Phenotypes, Developmental Basis, and Genetics of Pierre Robin Complex

 ,
,  ,
, 
Abstract
1. Introduction
2. Historical Perspective
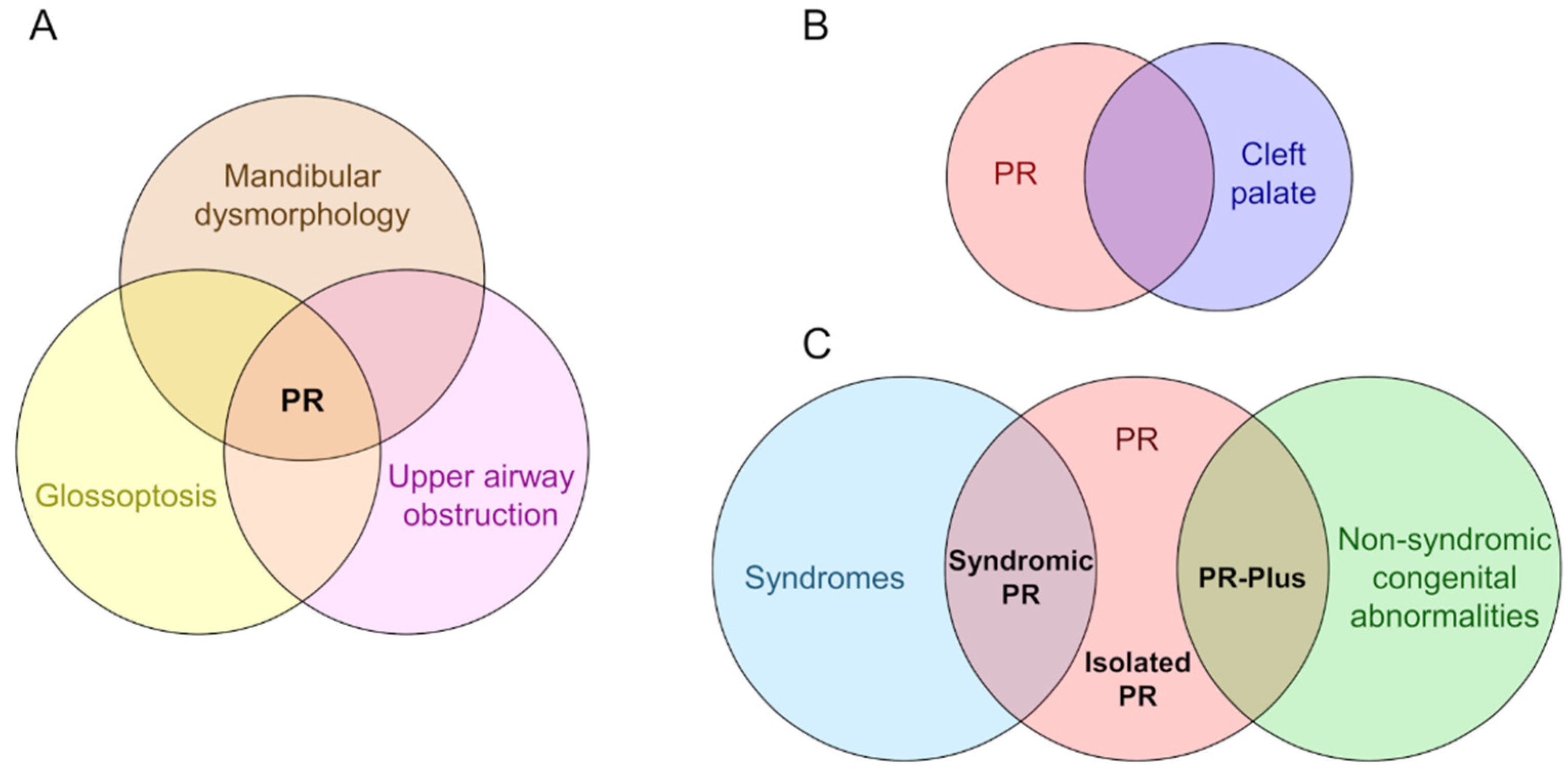
3. Epidemiology of PR
4. Uncertainty of Diagnosis
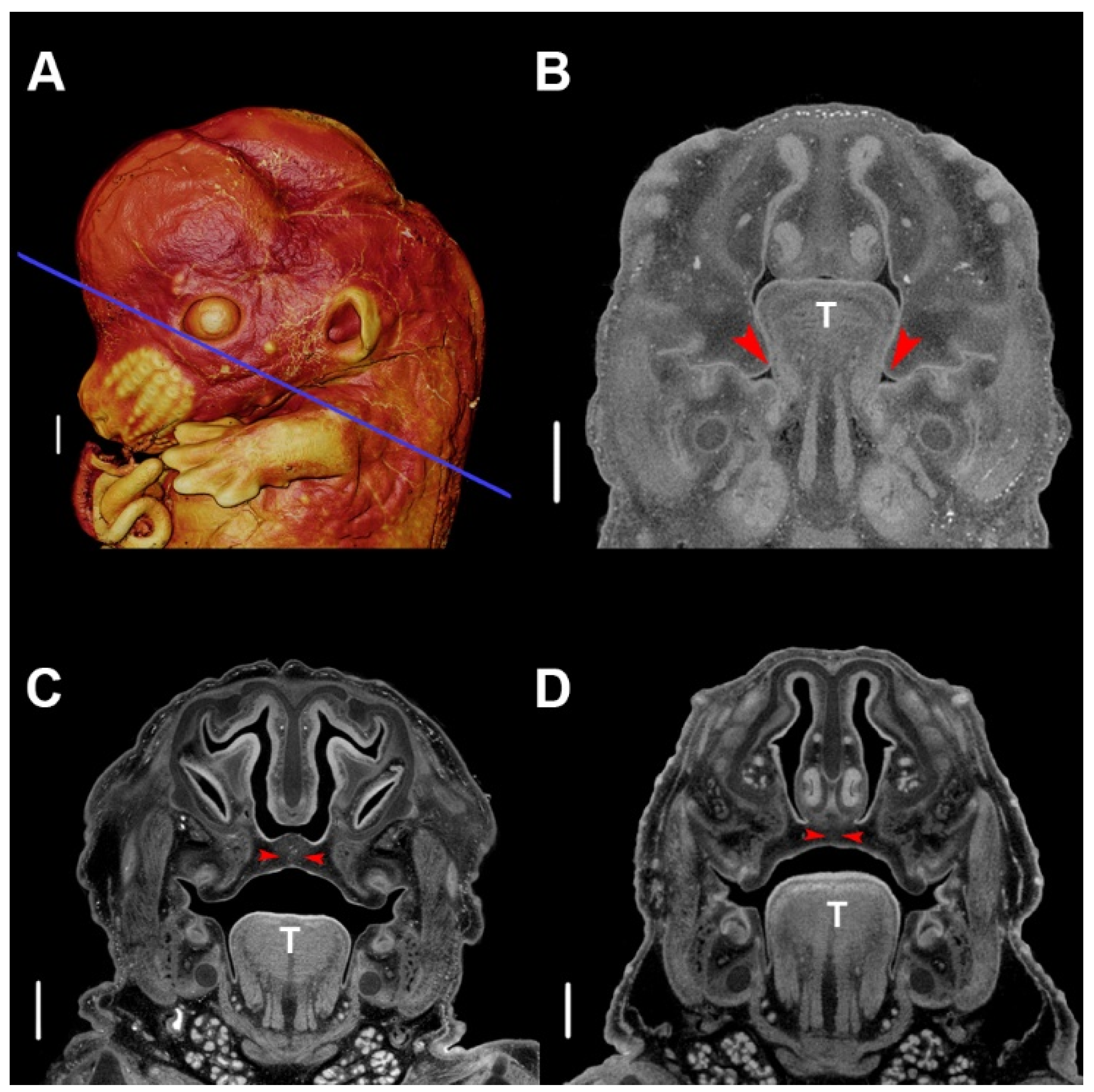
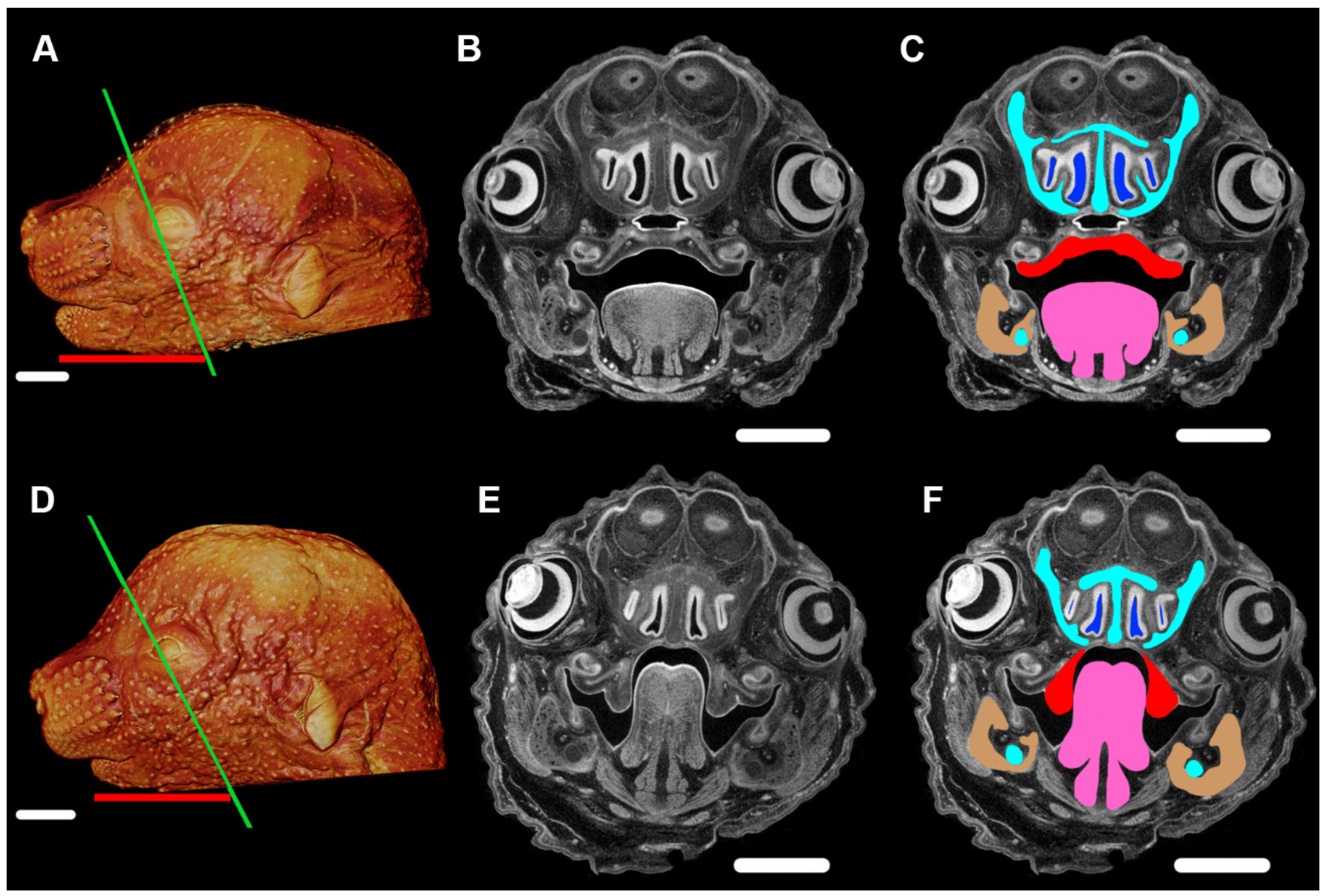
5. Development of PR Phenotypes
6. Genetics of PR
7. Animal Models as a Means for Understanding PR
8. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Robin, P. La glossoptose. Son diagnostic, ses consequences, son traitement. Bull. Acad. Natl. Med. 1923, 89, 37–41. [Google Scholar]
- Robin, P. Glossoptosis due to atresia and hypotrophy of the mandible. Am. J. Dis. Child 1934, 48, 541–547. [Google Scholar] [CrossRef]
- Shprintzen, D.R.J. The implications of the diagnosis of Robin Sequence. Cleft Palate-Craniofac. J. 1992, 29, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Hanson, J.W.; Smith, D.W. U-shaped palatal defect in the Robin anomalad: Developmental and clinical relevance. J. Pediatr. 1975, 87, 30–33. [Google Scholar] [CrossRef]
- Cohen, M.M., Jr. The Robin anomalad-its nonspecificity and associated syndromes. J. Oral Surg. 1976, 34, 587–593. [Google Scholar] [PubMed]
- Carey, J.C.; Fineman, R.M.; Ziter, F.A. The Robin sequence as a consequence of malformation, dysplasia, and neuromuscular syndromes. J. Pediatr. 1982, 101, 858–864. [Google Scholar] [CrossRef]
- Mackay, D.R. Controversies in the diagnosis and management of the Robin Sequence. J. Craniofac. Surg. 2011, 22, 415–420. [Google Scholar] [CrossRef]
- Smith, J.L.; Stowe, F.R. The Pierre Robin syndrome (glossoptosis, micrognathia, cleft palate): A review of 39 cases with emphasis on associated ocular lesions. Pediatrics 1961, 27, 128–133. [Google Scholar]
- Leung, A.K. Natal teeth. Am. J. Dis. Child. 1986, 140, 249–251. [Google Scholar] [CrossRef]
- St-Hilaire, H.; Buchbinder, D. Maxillofacial pathology and management of Pierre Robin sequence. Otolaryngol. Clin. N. Am. 2000, 33, 1241–1256. [Google Scholar] [CrossRef]
