
Development of SNP Set for the Marker-Assisted Selection of Guar (Cyamopsis tetragonoloba (L.) Taub.) Based on a Custom Reference Genome Assembly

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Results
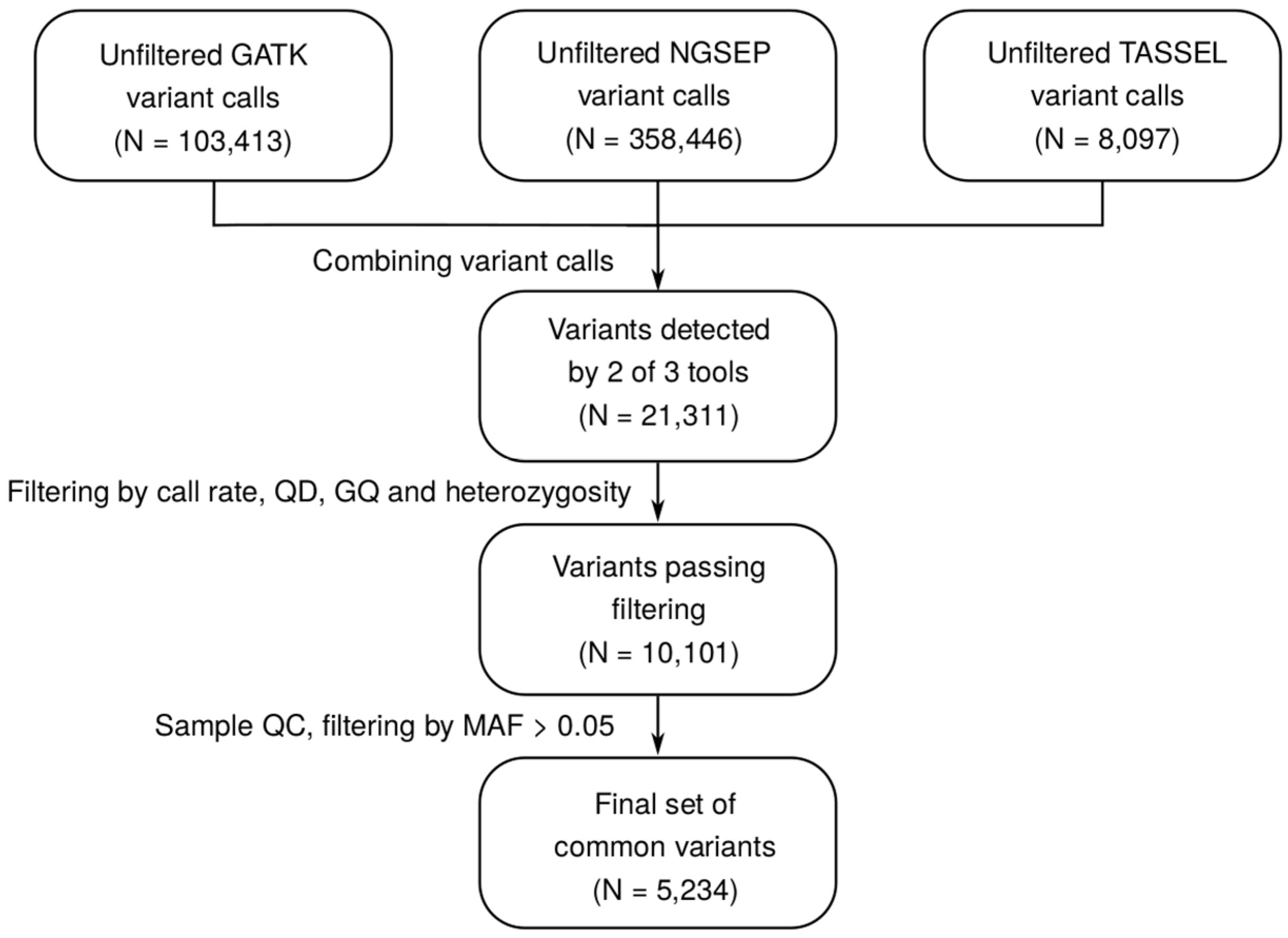
2.1. Construction of a Wide-Ranging SNP Dataset for Guar
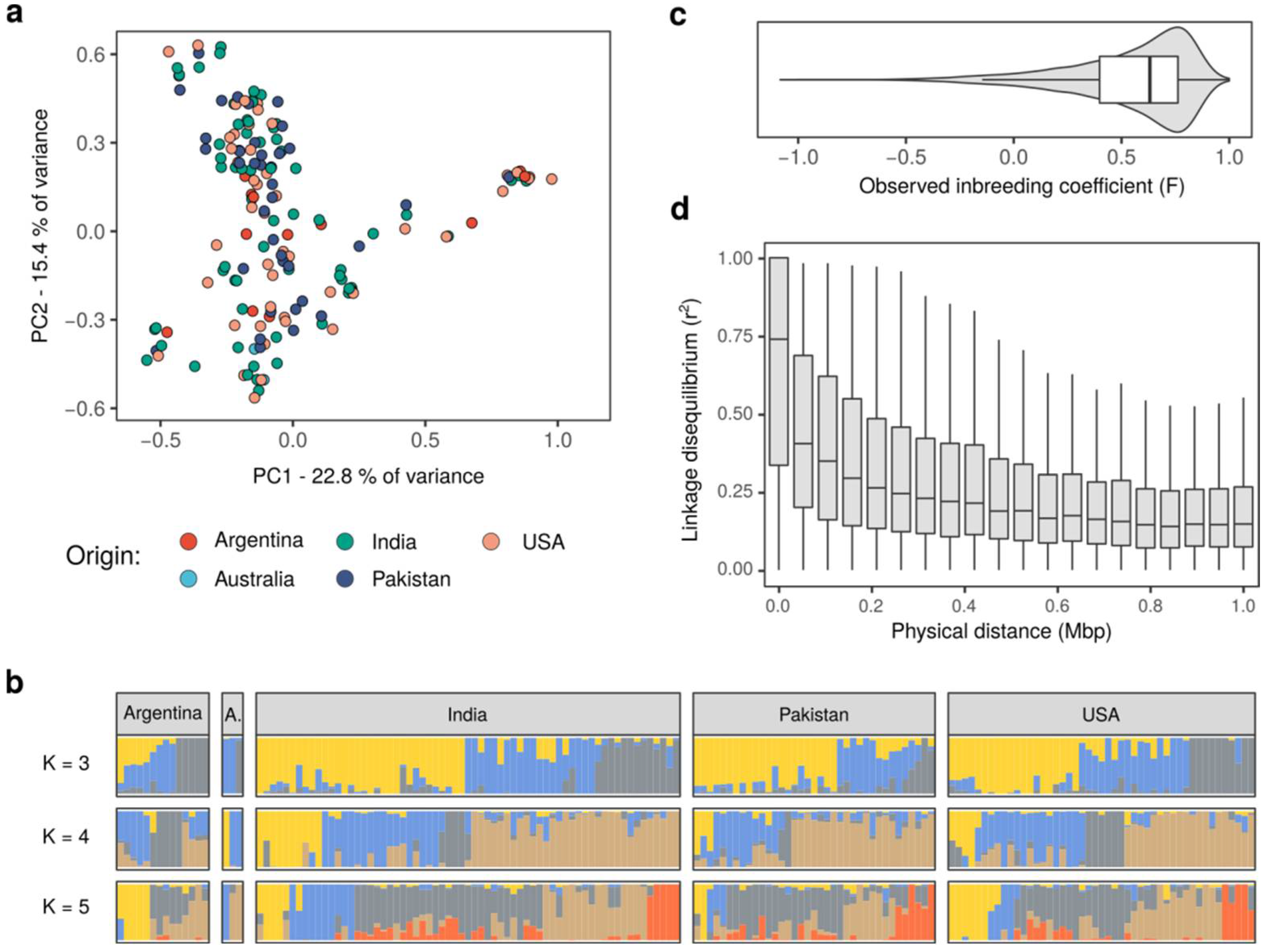
2.2. Genotype Data Provide Insights into the Genetic Structure of the Guar Populations
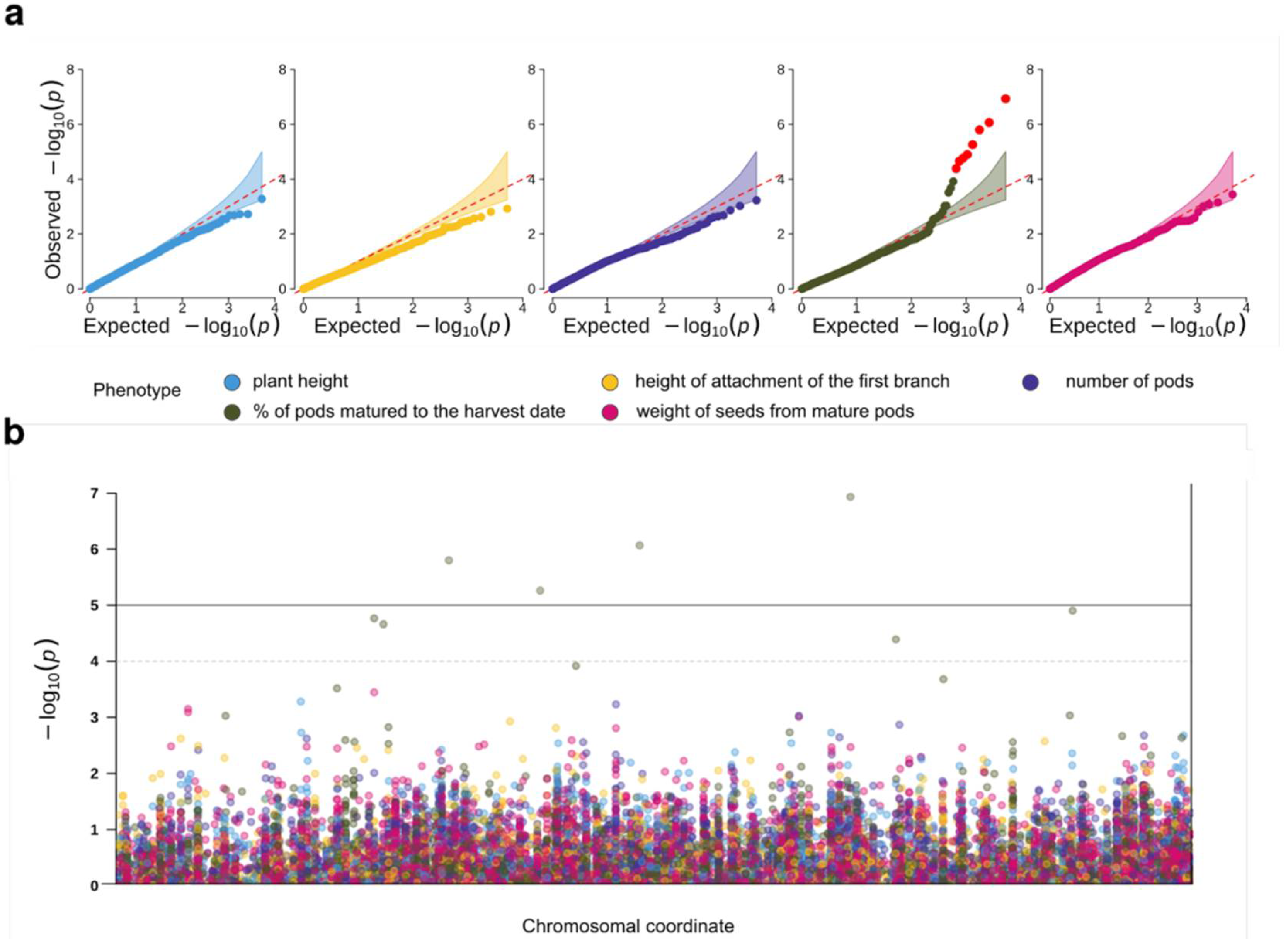
2.3. Genome-Wide Association Analysis for Productivity-Related Traits
2.4. PCR Validation of the Bioinformatics-Based SNP Prediction
3. Discussion
4. Materials and Methods
4.1. Sample and Phenotypes Collection
4.2. RAD-Seq Library Preparation and Sequencing
4.3. WGS Library Preparation and Custom Reference Genome Assembly
4.4. RAD-seq Data Analysis Pipelines and Construction of SNP Dataset
4.5. Population Structure Analysis
4.6. Phenotypic Data Preprocessing
4.7. Genome-Wide Association Analysis
4.8. SNP Validation
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jukanti, A.K.; Pathak, R.; Mushyam, C. Cluster Bean [Cyamopsis tetragonoloba (L.) Taub.] Breeding. In Advances in Plant Breeding Strategies: Legumes; Al-Khayri, J.M., Jain, S.M., Johnson, D.V., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 113–149. ISBN 978-3-030-23399-0. [Google Scholar]
- Punia, A.; Yadav, R.; Arora, P.; Chaudhury, A. Molecular and Morphophysiological Characterization of Superior Cluster Bean (Cymopsis tetragonoloba) Varieties. J. Crop Sci. Biotechnol. 2009, 12, 143–148. [Google Scholar] [CrossRef]
- Teplyakova, S.B.; Volkov, V.A.; Dzyubenko, E.A.; Potokina, E.K. Variability of the Photoperiod Response in Guar (Cyamopsis tetragonoloba (L.) Taub.) Genotypes of Different Geographic Origin. Vestn. VOGiS 2019, 23, 730–737. [Google Scholar] [CrossRef]
