
Comparative Analysis of Genotyping by Sequencing and Whole-Genome Sequencing Methods in Diversity Studies of Olea europaea L.

 , , and
, , and 
Abstract
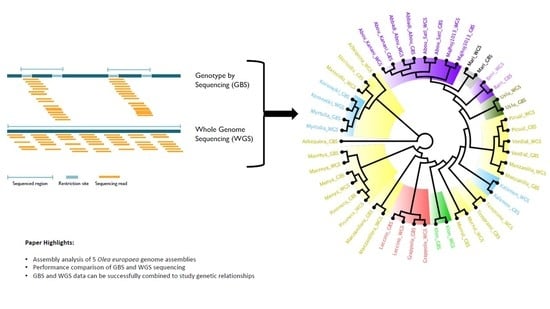
:
1. Introduction
2. Results and Discussion
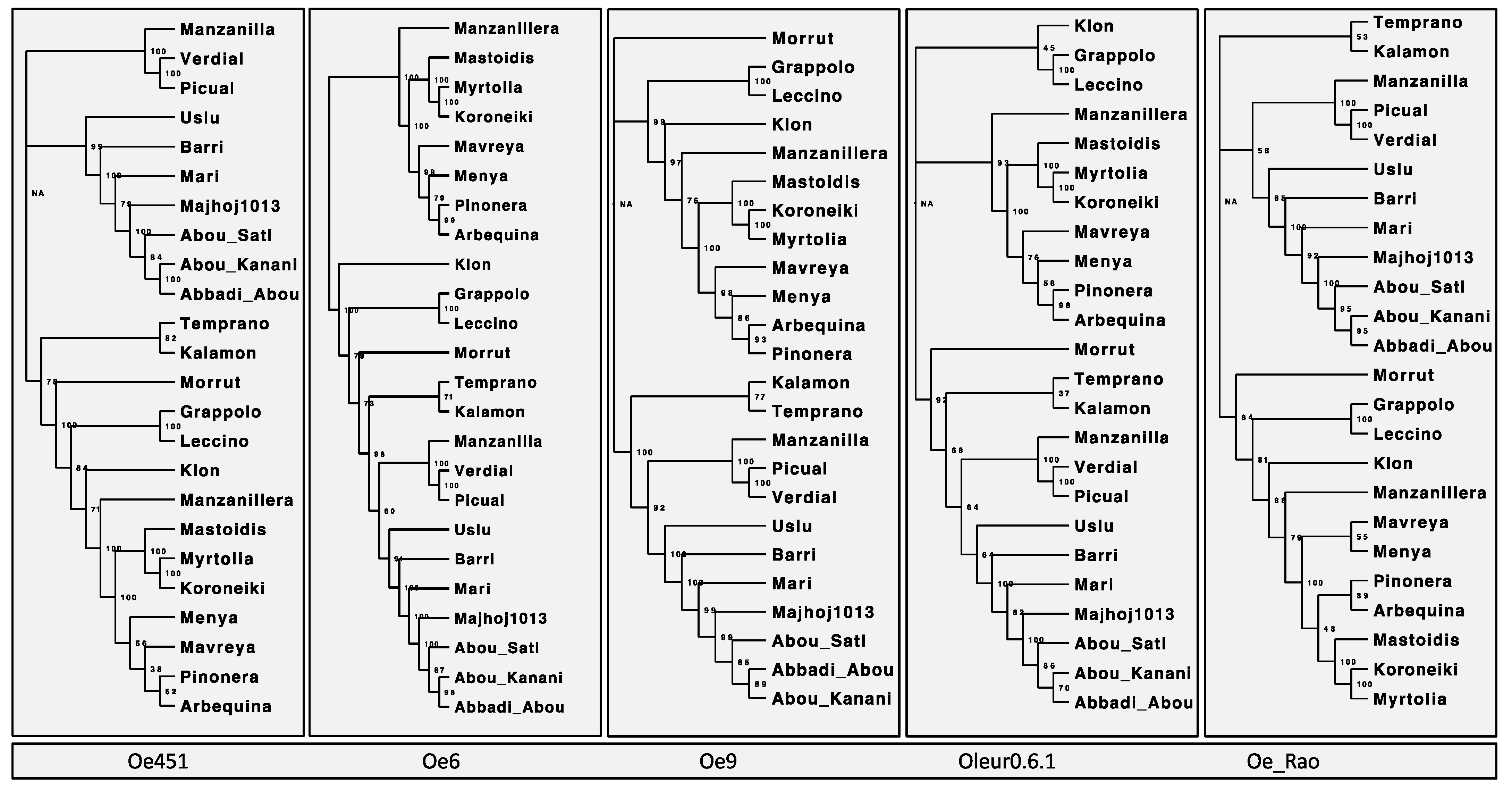
2.1. Genome Assembly Comparison
2.2. Effect of Reference Genome Choice on Population Analysis
2.2.1. Analysis GBS Read Mapping and Variant Calling
2.2.2. GBS Population Structure
2.3. Analysis of WGS Data
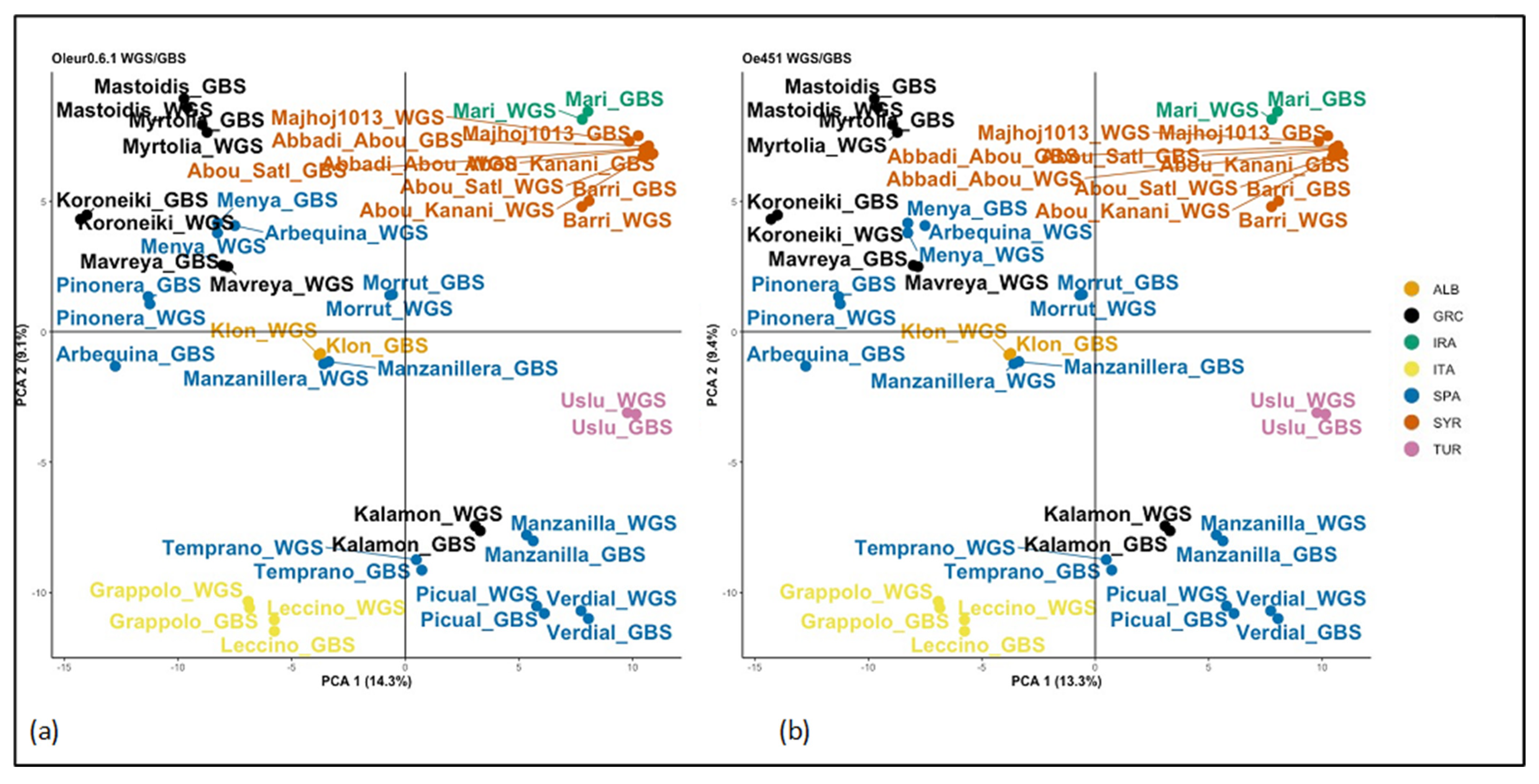
2.4. Analysis of WGS/GBS Merged Data
3. Conclusions
4. Materials and Methods
4.1. Plant Material and Genomic DNA Extraction
4.2. GBS Library Construction and Sequencing
4.3. WGS Library Construction and Sequencing
4.4. Sequence Assembly Assessment
4.5. Read Processing, Mapping, Filtering, and Variant Calling
4.6. Population Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Green, P.S. A Revision of Olea L. (Oleaceae). Kew Bull. 2002, 57, 91–140. [Google Scholar] [CrossRef]
- Wallander, E.; Albert, V.A. Phylogeny and Classification of Oleaceae Based on Rps16 and TrnL-F Sequence Data. Am. J. Bot. 2000, 87, 1827–1841. [Google Scholar] [CrossRef]
- Besnard, G.; Rubio de Casas, R.; Christin, P.-A.; Vargas, P. Phylogenetics of Olea (Oleaceae) based on plastid and nuclear ribosomal DNA sequences: Tertiary climatic shifts and lineage differentiation times. Ann. Bot. 2009, 104, 143–160. [Google Scholar] [CrossRef] [Green Version]
- Médail, F.; Quézel, P.; Besnard, G.; Khadari, B. Systematics, ecology and phylogeographic significance of Olea europaea L. ssp. maroccana (Greuter & Burdet) P. Vargas et al. a relictual olive tree in south-west Morocco. Bot. J. Linn. Soc. 2001, 137, 249–266. [Google Scholar] [CrossRef]
- Besnard, G.; Garcia-Verdugo, C.; De Casas, R.R.; Treier, U.A.; Galland, N.; Vargas, P. Polyploidy in the Olive Complex (Olea europaea): Evidence from Flow Cytometry and Nuclear Microsatellite Analyses. Ann. Bot. 2008, 101, 25–30. [Google Scholar] [CrossRef]
- Zohary, D.; Spiegel-Roy, P. Beginnings of Fruit Growing in the Old World. Science 1975, 187, 319–327. [Google Scholar] [CrossRef]
- Newton, C.; Lorre, C.; Sauvage, C.; Ivorra, S.; Terral, J.-F. On the origins and spread of Olea europaea L. (olive) domestication: Evidence for shape variation of olive stones at Ugarit, Late Bronze Age, Syria—A window on the Mediterranean Basin and on the westward diffusion of olive varieties. Veg. Hist. Archaeobotany 2014, 23, 567–575. [Google Scholar] [CrossRef]
