
Comparative Transcriptomic Analysis of Gene Expression Inheritance Patterns Associated with Cabbage Head Heterosis


 ,
,
Abstract
1. Introduction
2. Results
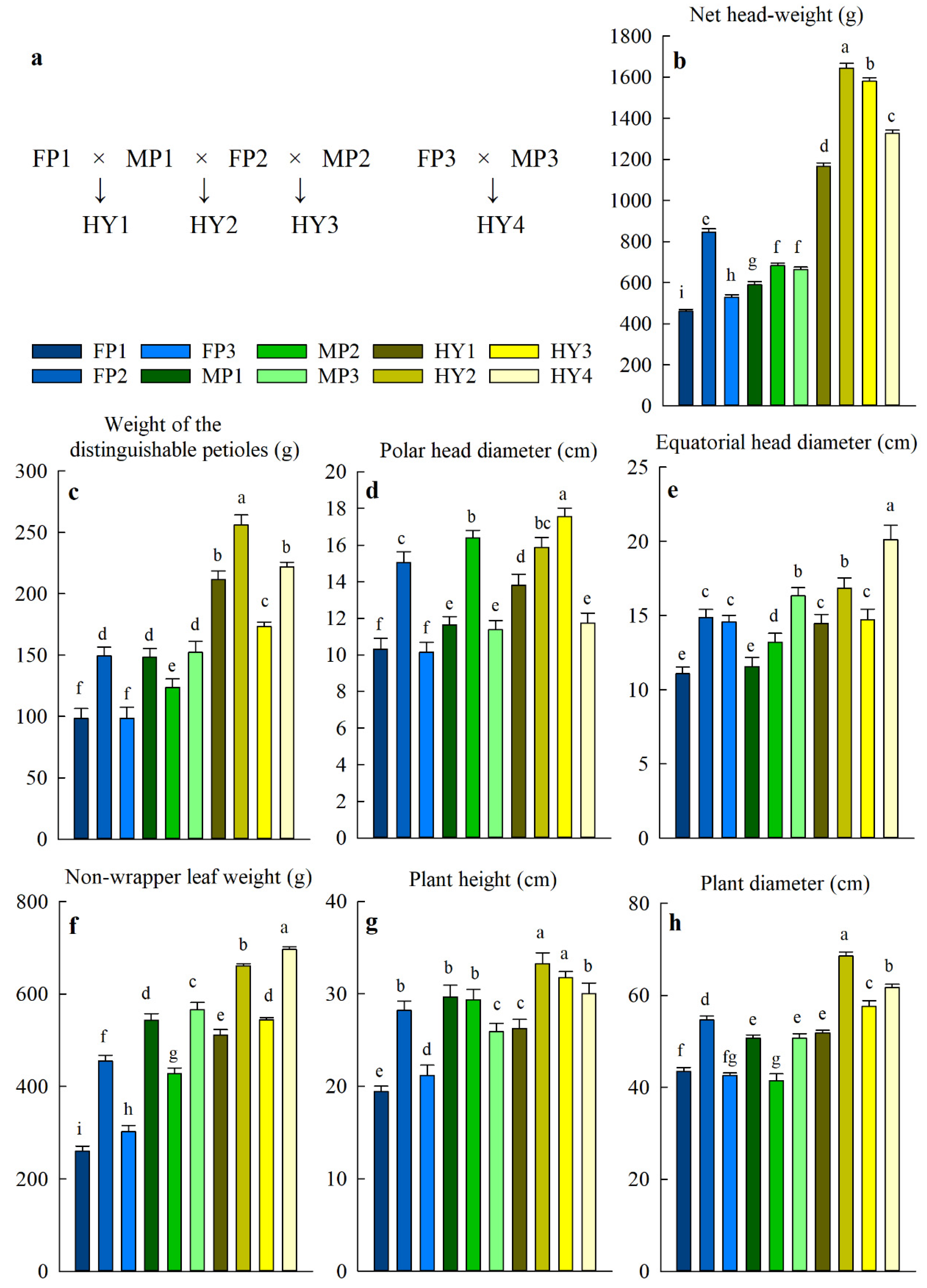
2.1. Cabbage Hybrids Show Heterosis in Several Horticultural Traits
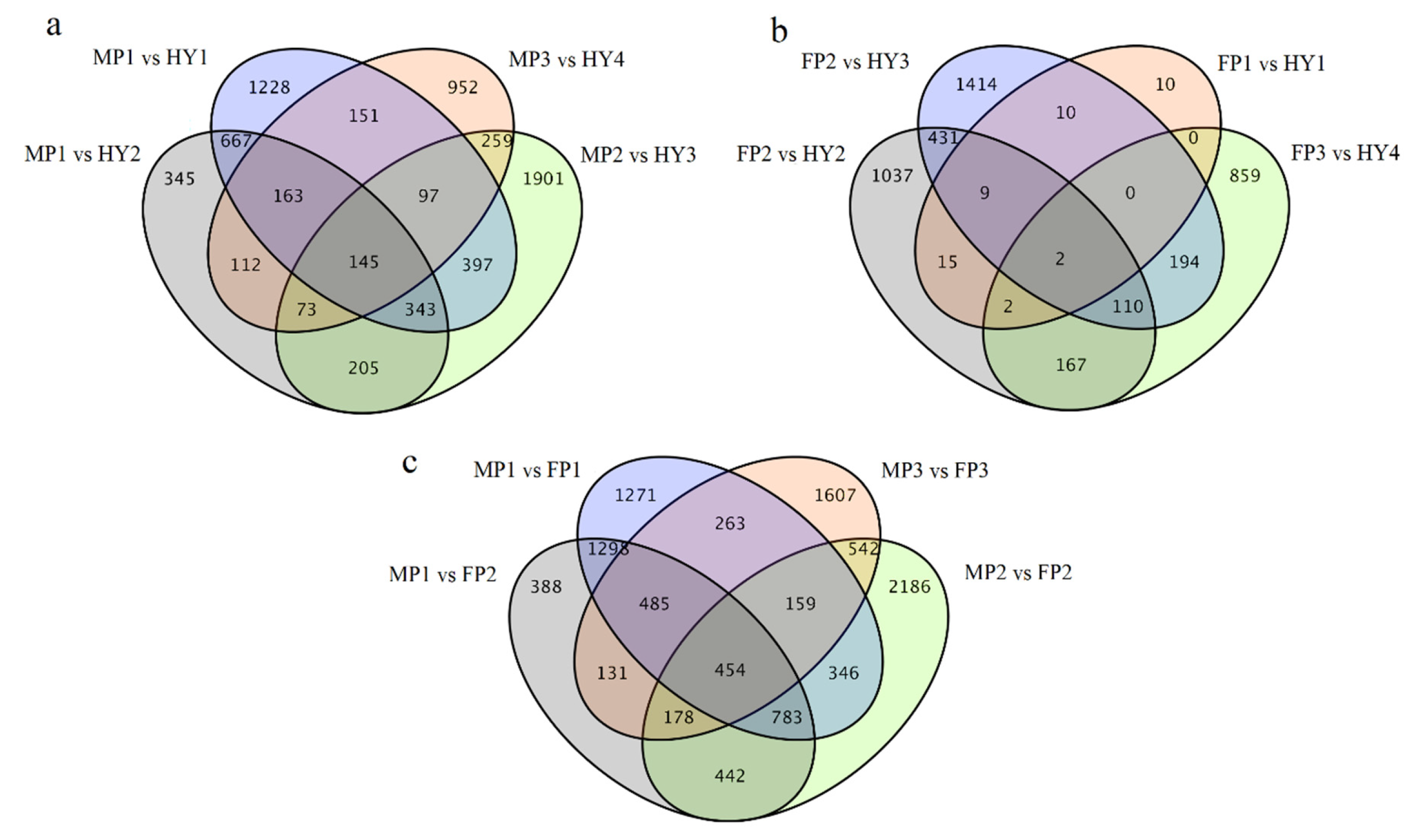
2.2. Genetic Variations and Gene Expression Divergence between Hybrids and Their Parents
2.3. Functions of DEGs Associated with Cabbage Head Heterosis
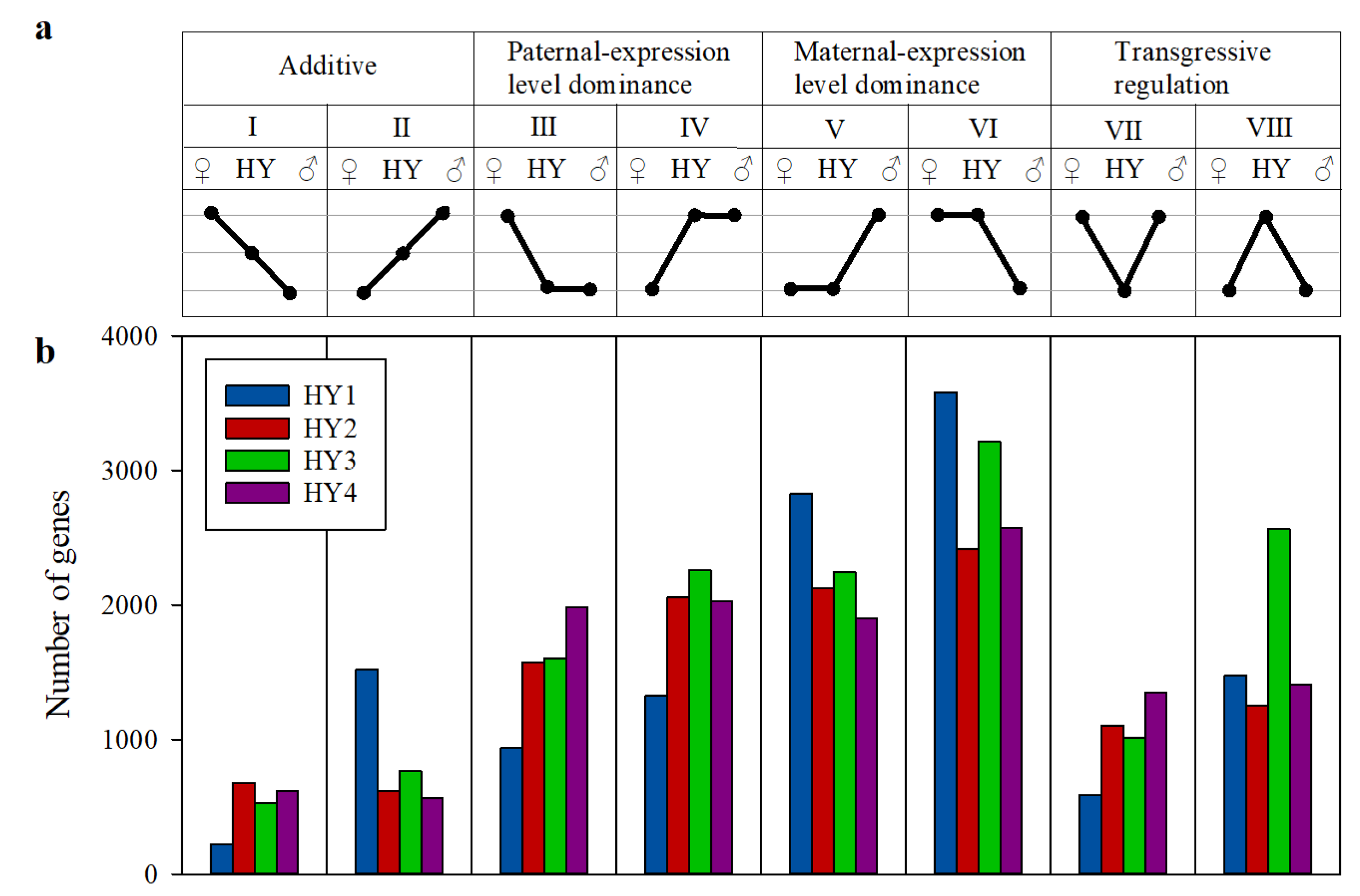
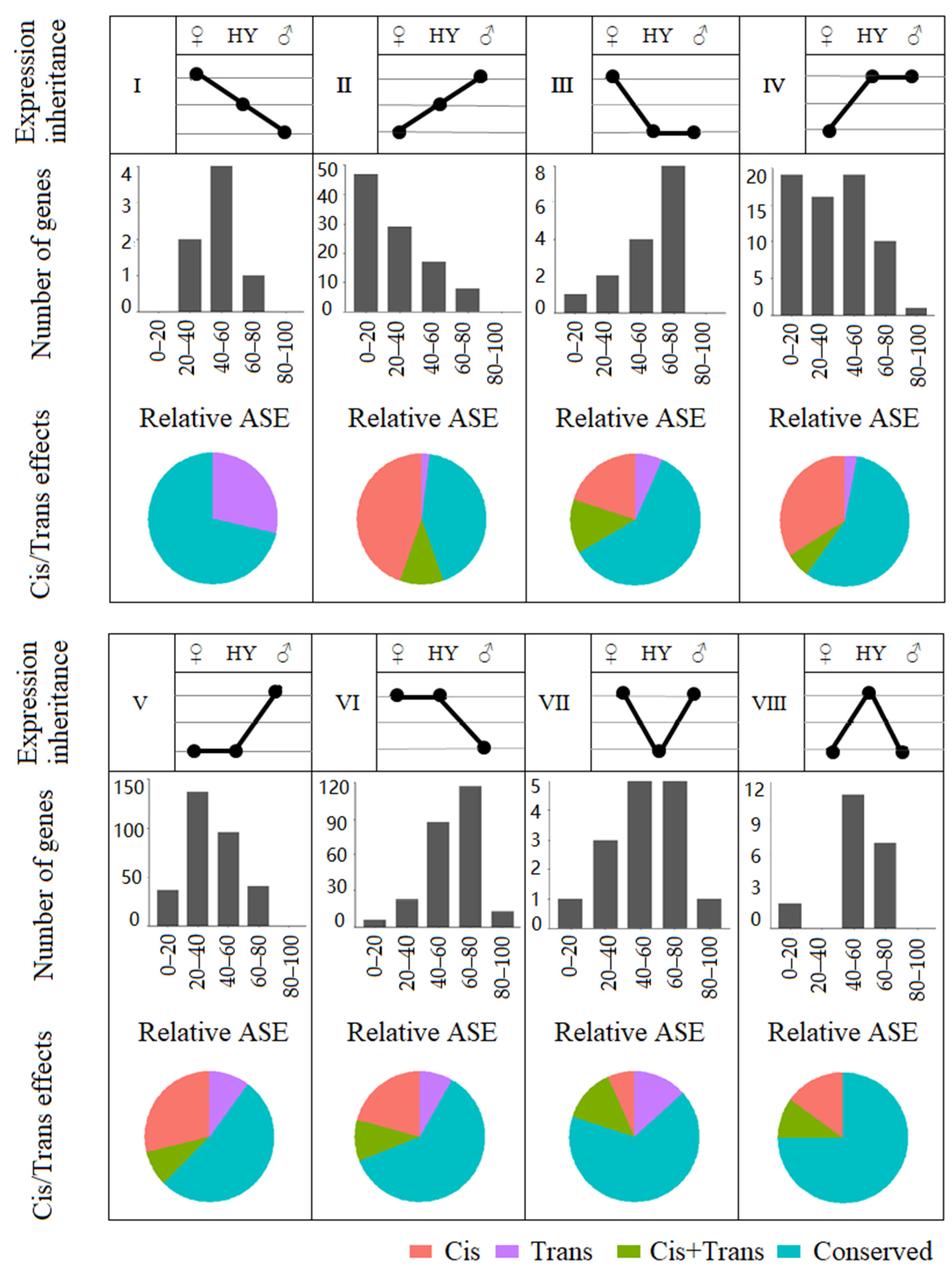
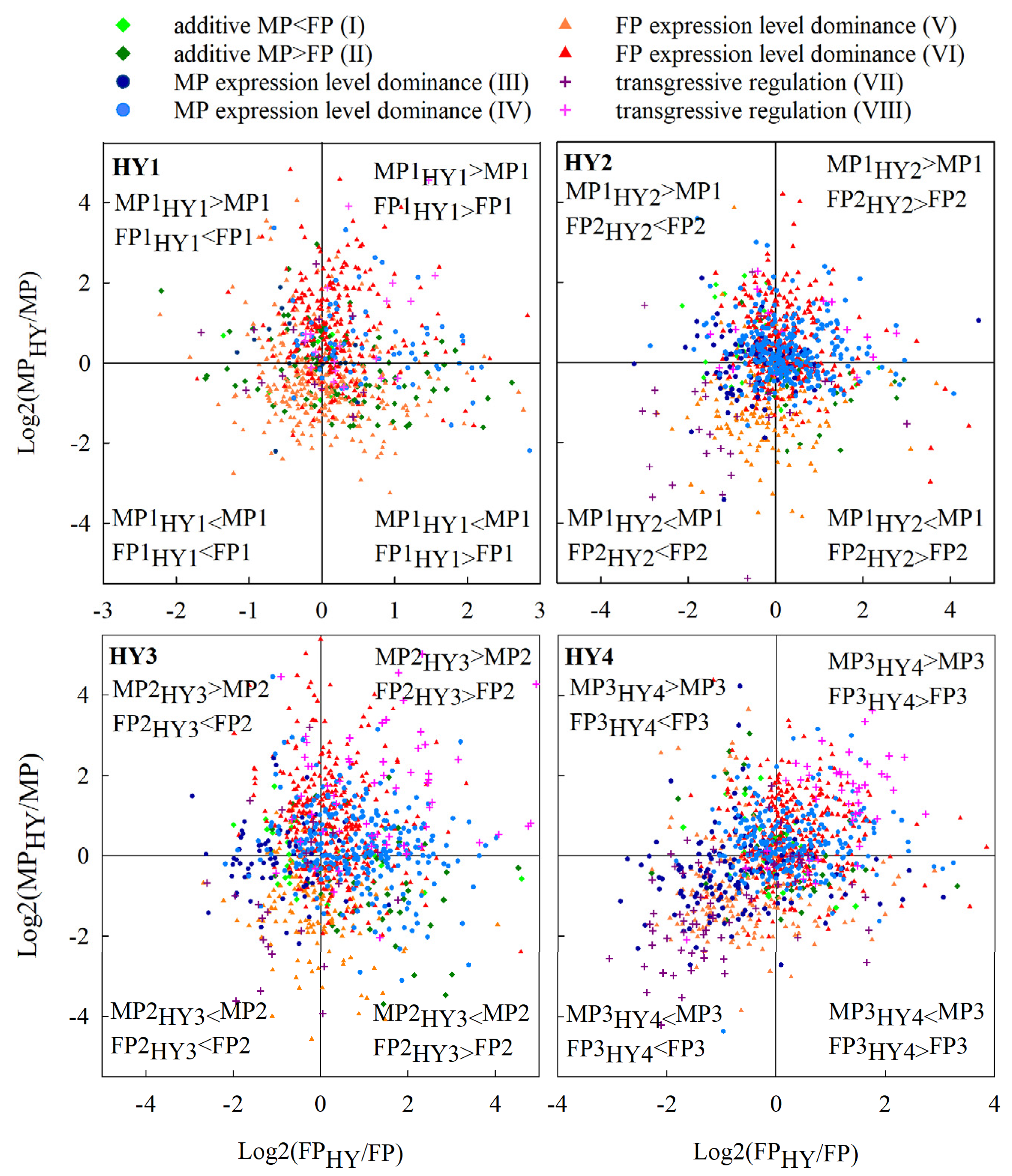
2.4. Gene Expression Inheritance Patterns in the Cabbage Hybrids
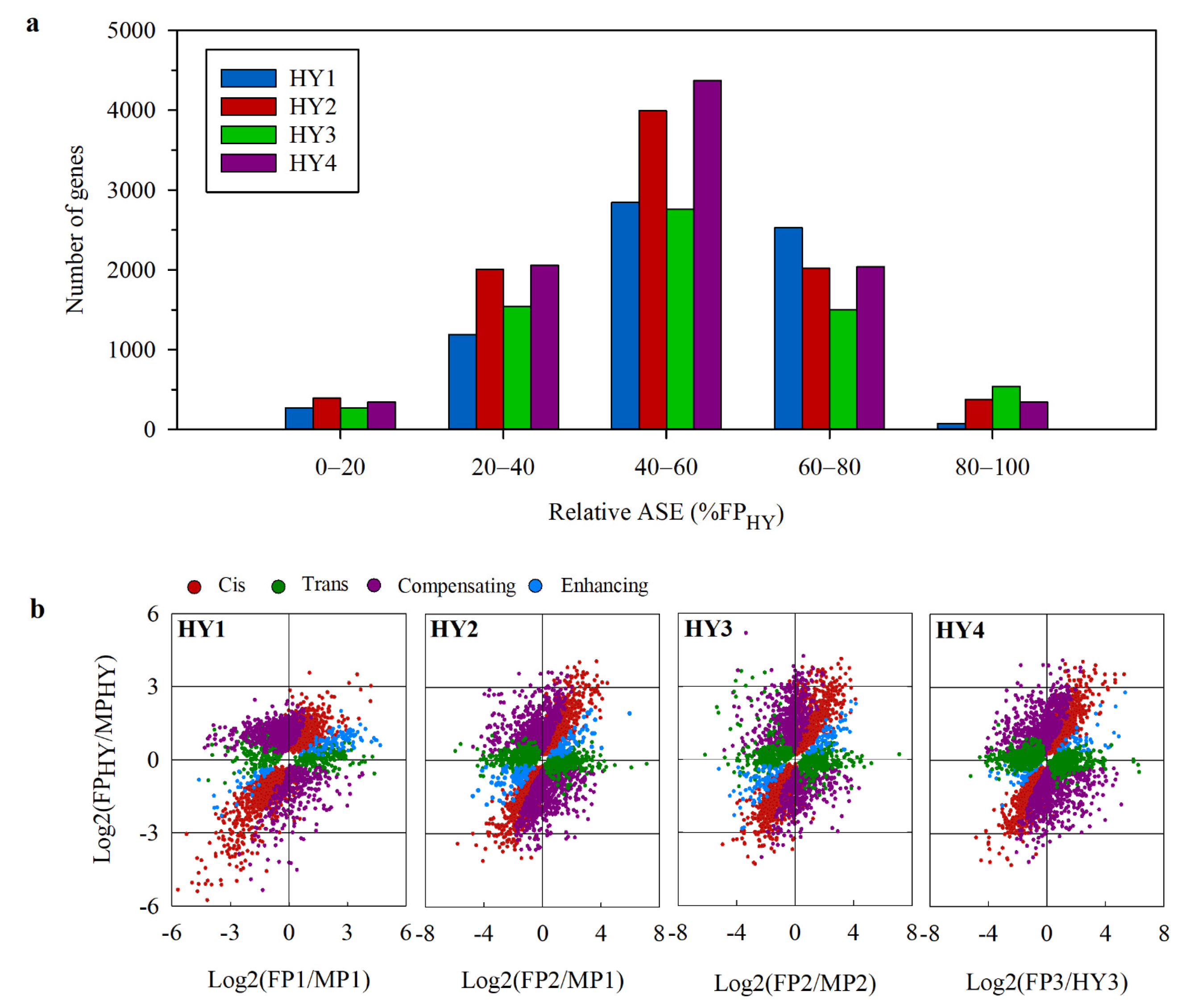
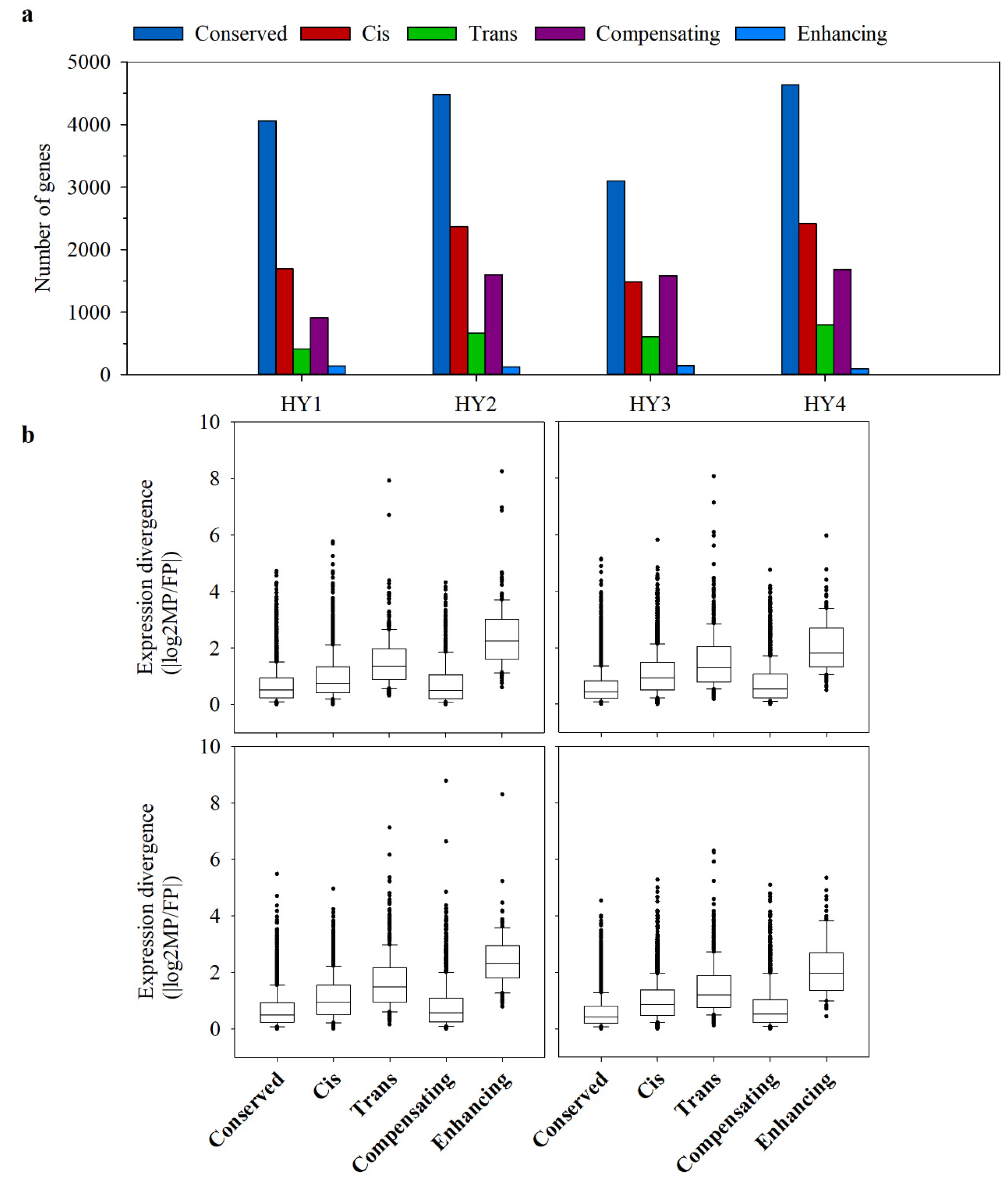
2.5. Cis- and Trans-Effects on Gene Expression Divergence
2.6. The Relationship between Allelic Expression Regulation and Expression Inheritance in Hybrids
3. Discussion
3.1. Gene Expression Divergence between Cabbage Hybrids and Their Parents
3.2. Maternal Expression Level Dominance is Predominant in Cabbage Hybrids
3.3. Cis-Regulation Predominates the Hybrid Gene Expression Divergence
4. Materials and Methods
4.1. Plant Materials and Measurement of Horticultural Traits
4.2. RNA Extraction and Sequencing
4.3. Quality Control and Read Mapping
4.4. Validation of RNA-Seq Data by qRT-PCR
4.5. Analysis of the SNP Sites and the DEGs
4.6. Inheritance Classification and Cis- and Trans-Regulatory Effects
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ryder, P.; McKeown, P.C.; Fort, A.; Spillane, C. Epigenetics and heterosis in crop plants. In Epigenetics in Plants of Agronomic Importance: Fundamentals and Applications; Springer: Cham, Switzeerland, 2014; pp. 129–147. [Google Scholar] [CrossRef]
- Gallais, A. Heterosis: Its genetic basis and its utilisation in plant breeding. Euphytica 1988, 39, 95–104. [Google Scholar] [CrossRef]
- Rokayya, S.; Li, C.-J.; Zhao, Y.; Li, Y.; Sun, C.-H. Cabbage (Brassica oleracea L. var. capitata) phytochemicals with antioxidant and anti-inflammatory potential. Asian Pac. J. Cancer Prev. 2013, 14, 6657–6662. [Google Scholar] [CrossRef] [PubMed]
- Hochholdinger, F.; Hoecker, N. Towards the molecular basis of heterosis. Trends Plant Sci. 2007, 12, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.J. Genomic and epigenetic insights into the molecular bases of heterosis. Nat. Rev. Genet. 2013, 14, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Goff, S.A. A unifying theory for general multigenic heterosis: Energy efficiency, protein metabolism, and implications for molecular breeding. New Phytol. 2011, 189, 923–937. [Google Scholar] [CrossRef] [PubMed]