- Tanwar, U.K.; Pruthi, V.; Randhawa, G.S. RNA-Seq of Guar (Cyamopsis tetragonoloba, (L.) Taub.) Leaves: De Novo Transcriptome Assembly, Functional Annotation and Development of Genomic Resources. Front. Plant Sci. 2017, 8, 91. [Google Scholar] [CrossRef] [Green Version]
- Rawal, H.; Kumar, S.; Mithra, S.V.; Solanke, A.; Nigam, D.; Saxena, S.; Tyagi, A.; Yadav, N.; Kalia, P.; Singh, N.P.; et al. High Quality Unigenes and Microsatellite Markers from Tissue Specific Transcriptome and Development of a Database in Clusterbean (Cyamopsis tetragonoloba (L.) Taub.). Genes 2017, 8, 313. [Google Scholar] [CrossRef] [Green Version]
- Thakur, O.; Randhawa, G.S. Identification and Characterization of SSR, SNP and InDel Molecular Markers from RNA-Seq Data of Guar (Cyamopsis tetragonoloba (L.) Taub.) Roots. BMC Genom. 2018, 19, 951. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Palve, A.S.; Patel, S.K.; Selvanayagam, S.; Sharma, R.; Rathore, A. Development of Genomic Microsatellite Markers in Cluster Bean Using Next-Generation DNA Sequencing and Their Utility in Diversity Analysis. Curr. Plant Biol. 2020, 21, 100134. [Google Scholar] [CrossRef]
- Sahu, S.; Sahu, T.K.; Ghosal, S.; Gaikwad, K.; Rao, A.R. Computational Analysis of SNPs and INDELs in Cluster Bean Cultivars Involved in Multiple Trait Expression. IJGPB 2020, 80, 80. [Google Scholar] [CrossRef]
- Gaikwad, K.; Ramakrishna, G.; Srivastava, H.; Saxena, S.; Kaila, T.; Tyagi, A.; Sharma, P.; Sharma, S.; Sharma, R.; Mahla, H.; et al. Chromosome Scale Reference Genome of Cluster Bean (Cyamopsis tetragonoloba (L.) Taub.). Genomics 2020. [Google Scholar]
- He, J.; Zhao, X.; Laroche, A.; Lu, Z.-X.; Liu, H.; Li, Z. Genotyping-by-Sequencing (GBS), an Ultimate Marker-Assisted Selection (MAS) Tool to Accelerate Plant Breeding. Front. Plant Sci. 2014, 5, 484. [Google Scholar] [CrossRef] [Green Version]