- Terral, J.-F.; Alonso, N.; Capdevila, R.B.I.; Chatti, N.; Fabre, L.; Fiorentino, G.; Marinval, P.; Jordà, G.P.; Pradat, B.; Rovira, N.; et al. Historical biogeography of olive domestication (Olea europaea L.) as revealed by geometrical morphometry applied to biological and archaeological material. J. Biogeogr. 2004, 31, 63–77. [Google Scholar] [CrossRef]
- Zohary, D.; Hopf, M.; Weiss, E. Domestication of Plants in the Old World: The Origin and Spread of Domesticated Plants in Southwest Asia, Europe, and the Mediterranean Basin, 3rd ed.; Oxford University Press: Oxford, UK, 2000; ISBN 978-0-19-181004-6. [Google Scholar]
- El Bakkali, A.; Essalouh, L.; Tollon, C.; Rivallan, R.; Mournet, P.; Moukhli, A.; Zaher, H.; Mekkaoui, A.; Hadidou, A.; Sikaoui, L.; et al. Characterization of Worldwide Olive Germplasm Banks of Marrakech (Morocco) and Córdoba (Spain): Towards management and use of olive germplasm in breeding programs. PLoS ONE 2019, 14, e0223716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Besnard, G.; Khadari, B.; Navascués, M.; Fernández-Mazuecos, M.; El Bakkali, A.; Arrigo, N.; Baali-Cherif, D.; De Caraffa, V.B.-B.; Santoni, S.; Vargas, P.; et al. The complex history of the olive tree: From Late Quaternary diversification of Mediterranean lineages to primary domestication in the northern Levant. Proc. R. Soc. B Boil. Sci. 2013, 280, 20122833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaniewski, D.; Van Campo, E.; Boiy, T.; Terral, J.-F.; Khadari, B.; Besnard, G. Primary domestication and early uses of the emblematic olive tree: Palaeobotanical, historical and molecular evidence from the Middle East. Biol. Rev. 2012, 87, 885–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiménez-Ruiz, J.; Ramírez-Tejero, J.A.; Fernández-Pozo, N.; Leyva-Pérez, M.d.l.O.; Yan, H.; Rosa, R.d.l.; Belaj, A.; Montes, E.; Rodríguez-Ariza, M.O.; Navarro, F.; et al. Transposon activation is a major driver in the genome evolution of cultivated olive trees (Olea europaea L.). Plant Genome 2020, 13, e20010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rallo, L.; Barranco, D.; Castro-García, S.; Connor, D.J.; Campo, M.G.; del Rallo, P. High-Density Olive Plantations. In Horticultural Reviews Volume 41; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2014; pp. 303–384. ISBN 978-1-118-70741-8. [Google Scholar]
