
Loss of the Extracellular Matrix Protein DIG-1 Causes Glial Fragmentation, Dendrite Breakage, and Dendrite Extension Defects

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Strains and Strain Maintenance
2.2. Forward Genetic Screens and Genetic Mapping
2.3. Recapitulation and Complementation Tests Using the n1321 Reference Allele
2.4. Microscopy and Image Processing
3. Results
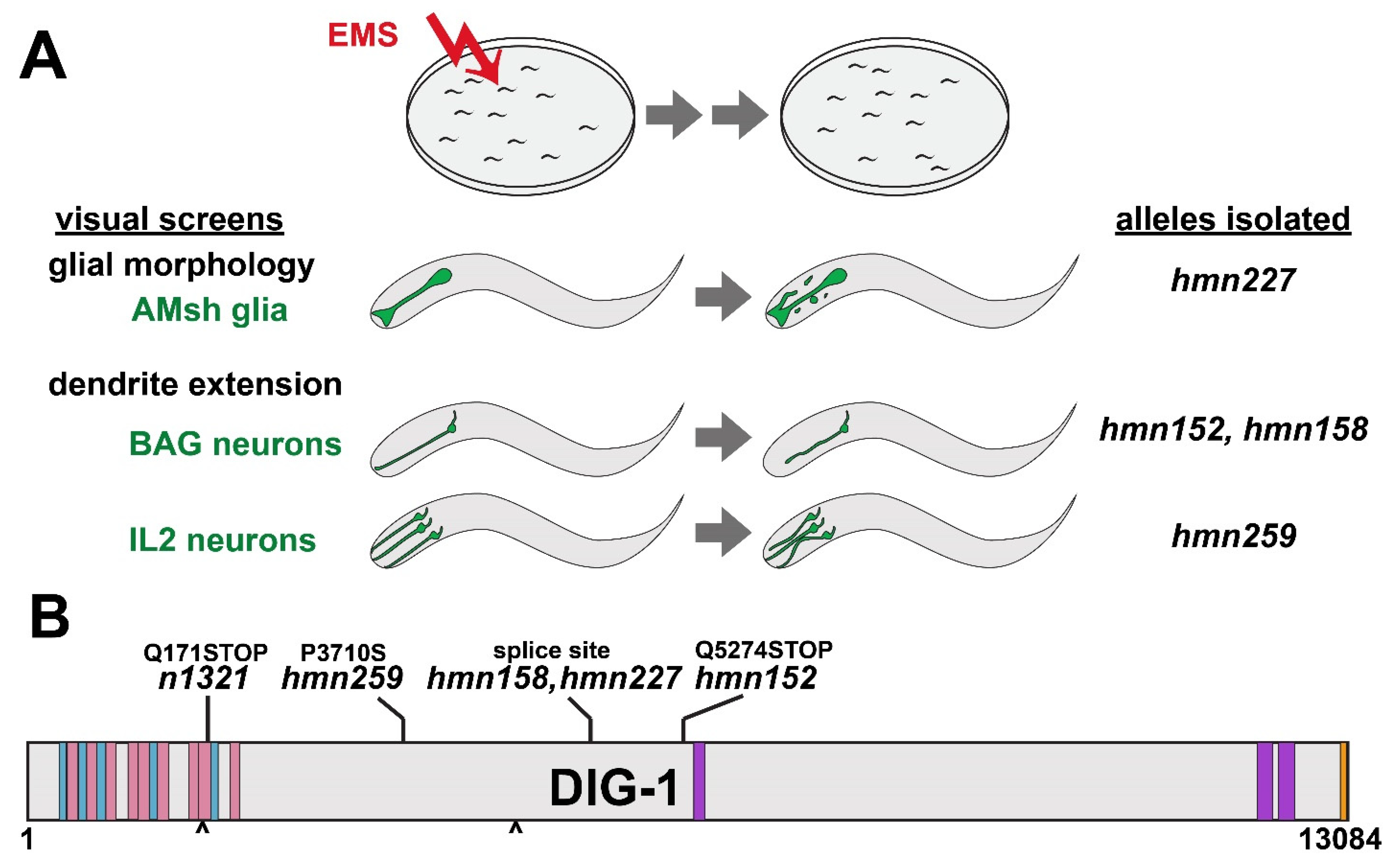
3.1. Genetic Screens for Disparate Glial and Neuronal Phenotypes Identify Lesions in DIG-1
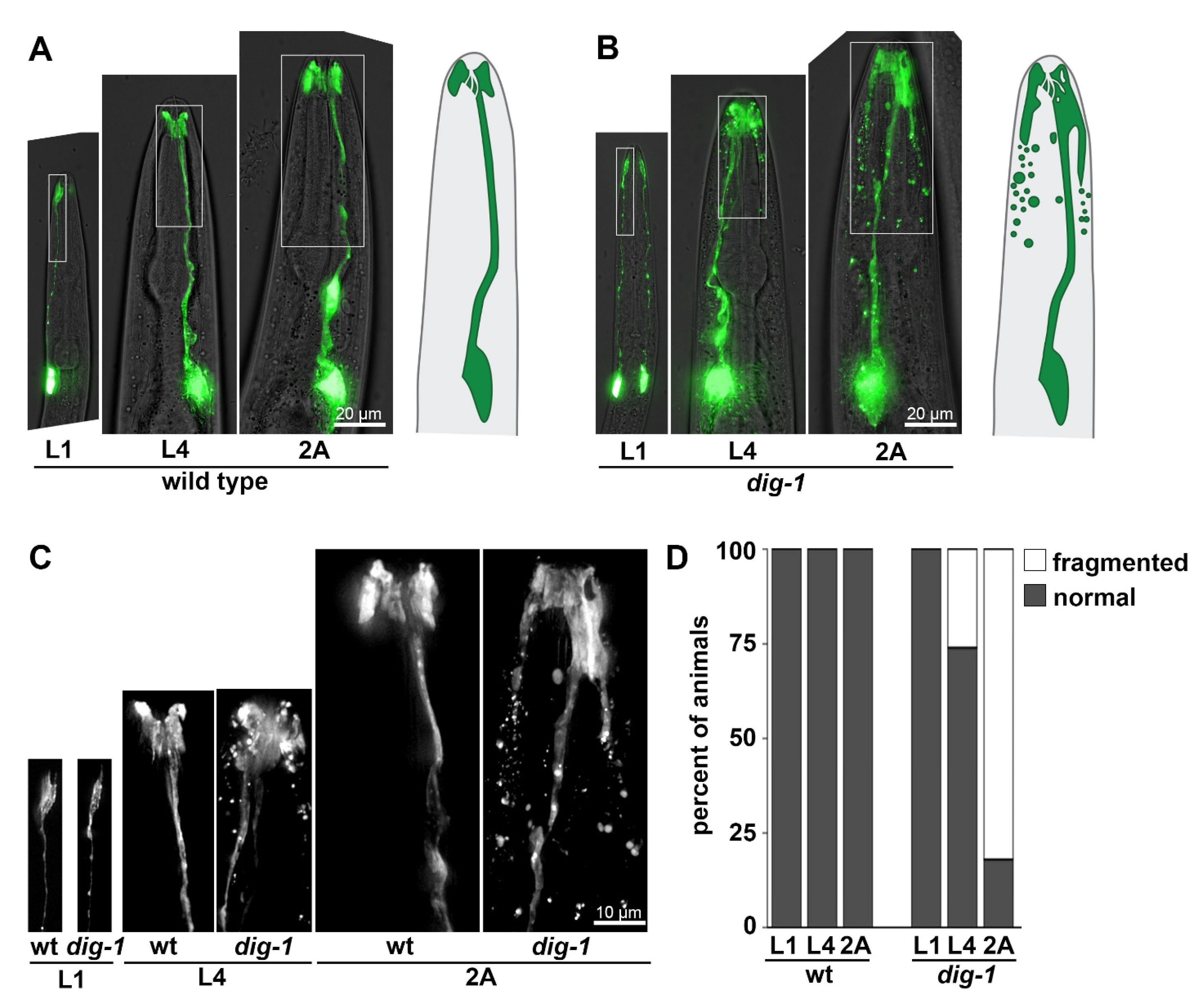
3.2. Loss of DIG-1 Causes Glial Fragmentation in Young Adults
3.3. Loss of DIG-1 Causes Breakage of BAG Dendrites in Young Adults
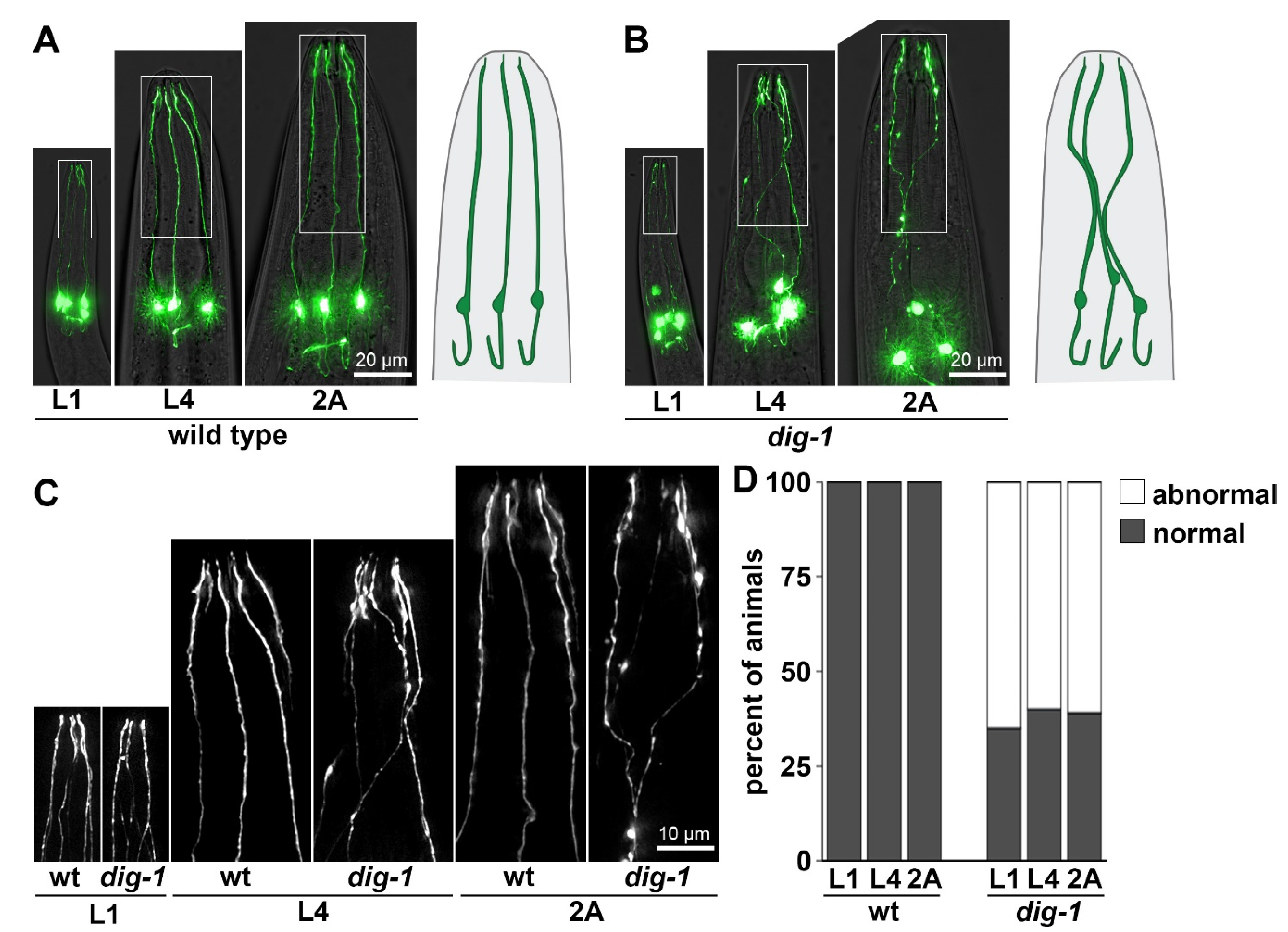
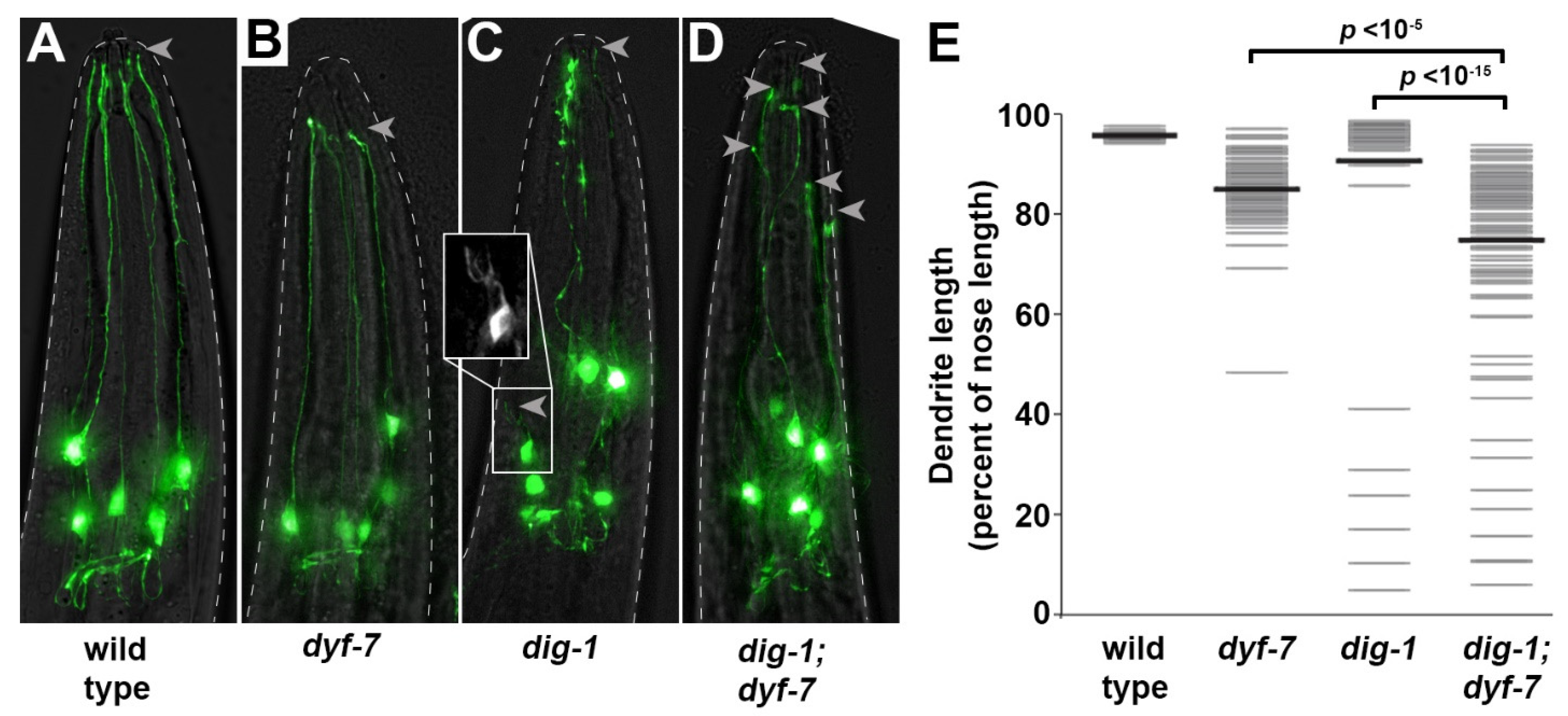
3.4. Loss of DIG-1 Causes Defasciculation and Dendrite Extension Defects in IL2 Neurons
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lamkin, E.R.; Heiman, M.G. Coordinated Morphogenesis of Neurons and Glia. Curr. Opin. Neurobiol. 2017, 47, 58–64. [Google Scholar] [CrossRef]
- Josephson, M.P.; Miltner, A.M.; Lundquist, E.A. Nonautonomous Roles of MAB-5/Hox and the Secreted Basement Membrane Molecule SPON-1/F-Spondin in Caenorhabditis elegans Neuronal Migration. Genetics 2016, 203, 1747–1762. [Google Scholar] [CrossRef]
- Lang, A.E.; Lundquist, E.A. The Collagens DPY-17 and SQT-3 Direct Anterior-Posterior Migration of the Q Neuroblasts in C. elegans. J. Dev. Biol. 2021, 9, 7. [Google Scholar] [CrossRef]