- Fiévet, J.B.; Nidelet, T.; Dillmann, C.; De Vienne, D. Heterosis is a systemic property emerging from non-linear genotype-phenotype relationships: Evidence from in vitro genetics and computer simulations. Front. Genet. 2018, 9, 159. [Google Scholar] [CrossRef] [PubMed]
- Kaeppler, S. Heterosis: One boat at a time, or a rising tide? New Phytol. 2011, 189, 900–902. [Google Scholar] [CrossRef]
- Fu, D.; Xiao, M.; Hayward, A.; Jiang, G.; Zhu, L.; Zhou, Q.; Li, J.; Zhang, M. What is crop heterosis: New insights into an old topic. J. Appl. Genet. 2015, 56, 1–13. [Google Scholar] [CrossRef]
- Li, D.; Zeng, R.; Li, Y.; Zhao, M.; Chao, J.; Li, Y.; Wang, K.; Zhu, L.; Tian, W.-M.; Liang, C. Gene expression analysis and SNP/InDel discovery to investigate yield heterosis of two rubber tree F1 hybrids. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef]
- Hu, X.; Wang, H.; Diao, X.; Liu, Z.; Li, K.; Wu, Y.; Liang, Q.; Wang, H.; Huang, C. Transcriptome profiling and comparison of maize ear heterosis during the spikelet and floret differentiation stages. BMC Genom. 2016, 17, 959. [Google Scholar] [CrossRef]
- Swanson-Wagner, R.A.; Jia, Y.; DeCook, R.; Borsuk, L.A.; Nettleton, D.; Schnable, P.S. All possible modes of gene action are observed in a global comparison of gene expression in a maize F1 hybrid and its inbred parents. Proc. Natl. Acad. Sci. USA 2006, 103, 6805–6810. [Google Scholar] [CrossRef] [PubMed]
- Yoo, M.-J.; Szadkowski, E.; Wendel, J.F. Homoeolog expression bias and expression level dominance in allopolyploid cotton. Heredity 2013, 110, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Dapp, M.; Reinders, J.; Bédiée, A.; Balsera, C.; Bucher, E.; Theiler, G.; Granier, C.; Paszkowski, J. Heterosis and inbreeding depression of epigenetic Arabidopsis hybrids. Nat. Plants 2015, 1, 15092. [Google Scholar] [CrossRef] [PubMed]
- Tian, M.; Nie, Q.; Li, Z.; Zhang, J.; Liu, Y.; Long, Y.; Wang, Z.; Wang, G.; Liu, R. Transcriptomic analysis reveals overdominance playing a critical role in nicotine heterosis in Nicotiana tabacum L. BMC Plant Biol. 2018, 18, 1–10. [Google Scholar] [CrossRef]
