press: Cambridge, UK, 1966. [Google Scholar]
- Murashige, T.; Skoog, F. A revised medium for rapid growth and bioassays with tobacco tissue cultures. Physiol. Plant. 1962, 15, 473–497. [Google Scholar] [CrossRef]
- Dalton, C.C.; Iqbal, K.; Turner, D.A. Iron phosphate precipitation in Murashige and Skoog media. Physiol. Plant. 1983, 57, 472–476. [Google Scholar] [CrossRef]
- Raizada, M.N.; Goron, T.L.; Bannerjee, O.; Mason, M.Q.; Pautler, M.; Brazolot, J.; Morris, A.D.; Kajenthira, A.; Dinka, S.J.; DiMeo, N. Loss of developmental pluripotency occurs in two stages during leaf aging in Arabidopsis thaliana. Vitr. Cell. Dev. Biol. Plant 2017, 53, 178–187. [Google Scholar] [CrossRef]
- Del Pozo, J.C.; Diaz-Trivino, S.; Cisneros, N.; Gutierrez, C. The balance between cell division and endoreplication depends on E2FC-DPB, transcription factors regulated by the ubiquitin-SCFSKP2A pathway in Arabidopsis. Plant Cell 2006, 18, 2224–2235. [Google Scholar] [PubMed]
- Zhao, J.; Morozova, N.; Williams, L.; Libs, L.; Avivi, Y.; Grafi, G. Two phases of chromatin decondensation during dedifferentiation of plant cells distinction between competence for cell fate switch and a commitment for S phase. J. Biol. Chem. 2001, 276, 22772–22778. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.D.; Cho, Y.H.; Sheen, J. Arabidopsis mesophyll protoplasts: A versatile cell system for transient gene expression analysis. Nat. Protoc. 2007, 2, 1565. [Google Scholar] [CrossRef] [PubMed]
- Menczel, L.; Nagy, F.; Kiss, Z.R.; Maliga, P. Streptomycin resistant and sensitive somatic hybrids of Nicotiana tabacum+ Nicotiana knightiana: Correlation of resistance to N. tabacum plastids. Theor. Appl. Genet. 1981, 59, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Kao, K.N.; Michayluk, M.R. Nutritional requirements for growth of Vicia hajastana cells and protoplasts at a very low population density in liquid media. Planta 1975, 126, 105–110. [Google Scholar] [CrossRef]
- Luo, Y.; Koop, H.U. Somatic embryogenesis in cultured immature zygotic embryos and leaf protoplasts of Arabidopsis thaliana ecotypes. Planta 1997, 202, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Franco-Navarro, J.D.; Brumos, J.; Rosales, M.A.; Cubero-Font, P.; Talón, M.; Colmenero-Flores, J.M. Chloride regulates leaf cell size and water relations in tobacco plants. J. Exp. Bot. 2016, 67, 873–891. [Google Scholar] [CrossRef]
- Kaiser, S.; Scheuring, D. To Lead or to Follow: Contribution of the Plant Vacuole to Cell Growth. Front. Plant Sci. 2020, 11, 553. [Google Scholar] [CrossRef]
- Pasternak, T.P.; Prinsen, E.; Ayaydin, F.; Miskolczi, P.; Potters, G.; Asard, H.; Van Onckelen, H.A.; Dudits, D.; Fehér, A. The role of auxin, pH, and stress in the activation of embryogenic cell division in leaf protoplast-derived cells of alfalfa. Plant Physiol. 2002, 129, 1807–1819. [Google Scholar] [CrossRef] [PubMed]
- Andreini, C.; Bertini, I.; Rosato, A. Metalloproteomes: A bioinformatic approach. Acc. Chem. Res. 2009, 42, 1471–1479. [Google Scholar] [PubMed]
- Stormea, N.D.; Masonb, A. Plant speciation through chromosome instability and ploidy change: Cellular mechanisms, molecular factors and evolutionary relevance. Curr. Plant Biol. 2014, 1, 10–33. [Google Scholar] [CrossRef]
- Rose, R.J. Medicago truncatula as a model for understanding plant interactions with other organisms, plant development and stress biology: Past, present and future. Funct. Plant Biol. 2008, 35, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Sheen, J. Signal transduction in maize and Arabidopsis mesophyll protoplasts. Plant Physiol. 2001, 127, 1466–1475. [Google Scholar] [PubMed]