- Sadewitz, V.L. Robin Sequence: Changes in thinking leading to changes in patient care. Cleft Palate-Craniofac. J. 1992, 29, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Shprintzen, R.; Siegel-Sadewitz, V. The relationship of communication disorders to syndrome identification. J. Speech Hear. Disord. 1982, 47, 338–354. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.W. Classification, nomenclature, and naming of morphologic defects. J. Pediatr. 1975, 87, 162–164. [Google Scholar] [CrossRef]
- Shprintzen, R.J. Pierre Robin, micrognathia, and airway obstruction: The dependency of treatment on accurate diagnosis. Int. Anesthesiol. Clin. 1988, 26, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Larson, M.; Hellquist, R.; Jakobsson, O.P. Dental abnormalities and ectopic eruption in patients with isolated cleft palate. Scand. J. Plast. Reconstr. Surg. Hand Surg. 1998, 32, 203–212. [Google Scholar]
- Cohen, M. Syndromes with cleft lip and cleft palate. Cleft Palate J. 1978, 15, 308. [Google Scholar]
- Vatlach, S.; Maas, C.; Poets, C.F. Birth prevalence and initial treatment of Robin sequence in Germany: A prospective epidemiologic study. Orphanet J. Rare Dis. 2014, 9, 9. [Google Scholar] [CrossRef]
- Printzlau, A.; Andersen, M. Pierre Robin Sequence in Denmark: A retrospective population-based epidemiological study. Cleft Palate-Cran J. 2004, 41, 47–52. [Google Scholar] [CrossRef]
- Amaratunga, N.A.D.S. A comparative clinical study of Pierre Robin syndrome and isolated cleft palate. Br. J. Oral Maxillofac. Surg. 1989, 27, 451–458. [Google Scholar] [CrossRef]
- Cahill, K.C.; Orr, D.J.A. Glossoptosis in Pierre Robin sequence. Arch. Dis. Child. 2019, 104, 693. [Google Scholar] [CrossRef]
- Pasyayan, H.M.; Lewis, M.B. Clinical experience with the Robin sequence. Cleft Palate-Craniofac. J. 1984, 21, 270–276. [Google Scholar]
- Wright, M.; Mehendale, F.; Urquhart, D.S. Epidemiology of Robin sequence with cleft palate in the East of Scotland between 2004 and 2013. Pediatr. Pulmonol. 2018, 53, 1040–1045. [Google Scholar] [CrossRef] [PubMed]