- Guo, M.; Rupe, M.A.; Zinselmeier, C.; Habben, J.; Bowen, B.A.; Smith, O.S. Allelic variation of gene expression in maize hybrids. Plant Cell. 2004, 16, 1707–1716. [Google Scholar] [CrossRef]
- Springer, N.M.; Stupar, R.M. Allele-specific expression patterns reveal biases and embryo-specific parent-of-origin effects in hybrid maize. Plant Cell. 2007, 19, 2391–2402. [Google Scholar] [CrossRef]
- Fontanillas, P.; Landry, C.R.; Wittkopp, P.J.; Russ, C.; Gruber, J.D.; Nusbaum, C.; Hartl, D.L. Key considerations for measuring allelic expression on a genomic scale using high-throughput sequencing. Mol. Ecol. 2010, 19, 212–227. [Google Scholar] [CrossRef]
- Stupar, R.M.; Springer, N.M. Cis-transcriptional variation in maize inbred lines B73 and Mo17 leads to additive expression patterns in the F1 hybrid. Genetics 2006, 173, 2199–2210. [Google Scholar] [CrossRef]
- Bell, G.D.; Kane, N.C.; Rieseberg, L.H.; Adams, K.L. RNA-Seq analysis of allele-specific expression, hybrid effects, and regulatory divergence in hybrids compared with their parents from natural populations. Genome Biol. Evol. 2013, 5, 1309–1323. [Google Scholar] [CrossRef]
- Shi, X.; Ng, D.W.-K.; Zhang, C.; Comai, L.; Ye, W.; Chen, Z.J. Cis- and trans-regulatory divergence between progenitor species determines gene-expression novelty in Arabidopsis allopolyploids. Nat. Commun. 2012, 3, 950–959. [Google Scholar] [CrossRef]
- Singh, B.; Sharma, S.; Singh, B. Heterosis for mineral elements in single cross-hybrids of cabbage (Brassica oleracea var. capitata L.). Sci. Hortic. 2009, 122, 32–36. [Google Scholar] [CrossRef]
- Singh, B.K.; Sharma, S.R.; Singh, B. Genetic combining ability for concentration of mineral elements in cabbage head (Brassica oleracea var. capitata L.). Euphytica 2012, 184, 265–273. [Google Scholar] [CrossRef]
- Zhang, X.; Su, Y.; Liu, Y.; Fang, Z.; Yang, L.; Zhuang, M.; Zhang, Y.; Li, Z.; Lv, H. Genetic analysis and QTL mapping of traits related to head shape in cabbage (Brassica oleracea var. capitata L.). Sci. Hortic. 2016, 207, 82–88. [Google Scholar] [CrossRef]
- Broadley, M.R.; Hammond, J.P.; King, G.J.; Astley, D.; Bowen, H.C.; Meacham, M.C.; Mead, A.; Pink, D.A.; Teakle, G.R.; Hayden, R.M.; et al. Shoot calcium and magnesium concentrations differ between subtaxa, are highly heritable, and associate with potentially pleiotropic loci in Brassica oleracea. Plant Physiol. 2008, 146, 1707–1720. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.-Y.; Ahmed, N.U.; Jung, H.-J.; Kim, H.-T.; Park, J.-I.; Nou, I.S. Discovery of candidate genes for heterosis breeding in Brassica oleracea L. Acta Physiol. Plant 2017, 39, 1–12. [Google Scholar] [CrossRef]