- Trujillo, I.; Ojeda, M.A.; Urdiroz, N.M.; Potter, D.; Barranco, D.; Rallo, L.; Diez, C.M. Identification of the Worldwide Olive Germplasm Bank of Córdoba (Spain) using SSR and morphological markers. Tree Genet. Genomes 2014, 10, 141–155. [Google Scholar] [CrossRef]
- IOOC International Olive Oil Council. 2020. Available online: https://www.Internationaloliveoil.Org/2020 (accessed on 10 February 2021).
- Cruz, F.; Julca, I.; Gómez-Garrido, J.; Loska, D.; Marcet-Houben, M.; Cano, E.; Galán, B.; Frias, L.; Ribeca, P.; Derdak, S.; et al. Genome sequence of the olive tree, Olea europaea. GigaScience 2016, 5, 29. [Google Scholar] [CrossRef] [PubMed]
- Julca, I.; Marcet-Houben, M.; Cruz, F.; Gómez-Garrido, J.; Gaut, B.S.; Díez, C.M.; Gut, I.G.; Alioto, T.S.; Vargas, P.; Gabaldón, T. Genomic evidence for recurrent genetic admixture during the domestication of Mediterranean olive trees (Olea europaea L.). BMC Biol. 2020, 18, 148. [Google Scholar] [CrossRef]
- Rao, G.; Zhang, J.; Liu, X.; Lin, C.; Xin, H.; Xue, L.; Wang, C. De novo assembly of a new Olea europaea genome accession using nanopore sequencing. Hortic. Res. 2021, 8, 64. [Google Scholar] [CrossRef]
- Unver, T.; Wu, Z.; Sterck, L.; Turktas, M.; Lohaus, R.; Li, Z.; Yang, M.; He, L.; Deng, T.; Escalante, F.J.; et al. Genome of wild olive and the evolution of oil biosynthesis. Proc. Natl. Acad. Sci. USA 2017, 114, E9413–E9422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muir, P.; Li, S.; Lou, S.; Wang, D.; Spakowicz, D.J.; Salichos, L.; Zhang, J.; Weinstock, G.M.; Isaacs, F.; Rozowsky, J.; et al. The real cost of sequencing: Scaling computation to keep pace with data generation. Genome Biol. 2016, 17, 53. [Google Scholar] [CrossRef] [Green Version]
- Ansorge, W.J. Next-generation DNA sequencing techniques. New Biotechnol. 2009, 25, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Metzker, M.L. Sequencing technologies—The next generation. Nat. Rev. Genet. 2010, 11, 31–46. [Google Scholar] [CrossRef] [Green Version]
- De Pristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; Del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef]
- Kyriakidou, M.; Tai, H.H.; Anglin, N.L.; Ellis, D.; Strömvik, M.V. Current Strategies of Polyploid Plant Genome Sequence Assembly. Front. Plant Sci. 2018, 9, 1660. [Google Scholar] [CrossRef] [PubMed]