- Wang, X.; Liu, J.; Zhu, Z.; Ou, G. The Heparan Sulfate-Modifying Enzyme Glucuronyl C5-Epimerase HSE-5 Controls Caenorhabditis elegans Q Neuroblast Polarization during Migration. Dev. Biol. 2015, 399, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Lee, W.S.; Tang, X.; Wadsworth, W.G. Extracellular Matrix Regulates UNC-6 (Netrin) Axon Guidance by Controlling the Direction of Intracellular UNC-40 (DCC) Outgrowth Activity. PLoS ONE 2014, 9, e97258. [Google Scholar] [CrossRef] [PubMed]
- Blanchette, C.R.; Perrat, P.N.; Thackeray, A.; Bénard, C.Y. Glypican Is a Modulator of Netrin-Mediated Axon Guidance. PLoS Biol. 2015, 13, e1002183. [Google Scholar] [CrossRef] [PubMed]
- Celestrin, K.; Díaz-Balzac, C.A.; Tang, L.T.H.; Ackley, B.D.; Bülow, H.E. Four Specific Immunoglobulin Domains in UNC-52/Perlecan Function with NID-1/Nidogen during Dendrite Morphogenesis in Caenorhabditis elegans. Development 2018, 145, dev158881. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Dong, X.; Moerman, D.G.; Shen, K.; Wang, X. Sarcomeres Pattern Proprioceptive Sensory Dendritic Endings through UNC-52/Perlecan in C. elegans. Dev. Cell 2015, 33, 388–400. [Google Scholar] [CrossRef]
- Low, I.I.C.; Williams, C.R.; Chong, M.K.; McLachlan, I.G.; Wierbowski, B.M.; Kolotuev, I.; Heiman, M.G. Morphogenesis of Neurons and Glia within an Epithelium. Development 2019, 146. [Google Scholar] [CrossRef]
- Yip, Z.C.; Heiman, M.G. Ordered Arrangement of Dendrites within a C. elegans Sensory Nerve Bundle. eLife 2018, 7, e35825. [Google Scholar] [CrossRef] [PubMed]