- Moon, J.; Hake, S. How a leaf gets its shape. Curr. Opin. Plant Biol. 2011, 14, 24–30. [Google Scholar] [CrossRef]
- Dubos, C.; Stracke, R.; Grotewold, E.; Weisshaar, B.; Martin, C.; Lepiniec, L. MYB transcription factors in Arabidopsis. Trends Plant Sci. 2010, 15, 573–581. [Google Scholar] [CrossRef]
- Blein, T.; Hasson, A.; Laufs, P. Leaf development: What it needs to be complex. Curr. Opin. Plant Biol. 2010, 13, 75–82. [Google Scholar] [CrossRef]
- Krogan, N.T.; Berleth, T. The identification and characterization of specific ARF-Aux/IAA regulatory modules in plant growth and development. Plant Signal Behav. 2015, 10, e992748. [Google Scholar] [CrossRef]
- Combes, M.-C.; Hueber, Y.; Dereeper, A.; Rialle, S.; Herrera, J.-C.; Lashermes, P. Regulatory divergence between parental alleles determines gene expression patterns in hybrids. Genome Biol. Evol. 2015, 7, 1110–1121. [Google Scholar] [CrossRef]
- Wittkopp, P.J.; Haerum, B.K.; Clark, A.G. Evolutionary changes in cis and trans gene regulation. Nature 2004, 430, 85–88. [Google Scholar] [CrossRef] [PubMed]
- Mcmanus, C.J.; Coolon, J.D.; Duff, M.O.; Eipper-Mains, J.; Graveley, B.R.; Wittkopp, P.J. Regulatory divergence in Drosophila revealed by mRNA-seq. Genome Res. 2010, 20, 816–825. [Google Scholar] [CrossRef] [PubMed]
- Wei, G.; Tao, Y.; Liu, G.; Chen, C.; Luo, R.; Xia, H.; Gan, Q.; Zeng, H.; Lu, Z.; Han, Y.; et al. A transcriptomic analysis of superhybrid rice LYP9 and its parents. Proc. Natl. Acad. Sci. USA 2009, 106, 7695–7701. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Yang, S.; Gong, J.; Zhao, Q.; Feng, Q.; Zhan, Q.; Zhao, Y.; Li, W.; Cheng, B.; Xia, J.; et al. Genomic architecture of heterosis for yield traits in rice. Nature 2016, 537, 629–633. [Google Scholar] [CrossRef]
- Botet, R.; Keurentjes, J.J. The role of transcriptional regulation in hybrid vigor. Front. Plant Sci. 2020, 11, 410. [Google Scholar] [CrossRef]
- Gehring, M. Genomic imprinting: Insights from plants. Annu. Rev. Genet. 2013, 47, 187–208. [Google Scholar] [CrossRef]
- Guo, M.; Rupe, M.A.; Yang, X.; Crasta, O.; Zinselmeier, C.; Smith, O.S.; Bowen, B. Genome-wide transcript analysis of maize hybrids: Allelic additive gene expression and yield heterosis. Theor. Appl. Genet. 2006, 113, 831–845. [Google Scholar] [CrossRef]