- Belaj, A.; Trujillo, I.; De La Rosa, R.; Rallo, L.; Giménez, M.J. Polymorphism and Discrimination Capacity of Randomly Amplified Polymorphic Markers in an Olive Germplasm Bank. J. Am. Soc. Hortic. Sci. 2001, 126, 64–71. [Google Scholar] [CrossRef] [Green Version]
- Marita, J.M.; Rodriguez, J.M.; Nienhuis, J. Development of an algorithm identifying maximally diverse core collections. Genet. Resour. Crop. Evol. 2000, 47, 515–526. [Google Scholar] [CrossRef]
- Angiolillo, A.; Mencuccini, M.; Baldoni, L. Olive genetic diversity assessed using amplified fragment length polymorphisms. Theor. Appl. Genet. 1999, 98, 411–421. [Google Scholar] [CrossRef]
- Wachira, F.; Tanaka, J.; Takeda, Y. Genetic variation and differentiation in tea (Camellia sinensis) germplasm revealed by RAPD and AFLP variation. J. Hortic. Sci. Biotechnol. 2001, 76, 557–563. [Google Scholar] [CrossRef]
- Belaj, A.; Šatović, Z.; Cipriani, G.; Baldoni, L.; Testolin, R.; Rallo, L.; Trujillo, I. Comparative study of the discriminating capacity of RAPD, AFLP and SSR markers and of their effectiveness in establishing genetic relationships in olive. Theor. Appl. Genet. 2003, 107, 736–744. [Google Scholar] [CrossRef]
- Gómez-Rodríguez, M.V.; Beuzon, C.; González-Plaza, J.J.; Fernández-Ocaña, A.M. Identification of an olive (Olea europaea L.) core collection with a new set of SSR markers. Genet. Resour. Crop. Evol. 2021, 68, 117–133. [Google Scholar] [CrossRef]
- Belaj, A.; del Carmen Dominguez-García, M.; Atienza, S.G.; Urdíroz, N.M.; De la Rosa, R.; Satovic, Z.; Martín, A.; Kilian, A.; Trujillo, I.; Valpuesta, V.; et al. Developing a core collection of olive (Olea europaea L.) based on molecular markers (DArTs, SSRs, SNPs) and agronomic traits. Tree Genet. Genomes 2012, 8, 365–378. [Google Scholar] [CrossRef]
- He, J.; Zhao, X.; Laroche, A.; Lu, Z.-X.; Liu, H.K.; Li, Z. Genotyping-by-sequencing (GBS), an ultimate marker-assisted selection (MAS) tool to accelerate plant breeding. Front. Plant Sci. 2014, 5. [Google Scholar] [CrossRef] [Green Version]
- Talavera, A.; Soorni, A.; Bombarely, A.; Matas, A.J.; Hormaza, J.I. Genome-Wide SNP discovery and genomic characterization in avocado (Persea americana Mill.). Sci. Rep. 2019, 9, 20137. [Google Scholar] [CrossRef]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A Robust, Simple Genotyping-by-Sequencing (GBS) Approach for High Diversity Species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wickland, D.P.; Battu, G.; Hudson, K.A.; Diers, B.W.; Hudson, M.E. A comparison of genotyping-by-sequencing analysis methods on low-coverage crop datasets shows advantages of a new workflow, GB-eaSy. BMC Bioinform. 2017, 18, 586. [Google Scholar] [CrossRef]