- Kondratenko, S.I.; Pasternak, T.P.; Samovol, O.P.; Mogilna, O.M.; Sergienko, O.V. Modeling of asymmetric division of somatic cell in protoplasts culture of higher plants. Regul. Mech. Biosyst. 2020, 11, 255–265. [Google Scholar] [CrossRef]
- Ferlini, C.; Biselli, R.; Nisini, R.; Fattorossi, A. Rhodamine 123: A useful probe for monitoring T cell activation. Cytom. J. Int. Soc. Anal. Cytol. 1995, 21, 284–293. [Google Scholar] [CrossRef]
- Cai, J.; Limke, T.L.; Ginis, I.; Rao, M.S. Identifying and tracking neural stem cells. Blood Cells Mol. Dis. 2003, 31, 18–27. [Google Scholar] [CrossRef]
- Pasternak, T.; Miskolczi, P.; Ayaydin, F.; Mészáros, T.; Dudits, D.; Fehér, A. Exogenous auxin and cytokinin dependent activation of CDKs and cell division in leaf protoplast-derived cells of alfalfa. Plant Growth Regul. 2000, 32, 129–141. [Google Scholar] [CrossRef]
- Pasternak, T.; Asard, H.; Potters, G.; Jansen, M.A. The thiol compounds glutathione and homoglutathione differentially affect cell development in alfalfa (Medicago sativa L.). Plant Physiol. Biochem. 2014, 74, 16–23. [Google Scholar] [CrossRef]
- Popescu, S.C.; Tumer, N.E. Silencing of ribosomal protein L3 genes in N. tabacum reveals coordinate expression and significant alterations in plant growth, development and ribosome biogenesis. Plant J. 2004, 39, 29–44. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Wang, Y.; Liu, X.; Sun, J.; Wang, Y.; Xu, Y.; Lv, J.; Long, W.; Zhu, X.; Guo, X.; et al. The RICE MINUTE-LIKE1 (RML1) gene, encoding a ribosomal large subunit protein L3B, regulates leaf morphology and plant architecture in rice. J. Exp. Bot. 2016, 67, 3457–3469. [Google Scholar] [CrossRef] [PubMed]
- Pasternak, T.; Dudits, D. Epigenetic Clues to Better Understanding of the Asexual Embryogenesis in planta and in vitro. Front. Plant Sci. 2019, 10, 778. [Google Scholar] [CrossRef] [PubMed]
- Wójcikowska, B.; Wójcik, A.M.; Gaj, M.D. Epigenetic Regulation of Auxin-Induced Somatic Embryogenesis in Plants. Int. J. Mol. Sci. 2020, 21, 2307. [Google Scholar] [CrossRef] [PubMed]
- Ljung, K.; Bhalerao, R.P.; Sandberg, G. Sites and homeostatic control of auxin biosynthesis in Arabidopsis during vegetative growth. Plant J. 2001, 28, 465–474. [Google Scholar] [CrossRef]
- Cheng, Y.; Dai, X.; Zhao, Y. Auxin synthesized by the YUCCA flavin monooxygenases is essential for embryogenesis and leaf formation in Arabidopsis. Plant Cell 2007, 19, 2430–2439. [Google Scholar] [CrossRef] [PubMed]
- Mashiguchi, K.; Tanaka, K.; Sakai, T.; Sugawara, S.; Kawaide, H.; Natsume, M.; Hanada, A.; Yaeno, T.; Shirasu, K.; Yao, H.; et al. The main auxin biosynthesis pathway in Arabidopsis. Proc. Natl. Acad. Sci. USA 2011, 108, 18512–18517. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Yang, H.; Shang, C.; Ma, S.; Liu, L.; Cheng, J. The Roles of Auxin Biosynthesis YUCCA Gene Family in Plants. Int. J. Mol. Sci. 2019, 20, 6343. [Google Scholar] [CrossRef]
- Mateo-Bonmatí, E.; Casanova-Sáez, R.; Ljung, K. Epigenetic regulation of auxin homeostasis. Biomolecules 2019, 9, 623. [Google Scholar] [CrossRef]
- Pasternak, T.; Potters, G.; Caubergs, R.; Jansen, M.A. Complementary interactions between oxidative stress and auxins control plant growth responses at plant, organ, and cellular level. J. Exp. Bot. 2005, 56, 1991–2001. [Google Scholar] [PubMed]
- Pasternak, T.; Palme, K.; Paponov, I.A. Glutathione Enhances Auxin Sensitivity in Arabidopsis Roots. Biomolecules 2020, 10, 1550. [Google Scholar] [CrossRef] [PubMed]
- Boisnard-Lorig, C.; Colon-Carmona, A.; Bauch, M.; Hodge, S.; Doerner, P.; Bancharel, E.; Dumas, C.; Haseloff, J.; Berger, F. Dynamic analyses of the expression of the HISTONE: YFP fusion protein in Arabidopsis show that syncytial endosperm is divided in mitotic domains. Plant Cell 2001, 13, 495–509. [Google Scholar]