- Lisec, J.; Steinfath, M.; Meyer, R.C.; Selbig, J.; Melchinger, A.E.; Willmitzer, L.; Altmann, T. Identification of heterotic metabolite QTL in Arabidopsis thaliana RIL and IL populations. Plant J. 2009, 59, 777–788. [Google Scholar] [CrossRef]
- Zhang, T.-F.; Li, B.; Zhang, D.-F.; Jia, G.-Q.; Li, Z.-Y.; Wang, S. Genome-wide transcriptional analysis of yield and heterosis-associated genes in maize (Zea mays L.). J. Integr. Agric. 2012, 11, 1245–1256. [Google Scholar] [CrossRef]
- Zhai, R.; Feng, Y.; Wang, H.; Zhan, X.; Shen, X.; Wu, W.; Cao, L.-Y.; Chen, D.; Dai, G.; Yang, Z.; et al. Transcriptome analysis of rice root heterosis by RNA-Seq. BMC Genom. 2013, 14, 19. [Google Scholar] [CrossRef]
- Zhang, H.-Y.; He, H.; Chen, L.; Li, L.; Liang, M.-Z.; Wang, X.; Liu, X.-G.; He, G.-M.; Chen, R.-S.; Ma, L.; et al. A genome-wide transcription analysis reveals a close correlation of promoter INDEL polymorphism and heterotic gene expression in rice hybrids. Mol. Plant 2008, 1, 720–731. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Zhang, D.-F.; Jia, G.-Q.; Dai, J.-R.; Wang, S.-C. Genome-wide comparisons of gene expression for yield heterosis in maize. Plant Mol. Biol. Rep. 2009, 27, 162–176. [Google Scholar] [CrossRef]
- Groszmann, M.; Gonzalez-Bayon, R.; Lyons, R.L.; Greaves, I.K.; Kazan, K.; Peacock, W.J.; Dennis, E.S. Hormone-regulated defense and stress response networks contribute to heterosis in Arabidopsis F1 hybrids. Proc. Natl. Acad. Sci. USA 2015, 112, E6397–E6406. [Google Scholar] [CrossRef] [PubMed]
- Rédei, G.P. Single locus heterosis. Mol. Gen. Genet. 1962, 93, 164–170. [Google Scholar]
- Ma, J.; Li, J.; Cao, Y.; Wang, L.; Wang, F.; Wang, H.; Li, H.-Y. Comparative study on the transcriptome of maize mature embryos from two China elite hybrids Zhengdan958 and Anyu5. PLoS ONE 2016, 11, e0158028. [Google Scholar] [CrossRef]
- Taliercio, E.; Eickholt, D.; Rouf, R.; Carter, T. Changes in gene expression between a soybean F1 hybrid and its parents are associated with agronomically valuable traits. PLoS ONE 2017, 12, e0177225. [Google Scholar] [CrossRef]
- Zhang, J.; Li, G.; Li, H.; Pu, X.; Jiang, J.; Chai, L.; Zheng, B.; Cui, C.; Yang, Z.; Zhu, Y.; et al. Transcriptome analysis of interspecific hybrid between Brassica napus and B. rapa reveals heterosis for oil rape improvement. Int. J. Genom. 2015, 2015, 230985. [Google Scholar]