- Paes, E.C.; van Nunen, D.P.F.; Basart, H.; Don Griot, J.P.W.; van Hagen, J.M.; van der Horst, C.M.A.M.; van den Boogaard, M.-J.H.; Breugem, C.C. Birth prevalence of Robin sequence in the Netherlands from 2000-2010: A retrospective population-based study in a large Dutch cohort and review of the literature. Am. J. Med. Genet. A 2015, 167A, 1972–1982. [Google Scholar] [CrossRef] [PubMed]
- Bütow, K.-W.; Zwahlen, R.A.; Morkel, J.A.; Naidoo, S. Pierre Robin sequence: Subdivision, data, theories, and treatment-Part 1: History, subdivisions, and data. Ann. Maxillofac. Surg. 2016, 6, 31–34. [Google Scholar] [PubMed]
- Evans, K.N.; Sie, K.C.; Hopper, R.A.; Glass, R.P.; Hing, A.V.; Cunningham, M.L. Robin Sequence: From diagnosis to development of an effective management plan. Pediatrics 2011, 127, 936–948. [Google Scholar] [CrossRef]
- Caouette-Laberge, L.; Bayet, B.; Larocque, Y. The Pierre Robin sequence: Review of 125 cases and volution of treatment modalities. Plast. Reconstr. Surg. 1994, 93, 934–942. [Google Scholar] [CrossRef]
- Holder-Espinasse, M.; Abadie, V.; Cormier-Daire, V.; Beyler, C.; Manach, Y.; Munnich, A.; Lyonnet, S.; Couly, G.; Amiel, J. Pierre Robin Sequence: A series of 117 consecutive cases. J. Pediatr. 2001, 139, 588–590. [Google Scholar] [CrossRef]
- van den Elzen, A.P.M.; Semmekrot, B.A.; Bongers, E.M.H.F.; Huygen, P.L.M.; Marres, H.A.M. Diagnosis and treatment of the Pierre Robin sequence: Results of a retrospective clinical study and review of the literature. Eur. J. Pediatr. 2001, 160, 47–53. [Google Scholar] [CrossRef]
- Marques, I.L.; de Sousa, T.V.; Carneiro, A.F.; Barbieri, M.A.; Bettiol, H.; Pereira Gutierrez, M.R. Clinical experience with infants with Robin sequence: A prospective study. Cleft Palate-Craniofac. J. 2001, 38, 171–178. [Google Scholar] [CrossRef]
- Isolated Pierre Robin Sequence. Available online: https://ghr.nlm.nih.gov/condition/isolated-pierre-robin-sequence (accessed on 13 August 2020).