- Burket, C.T.; Higgins, C.E.; Hull, L.C.; Berninsone, P.M.; Ryder, E.F. The C. elegans Gene Dig-1 Encodes a Giant Member of the Immunoglobulin Superfamily That Promotes Fasciculation of Neuronal Processes. Dev. Biol. 2006, 299, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Bénard, C.Y.; Boyanov, A.; Hall, D.H.; Hobert, O. DIG-1, a Novel Giant Protein, Non-Autonomously Mediates Maintenance of Nervous System Architecture. Development 2006, 133, 3329–3340. [Google Scholar] [CrossRef] [PubMed]
- Bénard, C.Y.; Blanchette, C.; Recio, J.; Hobert, O. The Secreted Immunoglobulin Domain Proteins ZIG-5 and ZIG-8 Cooperate with L1CAM/SAX-7 to Maintain Nervous System Integrity. PLoS Genet. 2012, 8, e1002819. [Google Scholar] [CrossRef] [PubMed]
- Blanchette, C.R.; Thackeray, A.; Perrat, P.N.; Hekimi, S.; Bénard, C.Y. Functional Requirements for Heparan Sulfate Biosynthesis in Morphogenesis and Nervous System Development in C. elegans. PLoS Genet. 2017, 13, e1006525. [Google Scholar] [CrossRef] [PubMed]
- Desse, V.E.; Blanchette, C.R.; Nadour, M.; Perrat, P.; Rivollet, L.; Khandekar, A.; Bénard, C.Y. Neuronal Post-Developmentally Acting SAX-7S/L1CAM Can Function as Cleaved Fragments to Maintain Neuronal Architecture in C. elegans. Genetics 2021, 218, iyab086. [Google Scholar] [CrossRef]
- Cizeron, M.; Granger, L.; Bülow, H.E.; Bessereau, J.-L. Specific Heparan Sulfate Modifications Stabilize the Synaptic Organizer MADD-4/Punctin at C. elegans Neuromuscular Junctions. Genetics 2021, 218, iyab073. [Google Scholar] [CrossRef]