- Jindal, S.K.; Thakur, J.C. Genetic architecture of quantitative characters in November maturity cauliflower. Crop Improv. 2005, 32, 92–94. [Google Scholar]
- Meyer, R.C.; Witucka-Wall, H.; Becher, M.; Blacha, A.; Boudichevskaia, A.; Dörmann, P.; Fiehn, O.; Friedel, S.; Von Korff, M.; Lisec, J.; et al. Heterosis manifestation during early Arabidopsis seedling development is characterized by intermediate gene expression and enhanced metabolic activity in the hybrids. Plant J. 2012, 71, 669–683. [Google Scholar] [CrossRef]
- Lemos, B.; Araripe, L.O.; Fontanillas, P.; Hartl, D.L. Dominance and the evolutionary accumulation of cis- and trans-effects on gene expression. Proc. Natl. Acad. Sci. USA 2008, 105, 14471–14476. [Google Scholar] [CrossRef]
- Vuylsteke, M.; Van Eeuwijk, F.; Van Hummelen, P.; Kuiper, M.; Zabeau, M. Genetic analysis of variation in gene expression in Arabidopsis thaliana. Genetics 2005, 171, 1267–1275. [Google Scholar] [CrossRef] [PubMed]
- A Signor, S.; Nuzhdin, S.V. The evolution of gene expression in cis and trans. Trends Genet. 2018, 34, 532–544. [Google Scholar] [CrossRef] [PubMed]
- Cox, M.P.; Dong, T.; Shen, G.; Dalvi, Y.B.; Scott, D.B.; Ganley, A.R.D. An interspecific fungal hybrid reveals cross-kingdom rules for allopolyploid gene expression patterns. PLoS Genet. 2014, 10, e1004180. [Google Scholar] [CrossRef] [PubMed]
- Hiroshi, O. Studies on the new male-sterility in Japanese radish, with special reference to the utilization of this sterility towerds the practical raising of hybrid seeds. Mem. Fac. Agric. Kagoshima Univ. 1968, 6, 40–75. [Google Scholar]
- Peng, Y.; Shi, D.; Zhang, T.; Li, X.; Fu, T.; Xu, Y.; Wan, Z. Development and utilization of an efficient cytoplasmic male sterile system for Cai-xin (Brassica rapa L.). Sci. Hortic. 2015, 190, 36–42. [Google Scholar] [CrossRef]
- Liu, S.; Liu, Y.; Yang, X.; Tong, C.; Edwards, D.; Parkin, I.A.P.; Zhao, M.; Ma, J.; Yu, J.; Huang, S.; et al. The Brassica oleracea genome reveals the asymmetrical evolution of polyploid genomes. Nat. Commun. 2014, 5, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Chan, C.-K.K. Analysis of RNA-Seq Data Using TopHat and Cufflinks; Humana Press Inc.: Totowa, NJ, USA, 2016; Volume 1374, pp. 339–361. [Google Scholar]