- Izumi, K.; Konczal, L.L.; Mitchell, A.L.; Jones, M.C. Underlying genetic diagnosis of Pierre Robin Sequence: Retrospective chart review at two children’s hospitals and a systematic literature review. J. Pediatr. 2012, 160, 645–650.e2. [Google Scholar] [CrossRef]
- Mossey, P.A.; Little, J.; Munger, R.G.; Dixon, M.J.; Shaw, W.C. Cleft lip and palate. Lancet 2009, 374, 13. [Google Scholar] [CrossRef]
- Wehby, G.; Cassell, C. The impact of orofacial clefts on quality of life and healthcare use and costs: Orofacial clefts, quality of life, and health care. Oral Dis. 2010, 16, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Fukami, M.; Tsuchiya, T.; Takada, S.; Kanbara, A.; Asahara, H.; Igarashi, A.; Kamiyama, Y.; Nishimura, G.; Ogata, T. Complex genomic rearrangement in the SOX9 5′ region in a patient with Pierre Robin sequence and hypoplastic left scapula. Am. J. Med. Genet. Part A 2012, 158A, 1529–1534. [Google Scholar] [CrossRef] [PubMed]
- Parada, C.; Chai, Y. Mandible and tongue development. Curr. Top. Dev. Biol. 2015, 115, 31–58. [Google Scholar]
- Yu, K.; Ornitz, D.M. Histomorphological study of palatal shelf elevation during murine secondary palate formation. Dev. Dyn. 2011, 240, 1737–1744. [Google Scholar] [CrossRef]
- Mina, M. Regulation of mandibular growth and morphogenesis. Crit. Rev. Oral Biol. Med. 2001, 12, 276–300. [Google Scholar] [CrossRef]
- Bjork, B.C.; Turbe-Doan, A.; Prysak, M.; Herron, B.J.; Beier, D.R. Prdm16 is required for normal palatogenesis in mice. Hum. Mol. Genet. 2010, 19, 774–789. [Google Scholar] [CrossRef]
- Strassman, A.; Schnütgen, F.; Dai, Q.; Jones, J.C.; Gomez, A.C.; Pitstick, L.; Holton, N.E.; Moskal, R.; Leslie, E.R.; von Melchner, H.; et al. Generation of a multipurpose Prdm16 mouse allele by targeted gene trapping. Dis. Model. Mech. 2017, 10, 909–922. [Google Scholar] [CrossRef]
- Shull, L.C.; Sen, R.; Menzel, J.; Goyama, S.; Kurokawa, M.; Artinger, K.B. The conserved and divergent roles of Prdm3 and Prdm16 in zebrafish and mouse craniofacial development. Dev. Biol. 2020, 461, 132–144. [Google Scholar] [CrossRef]