- Lemopoulos, A.; Prokkola, J.M.; Uusi-Heikkilä, S.; Vasemägi, A.; Huusko, A.; Hyvärinen, P.; Koljonen, M.; Koskiniemi, J.; Vainikka, A. Comparing RADseq and Microsatellites for Estimating Genetic Diversity and Relatedness—Implications for Brown Trout Conservation. Ecol. Evol. 2019, 9, 2106–2120. [Google Scholar] [CrossRef]
- Poland, J.A.; Rife, T.W. Genotyping-by-Sequencing for Plant Breeding and Genetics. Plant Genome 2012, 5, 92–102. [Google Scholar] [CrossRef] [Green Version]
- Tribhuvan, K.U.; Amitha Mithra, S.V.; Sharma, P.; Das, A.; Kumar, K.; Tyagi, A.; Solanke, A.U.; Sandhya; Sharma, R.; Jadhav, P.V.; et al. Identification of Genomic SSRs in Cluster Bean (Cyamopsis tetragonoloba (L.) Taub.) and Demonstration of Their Utility in Genetic Diversity Analysis. Ind. Crop. Prod. 2019, 133, 221–231. [Google Scholar] [CrossRef]
- Bresadola, L.; Link, V.; Buerkle, C.A.; Lexer, C.; Wegmann, D. Estimating and Accounting for Genotyping Errors in RAD-seq Experiments. Mol. Ecol. Resour. 2020, 20, 856–870. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce Framework for Analyzing next-Generation DNA Sequencing Data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Ye, X.; Zhao, L.; Li, D.; Guo, Z.; Zhuang, H. Genome-Wide RAD Sequencing Data Provide Unprecedented Resolution of the Phylogeny of Temperate Bamboos (Poaceae: Bambusoideae). Sci. Rep. 2017, 7, 11546. [Google Scholar] [CrossRef] [Green Version]
- Wright, B.; Farquharson, K.A.; McLennan, E.A.; Belov, K.; Hogg, C.J.; Grueber, C.E. From Reference Genomes to Population Genomics: Comparing Three Reference-Aligned Reduced-Representation Sequencing Pipelines in Two Wildlife Species. BMC Genom. 2019, 20, 453. [Google Scholar] [CrossRef]
- Perea, C.; De La Hoz, J.F.; Cruz, D.F.; Lobaton, J.D.; Izquierdo, P.; Quintero, J.C.; Raatz, B.; Duitama, J. Bioinformatic Analysis of Genotype by Sequencing (GBS) Data with NGSEP. BMC Genom. 2016, 17, 498. [Google Scholar] [CrossRef] [Green Version]