- Jiang, H.; Wong, W. Statistical inferences for isoform expression in RNA-Seq. Bioinformatics 2009, 25, 1026–1032. [Google Scholar] [CrossRef]
- Sims, D.W.; Sudbery, I.M.; Ilott, N.E.; Heger, A.; Ponting, C.P. Sequencing depth and coverage: Key considerations in genomic analyses. Nat. Rev. Genet. 2014, 15, 121–132. [Google Scholar] [CrossRef]
- Kumar, V.; Sharma, R.; Trivedi, P.C.; Vyas, G.K.; Khandelwal, V. Traditional and novel references towards systematic normalization of qRT-PCR data in plants. Aust. J. Crop Sci. 2011, 5, 1455–1468. [Google Scholar]
- Adnan, M.; Morton, G.; Hadi, S. Analysis of rpoS and bolA gene expression under various stress-induced environments in planktonic and biofilm phase using 2-△△CT method. Mol. Cell Biochem. 2011, 357, 275–282. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map (SAM) format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.B.; Daly, M.J.; et al. The genome analysis toolkit: A mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, 1. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hybrid | Net Head- Weight | Weight of the Distinguishable Petioles | Polar Head Diameter | Equatorial Head Diameter | Non- Wrapper Leaf Weight | Plant Height | Plant Diameter |
|---|---|---|---|---|---|---|---|
| MPH (%)╪ | |||||||
| HY1 | 121.9 ± 2.2 | 71.7 ± 4.9 | 25.9 ± 0.6 | 27.9 ± 0.7 | 27.3 ± 1.0 | 7.1 ± 1.4 | 10.2 ± 0.7 |
| HY2 | 129.2 ± 4.4 | 72.6 ± 6.8 | 18.9 ± 0.6 | 27.4 ± 0.7 | 32.4 ± 2.7 | 15.1 ± 1.3 | 30.2 ± 0.2 |
| HY3 | 106.7 ± 3.5 | 27.1 ± 4.8 | 11.8 ± 0.7 | 5.0 ± 0.8 | 23.3 ± 2.3 | 10.4 ± 1.8 | 19.8 ± 0.3 |
| HY4 | 122.6 ± 4.1 | 77.4 ± 10.0 | 9.1 ± 0.2 | 30.3 ± 2.2 | 60.5 ± 4.0 | 27.5 ± 0.6 | 32.3 ± 0.6 |
| HPH (%) | |||||||
| HY1 | 97.5 ± 2.3 | 42.6 ± 2.0 | 18.6 ± 0.7 | 25.1 ± 1.3 | −5.9 ± 0.2 | −11.3 ± 1.3 | 2.3 ± 0.3 |
| HY2 | 94.5 ± 3.2 | 72.1 ± 6.8 | 5.4 ± 0.7 | 13.2 ± 0.6 | 21.7 ± 2.4 | 12.3 ± 1.8 | 25.5 ± 0.5 |
| HY3 | 86.7 ± 3.1 | 16.1 ± 3.9 | 7.1 ± 0.3 | −0.9 ± 1.1 | 19.6 ± 2.2 | 7.9 ± 5.1 | 5.3 ± 0.8 |
| HY4 | 99.9 ± 3.2 | 45.9 ± 6.2 | 3.1 ± 0.3 | 23.3 ± 2.1 | 23.1 ± 2.3 | 15.8 ± 0.5 | 21.7 ± 0.9 |
| Genetic Variation | Hybrid | |||
|---|---|---|---|---|
| HY1 | HY2 | HY3 | HY4 | |
| SNPs | ||||
| Number of hybrid specific SNPs | 5180 | 11,057 | 22,290 | 9698 |
| Number of SNPs consistent with both parents | 44,795 | 47,246 | 46,570 | 45,092 |
| Number of SNPs consistent with the male parent | 54,757 | 72,233 | 62,763 | 72,271 |