- Long, H.K.; Osterwalder, M.; Welsh, I.C.; Hansen, K.; Davies, J.O.J.; Liu, Y.E.; Koska, M.; Adams, A.T.; Aho, R.; Arora, N.; et al. Loss of extreme long-range enhancers in human neural crest drives a craniofacial disorder. Cell Stem Cell 2020, 27, 765–783. [Google Scholar] [CrossRef]
- Jaalouk, D.E.; Lammerding, J. Mechanotransduction gone awry. Nat. Rev. Mol. Cell Biol. 2009, 10, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Stewart, S.; Darwood, A.; Masouros, S.; Higgins, C.; Ramasamy, A. Mechanotransduction in osteogenesis. Bone Jt. Res. 2020, 9, 1. [Google Scholar] [CrossRef] [PubMed]
- Cruz, M.J.; Kerschner, J.E.; Beste, D.J.; Conley, S.F. Pierre Robin sequences: Secondary respiratory difficulties and intrinsic feeding abnormalities. Laryngoscope 1999, 109, 1632–1636. [Google Scholar] [CrossRef] [PubMed]
- Sandow, R.; Kilpatrick, N.M.; Tan, T.Y.; Raj, S.; Forrest, L.E. Parental experiences and genetic counsellor roles in Pierre Robin sequence. J. Commun. Genet. 2020, 11, 475–484. [Google Scholar] [CrossRef]
- Goel, A.; Dave, N.; Shah, H.; Muneshwar, P. The troublesome triumvirate: Temporomandibular joint ankylosis, Pierre Robin syndrome and severe obstructive sleep apnoea. Indian J. Anaesth. 2020, 64, 800–803. [Google Scholar]
- Runyan, C.M.; Uribe-Rivera, A.; Tork, S.; Shikary, T.A.; Ehsan, Z.; Weaver, K.N.; Hossain, M.M.; Gordon, C.B.; Pan, B.S. Management of Airway Obstruction in Infants With Pierre Robin Sequence. Plast. Reconstr. Surg. Glob. Open 2018, 6, e1688. [Google Scholar] [CrossRef]
- Zhang, R.S.; Hoppe, I.C.; Taylor, J.A.; Bartlett, S.P. Surgical management and outcomes of Pierre Robin Sequence: A comparison of mandibular distraction osteogenesis and tongue-lip adhesion. Plast. Reconstr. Surg. 2018, 142, 480–509. [Google Scholar] [CrossRef]
- Breugem, C.C.; Mink van der Molen, A.B. What is ‘Pierre Robin sequence’? J. Plast. Reconstr. Aesthetic Surg. 2009, 62, 1555–1558. [Google Scholar] [CrossRef]
- Bütow, K.-W.; Zwahlen, R.A.; Morkel, J.A.; Naidoo, S. Pierre Robin sequence: Subdivision, data, theories, and treatment–Part 3: Prevailing controversial theories related to Pierre Robin sequence. Ann. Maxillofac. Surg. 2016, 6, 38–43. [Google Scholar]