- Glaubitz, J.C.; Casstevens, T.M.; Lu, F.; Harriman, J.; Elshire, R.J.; Sun, Q.; Buckler, E.S. TASSEL-GBS: A High Capacity Genotyping by Sequencing Analysis Pipeline. PLoS ONE 2014, 9, e90346. [Google Scholar] [CrossRef]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast Model-Based Estimation of Ancestry in Unrelated Individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Huang, M.; Fan, B.; Buckler, E.S.; Zhang, Z. Iterative Usage of Fixed and Random Effect Models for Powerful and Efficient Genome-Wide Association Studies. PLoS Genet. 2016, 12, e1005767. [Google Scholar] [CrossRef]
- Casanova, A.; Maroso, F.; Blanco, A.; Hermida, M.; Ríos, N.; García, G.; Manuzzi, A.; Zane, L.; Verissimo, A.; García-Marín, J.-L.; et al. Low Impact of Different SNP Panels from Two Building-Loci Pipelines on RAD-Seq Population Genomic Metrics: Case Study on Five Diverse Aquatic Species. BMC Genom. 2021, 22, 150. [Google Scholar] [CrossRef] [PubMed]
- Boghara, M.C.; Dhaduk, H.L.; Kumar, S.; Parekh, M.J.; Patel, N.J.; Sharma, R. Genetic Divergence, Path Analysis and Molecular Diversity Analysis in Cluster Bean (Cyamopsis tetragonoloba (L.) Taub.). Ind. Crop. Prod. 2016, 89, 468–477. [Google Scholar] [CrossRef]
- Kumar, S.; Parekh, M.J.; Patel, C.B.; Zala, H.N.; Sharma, R.; Kulkarni, K.S.; Fougat, R.S.; Bhatt, R.K.; Sakure, A.A. Development and Validation of EST-Derived SSR Markers and Diversity Analysis in Cluster Bean (Cyamopsis tetragonoloba (L.) Taub.). J. Plant Biochem. Biotechnol. 2016, 25, 263–269. [Google Scholar] [CrossRef]
- Hymowitz, T. The Trans-Domestication Concept as Applied to Guar. Econ. Bot. 1972, 26, 49–60. [Google Scholar] [CrossRef]
- Kuravadi, N.A.; Verma, S.; Pareek, S.; Gahlot, P.; Kumari, S.; Tanwar, U.K.; Bhatele, P.; Choudhary, M.; Gill, K.S.; Pruthi, V.; et al. Guar. In Agricultural Sustainability; Elsevier: Amsterdam, The Netherlands, 2013; pp. 47–60. ISBN 978-0-12-404560-6. [Google Scholar]