- Pasternak, T.; Tietz, O.; Rapp, K.; Begheldo, M.; Nitschke, R.; Ruperti, B.; Palme, K. Protocol: An improved and universal procedure for whole-mount immunolocalization in plants. Plant Methods 2015, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.; Schmittgen, T.D. Analysis of relative gene expression data using Real-Time Quantitative PCR and the 2—ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [PubMed]
- Sheahan, M.B.; Collings, D.A.; Rose, R.J. ACTIN7 Is Required for Perinuclear Clustering of Chloroplasts during Arabidopsis Protoplast Culture. Plants 2020, 9, 225. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | AM (½ MS) | TK1 |
|---|---|---|
| Macronutrients (mg/L) | ||
| NH4NO3 | 825 | - |
| KNO3 | 950 | 900 |
| NH4H2PO4 | - | 230 |
| MgSO4·7H2O | 180 | 200 |
| KH2PO4 | 68 | - |
| Ca(NO3)2 4H2O | - | 250 |
| CaCl2 2H2O | 220 | - |
| Fe-Chelate (*) | 2.5 ml | 2 ml |
| Micronutrients | ½ micro MS | ½ micro TK |
| Organic compounds (mg/L) | ||
| Bacto-tryptone | - | 150 |
| Vitamines (**) | ½ B5 vitamines | ½ B5 vitamines |
| Myoinositol | 50 | 50 |
| Sucrose | 10000 | 10000 |
| MES | 200 | 200 |
| pH | 5.6 | 5.6 |
| (a). Micronutrient concentrations in the medium (***). | ||
| Micronutrients (mg/L) | micro MS | micro TK |
| H3BO3 | 6.2 | 4 |
| MnCl2 4H2O | 16.8 | 18 |
| ZnSO4 7H2O | 30 | 1.8 |
| Na2MoO4 2H2O | 0.25 | 0.25 |
| NaI | 0.76 | 0.8 |
| CuSO4 5H2O | 0.025 | 0.2 |
| CoCl2 6H2O | 0.025 | 0.2 |
| Components | Concentrations (mg/L) |
|---|---|
| NH4H2PO4 | 200 |
| Ca(NO3)2·4H2O | 400 |
| KNO3 | 1500 |
| MgSO4·7H2O | 300 |
| KH2PO4 | 200 |
| CaCl2·2H2O | 80 |
| Fe-chelat (*) | 4 mL from 20 mM stock * |
| Glucose | 72000 |
| Mannitol | 18000 |
| Sucrose | 12000 |
| Micronutrients | ½ micro TK1 ** |
| Glutamine | 100 |
| Vitamins | 1 mL from the stock *** |
| Myoinositol | 200 |
| Bacto-tryptone | 150 |
| Na-citrate | 40 |
| Na-pyruvate | 20 |
| Na-fumarate | 40 |
| NH4 H2PO4 | 250 mg |
| (NH4 )2 SO4 | 400 mg |
| KNO3 | 2000 mg |
| MgSO4·7H2O | 450 mg |
| Ca(NO3)2 ·4H2O | 350 mg |
| Fe-chelat | 3 ml * |
| Micro TK (see Table 1(a)) | 1 ml ** |
| MES | 200 mg |
| B5 Vitamins | 1ml *** |
| Bacto tryptone | 200 mg |
| Myoinositol | 100 mg |
| Sucrose | 30 g |
| pH | 5.7 |
| Gene | Forward | Reverse |
|---|---|---|
| IAA2 | CGACGCTCCTGCTCTAGACT | AAAACCCCGAAGTTTCGTCT |
| TIR1 | AGATAAGGGACTGCCCGTTT | GACCAGCCACTGTTCGGTAT |
| ACT2 | CGCTATGTATGTCGCCA | CTTGCCCATCGGGTAA |
| EMB1 | AACGAGCTCTTCGTCGTCGCCGC | GAGAGGACACCACGATCACC |
| YUC1 | CCTAGAACGGTCGGATTCAA | AGTGGGAAGCGTAGGACTCA |
| YUC2 | TGCTCAAGTGGTTTCCAGTG | CCAACGTCCAAAACAGGAGT |
| YUC4 | TGGGCAATACCGACCTTTTA | AAAGAAATGGCACCGACATC |
| TAA1 | GATGAAGAATCGGTGGGAGAAGC | CGTCCCTAGCCACGCAAACGCAGG |
| TAR2 | CATGATTTGGCTTACTATTGGCCACAG | GTCTTTCACCAAAGCCCATCCAATC |
| RPL27 | ACCCACCTGCTGAGCTTGAGA | GGCAGTTTCCGCACACCACA |
| RPL3A | TTGGTGCGTGGCATCCTGCT | TGGCTGTGTGTGCCTCAGTACCA |
| RPS13A | GCTCATGGCCTTGCTCCTGAGA | GCGAGCGAGGCGGTGAATCC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pasternak, T.; Paponov, I.A.; Kondratenko, S. Optimizing Protocols for Arabidopsis Shoot and Root Protoplast Cultivation. Plants 2021, 10, 375. https://doi.org/10.3390/plants10020375
Pasternak T, Paponov IA, Kondratenko S. Optimizing Protocols for Arabidopsis Shoot and Root Protoplast Cultivation. Plants. 2021; 10(2):375. https://doi.org/10.3390/plants10020375
Chicago/Turabian StylePasternak, Taras, Ivan A. Paponov, and Serhii Kondratenko. 2021. "Optimizing Protocols for Arabidopsis Shoot and Root Protoplast Cultivation" Plants 10, no. 2: 375. https://doi.org/10.3390/plants10020375
APA StylePasternak, T., Paponov, I. A., & Kondratenko, S. (2021). Optimizing Protocols for Arabidopsis Shoot and Root Protoplast Cultivation. Plants, 10(2), 375. https://doi.org/10.3390/plants10020375