- Abadie, V.; Morisseau-Durand, M.-P.; Beyler, C.; Manach, Y.; Couly, G. Brainstem dysfunction: A possible neuroembryological pathogenesis of isolated Pierre Robin sequence. Eur. J. Pediatr. 2002, 161, 275–280. [Google Scholar] [CrossRef]
- Brugmann, S.A.; Tapadia, M.D.; Helms, J.A. The molecular origins of species-specific facial pattern. In Current Topics in Developmental Biology; Academic Press: Cambridge, MA, USA, 2006; Volume 73, pp. 1–42. [Google Scholar]
- Hu, D.; Marcucio, R.S. A SHH-responsive signaling center in the forebrain regulates craniofacial morphogenesis via the facial ectoderm. Development 2009, 136, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Depew, M.J.; Lufkin, T.; Rubenstein, J.L.R. Specification of jaw subdivisions by Dlx genes. Science 2002, 298, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Depew, M.J.; Simpson, C.A.; Morasso, M.; Rubenstein, J.L.R. Reassessing the Dlx code: The genetic regulation of branchial arch skeletal pattern and development. J. Anat. 2005, 207, 501–561. [Google Scholar] [CrossRef] [PubMed]
- Hooper, J.E.; Feng, W.; Li, H.; Leach, S.M.; Phang, T.; Siska, C.; Jones, K.L.; Spritz, R.A.; Hunter, L.E.; Williams, T. Systems biology of facial development: Contributions of ectoderm and mesenchyme. Dev. Biol. 2017, 426, 97–114. [Google Scholar] [CrossRef] [PubMed]
- Cobourne, M.T.; Iseki, S.; Birjandi, A.A.; Adel Al-Lami, H.; Thauvin-Robinet, C.; Xavier, G.M.; Liu, K.J. How to make a tongue: Cellular and molecular regulation of muscle and connective tissue formation during mammalian tongue development. Semin. Cell Dev. Biol. 2019, 91, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Dixon, M.J.; Marazita, M.L.; Beaty, T.H.; Murray, J.C. Cleft lip and palate: Understanding genetic and environmental influences. Nat. Rev. Genet. 2011, 12, 167–178. [Google Scholar] [CrossRef]
- Edwards, J.R.G.; Newall, D.R. The Pierre Robin syndrome reassessed in the light of recent research. Br. J. Plast. Surg. 1985, 38, 339–342. [Google Scholar] [CrossRef]