- Pinan-Lucarré, B.; Tu, H.; Pierron, M.; Cruceyra, P.I.; Zhan, H.; Stigloher, C.; Richmond, J.E.; Bessereau, J.-L. C. elegans Punctin Specifies Cholinergic versus GABAergic Identity of Postsynaptic Domains. Nature 2014, 511, 466–470. [Google Scholar] [CrossRef]
- Platsaki, S.; Zhou, X.; Pinan-Lucarré, B.; Delauzun, V.; Tu, H.; Mansuelle, P.; Fourquet, P.; Bourne, Y.; Bessereau, J.-L.; Marchot, P. The Ig-like Domain of Punctin/MADD-4 Is the Primary Determinant for Interaction with the Ectodomain of Neuroligin NLG-1. J. Biol. Chem. 2020, 295, 16267–16279. [Google Scholar] [CrossRef]
- Zhou, X.; Vachon, C.; Cizeron, M.; Romatif, O.; Bülow, H.E.; Jospin, M.; Bessereau, J.-L. The HSPG Syndecan Is a Core Organizer of Cholinergic Synapses. J. Cell Biol. 2021, 220, e202011144. [Google Scholar] [CrossRef]
- Lázaro-Peña, M.I.; Díaz-Balzac, C.A.; Bülow, H.E.; Emmons, S.W. Synaptogenesis Is Modulated by Heparan Sulfate in Caenorhabditis elegans. Genetics 2018, 209, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Ji, T.; Wang, K.; Huang, J.; Wang, M.; Manning, L.; Dong, X.; Shi, Y.; Zhang, X.; Shao, Z.; et al. A Muscle-Epidermis-Glia Signaling Axis Sustains Synaptic Specificity during Allometric Growth in Caenorhabditis elegans. eLife 2020, 9, e55890. [Google Scholar] [CrossRef]
- Heiman, M.G.; Shaham, S. DEX-1 and DYF-7 Establish Sensory Dendrite Length by Anchoring Dendritic Tips during Cell Migration. Cell 2009, 137, 344–355. [Google Scholar] [CrossRef] [PubMed]