- Health and Environment Risks from Oil and Gas Development. Hydraulic Fracturing Impacts and Technologies; Uddameri, V., Morse, A., Tindle, K.J., Eds.; CRC Press: Boca Raton, FL, USA, 2015; pp. 124–137. ISBN 978-0-429-15252-8. [Google Scholar]
- Grigoreva, E.; Tkachenko, A.; Arkhimandritova, S.; Beatovic, A.; Ulianich, P.; Volkov, V.; Karzhaev, D.; Ben, C.; Gentzbittel, L.; Potokina, E. Identification of Key Metabolic Pathways and Biomarkers Underlying Flowering Time of Guar (Cyamopsis tetragonoloba (L.) Taub.) via Integrated Transcriptome-Metabolome Analysis. Genes 2021, 12, 952. [Google Scholar] [CrossRef]
- Zhang, S.; Yang, W.; Zhao, Q.; Zhou, X.; Jiang, L.; Ma, S.; Liu, X.; Li, Y.; Zhang, C.; Fan, Y.; et al. Analysis of Weighted Co-Regulatory Networks in Maize Provides Insights into New Genes and Regulatory Mechanisms Related to Inositol Phosphate Metabolism. BMC Genom. 2016, 17, 129. [Google Scholar] [CrossRef] [Green Version]
- Raboy, V. Approaches and Challenges to Engineering Seed Phytate and Total Phosphorus. Plant Sci. 2009, 177, 281–296. [Google Scholar] [CrossRef]
- Zhang, W.; Gruszewski, H.A.; Chevone, B.I.; Nessler, C.L. An Arabidopsis Purple Acid Phosphatase with Phytase Activity Increases Foliar Ascorbate. Plant Physiol. 2008, 146, 431–440. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.K.; Choi, J.W.; Lim, S.; Kwon, O.; Seo, J.K.; Ryu, S.H.; Suh, P.-G. Phospholipase C-H1 Is Activated by Intracellular Ca2+ Mobilization and Enhances GPCRs/PLC/Ca2+ Signaling. Cell. Signal. 2011, 23, 1022–1029. [Google Scholar] [CrossRef]
- Majerus, P.W. Inositol Phosphate Biochemistry. Annu. Rev. Biochem. 1992, 61, 225–250. [Google Scholar] [CrossRef]