- Hsieh, S.T.; Woo, A.S. Pierre Robin sequence. Clin. Plast. Surg. 2019, 46, 249–259. [Google Scholar] [CrossRef]
- Gritli-Linde, A. Molecular control of secondary palate development. Dev. Biol. 2007, 301, 309–326. [Google Scholar] [CrossRef]
- Parada, C.; Han, D.; Grimaldi, A.; Sarrión, P.; Park, S.S.; Pelikan, R.; Sanchez-Lara, P.A.; Chai, Y. Disruption of the ERK/MAPK pathway in neural crest cells as a potential cause of Pierre Robin sequence. Development 2015, 142, 3734–3745. [Google Scholar] [CrossRef]
- Li, H.; Jones, K.L.; Hooper, J.E.; Williams, T. The molecular anatomy of mammalian upper lip and primary palate fusion at single cell resolution. Development 2019, 146–160. [Google Scholar] [CrossRef] [PubMed]
- Foster, J.W.; Dominguez-Steglich, M.A.; Guioli, S.; Kwok, C.; Weller, P.A.; Stevanović, M.; Weissenbach, J.; Mansour, S.; Young, I.D.; Goodfellow, P.N.; et al. Campomelic dysplasia and autosomal sex reversal caused by mutations in an SRY -related gene. Nature 1994, 372, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Houston, C.S.; Opitz, J.M.; Spranger, J.W.; Macpherson, R.I.; Reed, M.H.; Gilbert, E.F.; Herrmann, J.; Schinzel, A. The campomelic syndrome: Review, report of 17 cases, and follow-up on the currently 17-year-old boy first reported by Maroteaux et al in 1971. Am. J. Med. Genet. 1983, 15, 3–28. [Google Scholar] [CrossRef] [PubMed]
- Benko, S.; Fantes, J.A.; Amiel, J.; Kleinjan, D.-J.; Thomas, S.; Ramsay, J.; Jamshidi, N.; Essafi, A.; Heaney, S.; Gordon, C.T.; et al. Highly conserved non-coding elements on either side of SOX9 associated with Pierre Robin sequence. Nat. Genet. 2009, 41, 359–364. [Google Scholar] [CrossRef]
- Gordon, C.T.; Attanasio, C.; Bhatia, S.; Benko, S.; Ansari, M.; Tan, T.Y.; Munnich, A.; Pennacchio, L.A.; Abadie, V.; Temple, I.K.; et al. Identification of novel craniofacial regulatory domains located far upstream of SOX9 and disrupted in Pierre Robin sequence. Hum. Mutat. 2014, 35, 1011–1020. [Google Scholar] [CrossRef]