- Cebul, E.R.; McLachlan, I.G.; Heiman, M.G. Dendrites with Specialized Glial Attachments Develop by Retrograde Extension Using SAX-7 and GRDN-1. Development 2020, 147, dev180448. [Google Scholar] [CrossRef]
- Cohn, J.A.; Cebul, E.R.; Valperga, G.; Brose, L.; de Bono, M.; Heiman, M.G.; Pierce, J.T. Long-Term Activity Drives Dendritic Branch Elaboration of a C. elegans Sensory Neuron. Dev. Biol. 2020, 461, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Kovacevic, I.; Heiman, M.G.; Bao, Z. A Multicellular Rosette-Mediated Collective Dendrite Extension. eLife 2019, 8, e38065. [Google Scholar] [CrossRef] [PubMed]
- Nechipurenko, I.V.; Olivier-Mason, A.; Kazatskaya, A.; Kennedy, J.; McLachlan, I.G.; Heiman, M.G.; Blacque, O.E.; Sengupta, P. A Conserved Role for Girdin in Basal Body Positioning and Ciliogenesis. Dev. Cell 2016, 38, 493–506. [Google Scholar] [CrossRef]
- Ward, S.; Thomson, N.; White, J.G.; Brenner, S. Electron Microscopical Reconstruction of the Anterior Sensory Anatomy of the Nematode Caenorhabditis elegans. J. Comp. Neurol. 1975, 160, 313–337. [Google Scholar] [CrossRef]
- Johnson, R.P.; Kramer, J.M. Neural Maintenance Roles for the Matrix Receptor Dystroglycan and the Nuclear Anchorage Complex in Caenorhabditis elegans. Genetics 2012, 190, 1365–1377. [Google Scholar] [CrossRef]
- Zallen, J.A.; Kirch, S.A.; Bargmann, C.I. Genes Required for Axon Pathfinding and Extension in the C. elegans Nerve Ring. Development 1999, 126, 3679–3692. [Google Scholar] [CrossRef]