- Marchese, A.; Marra, F.P.; Caruso, T.; Mhelembe, K.; Costa, F.; Fretto, S.; Sargent, D.J. The First High-Density Sequence Characterized SNP-Based Linkage Map of Olive (“Olea Europaea” L. Subsp. ’Europaea’) Developed Using Genotyping by Sequencing. Aust. J. Crop Sci. 2016, 10, 857–863. [Google Scholar] [CrossRef]
- Zhu, S.; Niu, E.; Shi, A.; Mou, B. Genetic Diversity Analysis of Olive Germplasm (Olea europaea L.) With Genotyping-by-Sequencing Technology. Front. Genet. 2019, 10, 755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaya, H.B.; Akdemir, D.; Lozano, R.; Cetin, O.; Kaya, H.S.; Sahin, M.; Smith, J.L.; Tanyolac, B.; Jannink, J.-L. Genome wide association study of 5 agronomic traits in olive (Olea europaea L.). Sci. Rep. 2019, 9, 18764. [Google Scholar] [CrossRef] [Green Version]
- Whibley, A.; Kelley, J.L.; Narum, S.R. The changing face of genome assemblies: Guidance on achieving high-quality reference genomes. Mol. Ecol. Resour. 2021, 21, 641–652. [Google Scholar] [CrossRef]
- Amarasinghe, S.L.; Su, S.; Dong, X.; Zappia, L.; Ritchie, M.E.; Gouil, Q. Opportunities and challenges in long-read sequencing data analysis. Genome Biol. 2020, 21, 30. [Google Scholar] [CrossRef] [Green Version]
- Ekblom, R.; Wolf, J.B. A field guide to whole-genome sequencing, assembly and annotation. Evol. Appl. 2014, 7, 1026–1042. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [Green Version]
- Seppey, M.; Manni, M.; Zdobnov, E.M. BUSCO: Assessing Genome Assembly and Annotation Completeness. In Gene Prediction: Methods and Protocols; Kollmar, M., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2019; pp. 227–245. ISBN 978-1-4939-9173-0. [Google Scholar]
- Rhie, A.; Walenz, B.P.; Koren, S.; Phillippy, A.M. Merqury: Reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 2020, 21, 245. [Google Scholar] [CrossRef] [PubMed]
- Schadt, E.E.; Turner, S.; Kasarskis, A. A window into third-generation sequencing. Hum. Mol. Genet. 2010, 19, R227–R240. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.; Chen, J.; Jiang, N. Assessing genome assembly quality using the LTR Assembly Index (LAI). Nucleic Acids Res. 2018, 46, e126. [Google Scholar] [CrossRef] [PubMed]
- Besnard, G.; De Casas, R.R. Single vs multiple independent olive domestications: The jury is (still) out. New Phytol. 2016, 209, 466–470. [Google Scholar] [CrossRef] [Green Version]