| Number of SNPs consistent with the female parent | 75,299 | 77,013 | 76,463 | 72,426 |
| InDels | ||||
| Number of hybrid specific InDels | 457 | 677 | 921 | 591 |
| Number of InDels consistent with both parents | 1309 | 1415 | 1433 | 1380 |
| Number of InDels consistent with the male parent | 1642 | 2124 | 2007 | 2189 |
| Number of InDels consistent with the female parent | 2359 | 2428 | 2398 | 2258 |
| Comparison Group | Number of DEGs | Up-Regulated | Down-Regulated |
|---|---|---|---|
| FP1 × MP1→HY1 | |||
| FP1 vs. HY1 | 48 | 25 | 23 |
| MP1 vs. HY1 | 3191 | 1562 | 1629 |
| FP2 × MP1→HY2 | |||
| FP2 vs. HY2 | 1773 | 1142 | 631 |
| MP1 vs. HY2 | 2053 | 1272 | 781 |
| FP2 × MP2→HY3 | |||
| FP2 vs. HY3 | 2170 | 1454 | 716 |
| MP2 vs. HY3 | 3420 | 2098 | 1322 |
| FP3 × MP3→HY4 | |||
| FP3 vs. HY4 | 1334 | 874 | 460 |
| MP3 vs. HY4 | 1952 | 1118 | 834 |
| Hybrids/parents | |||
| HY1 vs. HY2 | 948 | 523 | 425 |
| HY1 vs. HY3 | 1294 | 870 | 424 |
| HY1 vs. HY4 | 1991 | 1082 | 909 |
| HY2 vs. HY3 | 2342 | 1293 | 1049 |
| HY2 vs. HY4 | 2872 | 1208 | 1664 |
| HY3 vs. HY4 | 2475 | 1053 | 1422 |
| MP1 vs. MP2 | 5139 | 2369 | 2770 |
| MP1 vs. MP3 | 4391 | 2047 | 2344 |
| MP2 vs. MP3 | 4915 | 2503 | 2412 |
| FP1 vs. FP2 | 2011 | 1132 | 879 |
| FP1 vs. FP3 | 3683 | 1933 | 1750 |
| FP2 vs. FP3 | 3699 | 1792 | 1907 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, S.; Jayasinghege, C.P.A.; Guo, J.; Zhang, E.; Wang, X.; Xu, Z. Comparative Transcriptomic Analysis of Gene Expression Inheritance Patterns Associated with Cabbage Head Heterosis. Plants 2021, 10, 275. https://doi.org/10.3390/plants10020275
Li S, Jayasinghege CPA, Guo J, Zhang E, Wang X, Xu Z. Comparative Transcriptomic Analysis of Gene Expression Inheritance Patterns Associated with Cabbage Head Heterosis. Plants. 2021; 10(2):275. https://doi.org/10.3390/plants10020275
Chicago/Turabian StyleLi, Shengjuan, Charitha P. A. Jayasinghege, Jia Guo, Enhui Zhang, Xingli Wang, and Zhongmin Xu. 2021. "Comparative Transcriptomic Analysis of Gene Expression Inheritance Patterns Associated with Cabbage Head Heterosis" Plants 10, no. 2: 275. https://doi.org/10.3390/plants10020275
APA StyleLi, S., Jayasinghege, C. P. A., Guo, J., Zhang, E., Wang, X., & Xu, Z. (2021). Comparative Transcriptomic Analysis of Gene Expression Inheritance Patterns Associated with Cabbage Head Heterosis. Plants, 10(2), 275. https://doi.org/10.3390/plants10020275