- Doitsidou, M.; Poole, R.J.; Sarin, S.; Bigelow, H.; Hobert, O. C. Elegans Mutant Identification with a One-Step Whole-Genome-Sequencing and SNP Mapping Strategy. PLoS ONE 2010, 5, e15435. [Google Scholar] [CrossRef]
- Minevich, G.; Park, D.S.; Blankenberg, D.; Poole, R.J.; Hobert, O. CloudMap: A Cloud-Based Pipeline for Analysis of Mutant Genome Sequences. Genetics 2012, 192, 1249–1269. [Google Scholar] [CrossRef]
- Chen, H.; Hughes, D.D.; Chan, T.A.; Sedat, J.W.; Agard, D.A. IVE (Image Visualization Environment): A Software Platform for All Three-Dimensional Microscopy Applications. J. Struct. Biol. 1996, 116, 56–60. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Thomas, J.H.; Stern, M.J.; Horvitz, H.R. Cell Interactions Coordinate the Development of the C. Elegans Egg-Laying System. Cell 1990, 62, 1041–1052. [Google Scholar] [CrossRef]
- Doroquez, D.B.; Berciu, C.; Anderson, J.R.; Sengupta, P.; Nicastro, D. A High-Resolution Morphological and Ultrastructural Map of Anterior Sensory Cilia and Glia in Caenorhabditis elegans. eLife 2014, 3, e01948. [Google Scholar] [CrossRef] [PubMed]
- Mizeracka, K.; Rogers, J.M.; Shaham, S.; Bulyk, M.L.; Heiman, M.G. Lineage-Specific Control of Convergent Differentiation by a Forkhead Repressor. Development 2021, 148, dev199493. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Noma, K.; Yan, D. Regulation of Gliogenesis by Lin-32/Atoh1 in Caenorhabditis elegans. G3 (Bethesda) 2020, 10, 3271–3278. [Google Scholar] [CrossRef]
- Zhang, A.; Guan, Z.; Ockerman, K.; Dong, P.; Guo, J.; Wang, Z.; Yan, D. Regulation of Glial Size by Eicosapentaenoic Acid through a Novel Golgi Apparatus Mechanism. PLoS Biol. 2020, 18, e3001051. [Google Scholar] [CrossRef]