- Gage, J.L.; Vaillancourt, B.; Hamilton, J.P.; Manrique-Carpintero, N.C.; Gustafson, T.J.; Barry, K.; Lipzen, A.; Tracy, W.F.; Mikel, M.A.; Kaeppler, S.M.; et al. Multiple Maize Reference Genomes Impact the Identification of Variants by Genome-Wide Association Study in a Diverse Inbred Panel. Plant Genome 2019, 12, 180069. [Google Scholar] [CrossRef] [Green Version]
- Della Coletta, R.; Qiu, Y.; Ou, S.; Hufford, M.B.; Hirsch, C.N. How the pan-genome is changing crop genomics and improvement. Genome Biol. 2021, 22, 3. [Google Scholar] [CrossRef] [PubMed]
- Hakim, I.R.; Kammoun, N.G.; Makhloufi, E.; Rebaï, A. Discovery and Potential of SNP Markers in Characterization of Tunisian Olive Germplasm. Diversity 2010, 2, 17–27. [Google Scholar] [CrossRef] [Green Version]
- Aronesty, E. Comparison of Sequencing Utility Programs. Open Bioinform. J. 2013, 7, 1–8. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Garrison, E.; Marth, G. Haplotype-based variant detection from short-read sequencing. arXiv 2012, arXiv:1207.3907. [Google Scholar]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef] [PubMed]
- Jombart, T.; Devillard, S.; Balloux, F. Discriminant analysis of principal components: A new method for the analysis of genetically structured populations. BMC Genet. 2010, 11, 94. [Google Scholar] [CrossRef] [Green Version]
- Raj, A.; Stephens, M.; Pritchard, J.K. fastSTRUCTURE: Variational Inference of Population Structure in Large SNP Data Sets. Genetics 2014, 197, 573–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frichot, E.; François, O. LEA: An R package for landscape and ecological association studies. Methods Ecol. Evol. 2015, 6, 925–929. [Google Scholar] [CrossRef]
- Jombart, T.; Ahmed, I. adegenet 1.3-1: New tools for the analysis of genome-wide SNP data. Bioinformatics 2011, 27, 3070–3071. [Google Scholar] [CrossRef] [Green Version]
- Kamvar, Z.N.; Brooks, J.C.; GrãNwald, N.J. Novel R tools for analysis of genome-wide population genetic data with emphasis on clonality. Front. Genet. 2015, 6, 208. [Google Scholar] [CrossRef] [Green Version]
- Paradis, E.; Schliep, K. Ape 5.0: An environment for modern phylogenetics and evolutionary analyses in R. Bioinforma. Oxf. Engl. 2019, 35, 526–528. [Google Scholar] [CrossRef]
- Ramaut, A. FigTree. 2018. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 13 June 2020).