- Volynkin, V.; Vasylyk, I.; Volodin, V.; Grigoreva, E.; Karzhaev, D.; Lushchay, E.; Ulianich, P.; Volkov, V.; Risovannaya, V.; Blinova, S.; et al. The Assessment of Agrobiological and Disease Resistance Traits of Grapevine Hybrid Populations (Vitis Vinifera L. × Muscadinia Rotundifolia Michx.) in the Climatic Conditions of Crimea. Plants 2021, 10, 1215. [Google Scholar] [CrossRef] [PubMed]
- Zhigunov, A.V.; Ulianich, P.S.; Lebedeva, M.V.; Chang, P.L.; Nuzhdin, S.V.; Potokina, E.K. Development of F1 Hybrid Population and the High-Density Linkage Map for European Aspen (Populus tremula L.) Using RADseq Technology. BMC Plant Biol. 2017, 17, 180. [Google Scholar] [CrossRef]
- Grigoreva, E.; Ulianich, P.; Ben, C.; Gentzbittel, L.; Potokina, E. First Insights into the Guar (Cyamopsis tetragonoloba (L.) Taub.) Genome of the ‘Vavilovskij 130′ Accession, Using Second and Third-Generation Sequencing Technologies. Russ. J. Genet. 2019, 55, 1406–1416. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Goh, L.; Yap, V.B. Effects of Normalization on Quantitative Traits in Association Test. BMC Bioinform. 2009, 10, 415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M. De Novo Transcript Sequence Reconstruction from RNA-Seq Using the Trinity Platform for Reference Generation and Analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Minimap2: Pairwise Alignment for Nucleotide Sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Variant Position | p-Value * | MAF | Function of the Nearest Transcript Match in A. thaliana | Function of the Nearest Transcript Match in G. max |
|---|---|---|---|---|
| Contig_59:14389378 | 1.2 × 10−7 | 0.23 | MYB domain protein, MYB43 | Transcription factor MYB20 |
| Contig_37:351180 | 8.5 × 10−7 | 0.36 | ANU1, SECA2, Component of the thylakoid-localized Sec system | SECA2, Protein translocase subunit |
| Contig_229:330388 | 1.6 × 10−6 | 0.22 | Single hybrid motif superfamily protein | Glycine cleavage system H protein 2 |