- Razzauti, A.; Laurent, P. Ectocytosis prevents accumulation of ciliary cargo in C. elegans sensory neurons. eLife 2021, 10, e67670. [Google Scholar] [CrossRef]
- Sundaram, M.V.; Cohen, J.D. Time to Make the Doughnuts: Building and Shaping Seamless Tubes. Semin. Cell Dev. Biol. 2017, 67, 123–131. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chong, M.K.; Cebul, E.R.; Mizeracka, K.; Heiman, M.G. Loss of the Extracellular Matrix Protein DIG-1 Causes Glial Fragmentation, Dendrite Breakage, and Dendrite Extension Defects. J. Dev. Biol. 2021, 9, 42. https://doi.org/10.3390/jdb9040042
Chong MK, Cebul ER, Mizeracka K, Heiman MG. Loss of the Extracellular Matrix Protein DIG-1 Causes Glial Fragmentation, Dendrite Breakage, and Dendrite Extension Defects. Journal of Developmental Biology. 2021; 9(4):42. https://doi.org/10.3390/jdb9040042
Chicago/Turabian StyleChong, Megan K., Elizabeth R. Cebul, Karolina Mizeracka, and Maxwell G. Heiman. 2021. "Loss of the Extracellular Matrix Protein DIG-1 Causes Glial Fragmentation, Dendrite Breakage, and Dendrite Extension Defects" Journal of Developmental Biology 9, no. 4: 42. https://doi.org/10.3390/jdb9040042
APA StyleChong, M. K., Cebul, E. R., Mizeracka, K., & Heiman, M. G. (2021). Loss of the Extracellular Matrix Protein DIG-1 Causes Glial Fragmentation, Dendrite Breakage, and Dendrite Extension Defects. Journal of Developmental Biology, 9(4), 42. https://doi.org/10.3390/jdb9040042