- Gruber, B.; Unmack, P.J.; Berry, O.F.; Georges, A. dartr: An r package to facilitate analysis of SNP data generated from reduced representation genome sequencing. Mol. Ecol. Resour. 2018, 18, 691–699. [Google Scholar] [CrossRef]
- Pfeifer, B.; Wittelsbürger, U.; Ramos-Onsins, S.E.; Lercher, M.J. PopGenome: An Efficient Swiss Army Knife for Population Genomic Analyses in R. Mol. Biol. Evol. 2014, 31, 1929–1936. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sequenced Cultivar | Country of Origin | Country Code | Available Data |
|---|---|---|---|
| Klon-14-1812 | Albania | ALB | GBS/WGS |
| Kalamon | Greece | GRC | GBS/WGS |
| Koroneiki | Greece | GRC | GBS/WGS |
| Mastoidis | Greece | GRC | GBS/WGS |
| Mavreya | Greece | GRC | GBS/WGS |
| Myrtolia | Greece | GRC | GBS/WGS |
| Mari | Iran | IRA | GBS/WGS |
| Grappolo | Italy | ITA | GBS/WGS |
| Leccino | Italy | ITA | GBS/WGS |
| Arberquina | Spain | SPA | GBS/WGS |
| Manzanilla de Sevilla | Spain | SPA | GBS/WGS |
| Manzanillera de Huercal Overa | Spain | SPA | GBS/WGS |
| Menya | Spain | SPA | GBS/WGS |
| Morrut | Spain | SPA | GBS/WGS |
| Picual | Spain | SPA | GBS/WGS |
| Piñonera | Spain | SPA | GBS/WGS |
| Temprano | Spain | SPA | GBS/WGS |
| Verdial de Velez-Malaga-1 | Spain | SPA | GBS/WGS |
| Abbadi Abou Gabra-842 | Syria | SYR | GBS/WGS |
| Abou Kanani | Syria | SYR | GBS/WGS |
| Abou Satl Mohazam | Syria | SYR | GBS/WGS |
| Barri | Syria | SYR | GBS/WGS |
| Majhoj-1013 | Syria | SYR | GBS/WGS |
| Uslu | Turkey | TUR | GBS/WGS |
| Removed Cultivars | |||
| Chemlal de Kabylie | Algeria | DZA | GBS |
| Megaritiki | Greece | GRC | GBS |
| Shengeh | Iran | IRA | GBS |
| Barnea | Israel | ISR | GBS |
| Frantoio | Italy | ITA | GBS |
| Forastera de Tortosa | Spain | SPA | GBS |
| Llumeta | Spain | SPA | GBS/WGS |
| Picudo | Spain | SPA | GBS/WGS |
| Jabali | Syria | SYR | GBS |
| Maarri | Syria | SYR | GBS |
| Majhoj-152 | Syria | SYR | GBS |
| Dokkar | Tunisia | TUN | GBS/WGS |
| Cultivar | Wild Olive | Farga | Farga | Picual | Arbequina |
|---|---|---|---|---|---|
| Assembly Stats | Oe451 GCA_002742605.1 (Unver et al., 2017) | Oe6 GCA_900603015.1 (Cruz et al., 2016) | Oe9 GCA_902713445.1 (Julca et al., 2020) | Oleur0.6.1 (Jiménez-Ruiz et al., 2020) | Oe_Rao GWHAOPM00000962 (Rao et al., 2021) |
| Chromosome assembly | Yes | No | yes | no | yes |
| Assembly size (Gb) | 1.14 | 1.32 | 1.31 | 1.68 | 1.1 |
| Scaffolds | 41,256 | 11,038 | 9753 | 9174 | 962 |
| Longest seq (Mb) | 46.03 | 2.58 | 36.44 | 4.14 | 68.07 |
| Shortest seq (bp) | 452 | 500 | 500 | 1017 | 10,772 |
| Average seq length (Kb) | 27.69 | 119.47 | 135.00 | 183.12 | 1146.54 |
| N90, number of seq | 3410 | 3099 | 2019 | 4503 | 320 |
| L90 (Kb) | 23 | 111 | 116 | 86,918 | 279,924 |
| N50, number of seq | 23 | 901 | 162 | 1145 | 11 |
| L50 (Mb) | 12.57 | 0.44 | 0.73 | 0.41 | 42.60 |
| % BUSCO complete | 85.9 | 96.6 | 96.6 | 96.6 | 93.4 |
| % BUSCO duplicated | 13.4 | 23.6 | 23.6 | 51.5 | 18.3 |