| Contig_88:4931265 | 1.3 × 10−5 | 0.33 | PGSIP7, plant glycogenin-like starch initiation protein 7 | PIGSIP7/PIGSIP8 Putative glucuronosyltransferase |
| Contig_182:841660 | 1.7 × 10−5 | 0.21 | ABCG40, ATP-Binding cassete G40 ** | ABC transporter G family member 39 ** |
| Contig_188:3026232 | 2.2 × 10−5 | 0.06 | RAE1, Regulation of ATALMT1 expression | F-box/LRR-repeat protein 3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grigoreva, E.; Barbitoff, Y.; Changalidi, A.; Karzhaev, D.; Volkov, V.; Shadrina, V.; Safronycheva, E.; Ben, C.; Gentzbittel, L.; Potokina, E. Development of SNP Set for the Marker-Assisted Selection of Guar (Cyamopsis tetragonoloba (L.) Taub.) Based on a Custom Reference Genome Assembly. Plants 2021, 10, 2063. https://doi.org/10.3390/plants10102063
Grigoreva E, Barbitoff Y, Changalidi A, Karzhaev D, Volkov V, Shadrina V, Safronycheva E, Ben C, Gentzbittel L, Potokina E. Development of SNP Set for the Marker-Assisted Selection of Guar (Cyamopsis tetragonoloba (L.) Taub.) Based on a Custom Reference Genome Assembly. Plants. 2021; 10(10):2063. https://doi.org/10.3390/plants10102063
Chicago/Turabian StyleGrigoreva, Elizaveta, Yury Barbitoff, Anton Changalidi, Dmitry Karzhaev, Vladimir Volkov, Veronika Shadrina, Elizaveta Safronycheva, Cécile Ben, Laurent Gentzbittel, and Elena Potokina. 2021. "Development of SNP Set for the Marker-Assisted Selection of Guar (Cyamopsis tetragonoloba (L.) Taub.) Based on a Custom Reference Genome Assembly" Plants 10, no. 10: 2063. https://doi.org/10.3390/plants10102063
APA StyleGrigoreva, E., Barbitoff, Y., Changalidi, A., Karzhaev, D., Volkov, V., Shadrina, V., Safronycheva, E., Ben, C., Gentzbittel, L., & Potokina, E. (2021). Development of SNP Set for the Marker-Assisted Selection of Guar (Cyamopsis tetragonoloba (L.) Taub.) Based on a Custom Reference Genome Assembly. Plants, 10(10), 2063. https://doi.org/10.3390/plants10102063