| % BUSCO fragmented | 5.9 | 1.9 | 2.0 | 1.9 | 2.1 |
| % BUSCO missing | 8.2 | 1.5 | 1.4 | 1.5 | 4.5 |
| Merqury completeness | 72.62 | 78.39 | 78.73 | 89.41 | NA |
| Merqury QV | 43.00 | 34.78 | 35.68 | 33.04 | NA |
| Merqury error | 5.00867 × 10−5 | 0.00033 | 0.00027 | 0.00050 | NA |
| LTR_Retriever LAI | 4.14 | 5.10 | 4.34 | 11.52 | 8.80 |
| Cultivar | O europaea var. sylvestris | Farga | Farga | Picual | Arbequina |
|---|---|---|---|---|---|
| Oe451 GCA_002742605.1 (Unver et al., 2017) | Oe6 GCA_900603015.1 (Cruz et al., 2016) | Oe9 GCA_902713445.1 (Julca et al., 2020) | Oleur0.6.1 (Jiménez-Ruiz et al., 2020) | Oe_Rao GWHAOPM00000962 (Rao et al., 2021) | |
| Total GBS reads mapped (M) | 294.29 | 298.86 | 299.56 | 300.28 | 297.58 |
| Total WGS reads mapped (M) | 8767 | NA | NA | 8789 | NA |
| % of total GBS reads | 94.4 | 95.8 | 96.0 | 96.3 | 95.4 |
| % of total WGS reads | 100.4 | NA | NA | 107.14 | NA |
| Avg number of sites GBS | 325,680 | 350,581 | 348,652 | 432,644 | 340,715 |
| Avg number of sites WGS | 493,618 | NA | NA | 1,073,623 | NA |
| GBS SNP before filtering | 7,722,425 | 7,415,201 | 23,651,384 | 4,401,892 | 1,346,358 |
| GBS SNP after filtering | 13,343 | 12,507 | 13,410 | 7023 | 10,177 |
| GBS unfiltered SNPs per site | 1.01 | 0.90 | 2.91 | 2.39 | 0.89 |
| GBS filtered SNPs per site | 0.02 | 0.02 | 0.02 | 0.01 | 0.02 |
| Average Het/site GBS | 0.36 | 0.34 | 0.35 | 0.31 | 0.35 |
| Average Het/site WGS | 0.42 | NA | NA | NA | 0.34 |
| WGS SNP before filtering | 144,579,296 | NA | NA | 128,172,089 | NA |
| WGS SNP after filtering | 119,190 | NA | NA | 114,169 | NA |
| WGS/GBS SNP before filtering | 150,952,260 | NA | NA | 11,679,534 | NA |
| WGS/GBS SNP after filtering | 9537 | NA | NA | 4004 | NA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Friel, J.; Bombarely, A.; Fornell, C.D.; Luque, F.; Fernández-Ocaña, A.M. Comparative Analysis of Genotyping by Sequencing and Whole-Genome Sequencing Methods in Diversity Studies of Olea europaea L. Plants 2021, 10, 2514. https://doi.org/10.3390/plants10112514
Friel J, Bombarely A, Fornell CD, Luque F, Fernández-Ocaña AM. Comparative Analysis of Genotyping by Sequencing and Whole-Genome Sequencing Methods in Diversity Studies of Olea europaea L. Plants. 2021; 10(11):2514. https://doi.org/10.3390/plants10112514
Chicago/Turabian StyleFriel, James, Aureliano Bombarely, Carmen Dorca Fornell, Francisco Luque, and Ana Maria Fernández-Ocaña. 2021. "Comparative Analysis of Genotyping by Sequencing and Whole-Genome Sequencing Methods in Diversity Studies of Olea europaea L." Plants 10, no. 11: 2514. https://doi.org/10.3390/plants10112514
APA StyleFriel, J., Bombarely, A., Fornell, C. D., Luque, F., & Fernández-Ocaña, A. M. (2021). Comparative Analysis of Genotyping by Sequencing and Whole-Genome Sequencing Methods in Diversity Studies of Olea europaea L. Plants, 10(11), 2514. https://doi.org/10.3390/plants10112514