- Smyk, M.; Roeder, E.; Cheung, S.W.; Szafranski, P.; Stankiewicz, P. A de novo 1.58 Mb deletion, including MAP2K6 and mapping 1.28 Mb upstream to SOX9, identified in a patient with Pierre Robin sequence and osteopenia with multiple fractures. Am. J. Med. Genet. Part A 2015, 167, 1842–1850. [Google Scholar] [CrossRef]
- Akiyama, H.; Lyons, J.P.; Mori-Akiyama, Y.; Yang, X.; Zhang, R.; Zhang, Z.; Deng, J.M.; Taketo, M.M.; Nakamura, T.; Behringer, R.R.; et al. Interactions between Sox9 and β-catenin control chondrocyte differentiation. Genes Dev. 2004, 18, 1072–1087. [Google Scholar] [CrossRef]
- Bi, W.; Deng, J.M.; Zhang, Z.; Behringer, R.R.; de Crombrugghe, B. Sox9 is required for cartilage formation. Nat. Genet. 1999, 22, 85–89. [Google Scholar] [CrossRef]
- Yamashita, S.; Kataoka, K.; Yamamoto, H.; Kato, T.; Hara, S.; Yamaguchi, K.; Renard-Guillet, C.; Katou, Y.; Shirahige, K.; Ochi, H.; et al. Comparative analysis demonstrates cell type-specific conservation of SOX9 targets between mouse and chicken. Sci. Rep. 2019, 9, 12560. [Google Scholar] [CrossRef]
- Karempelis, P.; Hagen, M.; Morrell, N.; Roby, B.B. Associated syndromes in patients with Pierre Robin Sequence. Int. J. Pediatric Otorhinolaryngol. 2020, 131, 109842. [Google Scholar] [CrossRef]
- Logjes, R.J.H.; Breugem, C.C.; Haaften, G.V.; Paes, E.C.; Sperber, G.H.; van den Boogaard, M.-J.H.; Farlie, P.G. The ontogeny of Robin sequence. Am. J. Med. Genet. Part A 2018, 176, 1349–1368. [Google Scholar] [CrossRef] [PubMed]
- Tan, T.Y.; Kilpatrick, N.; Farlie, P.G. Developmental and genetic perspectives on Pierre Robin sequence. Am. J. Med. Genet. C Semin. Med. Genet. 2013, 163C, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.X.; Kilpatrick, N.; Baker, N.L.; Penington, A.; Farlie, P.G.; Tan, T.Y. Clinical and molecular characterisation of children with Pierre Robin sequence and additional anomalies. Mol. Syndr. 2016, 7, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Ospina, N.; Bernstein, J.A. Clinical, cytogenetic, and molecular outcomes in a series of 66 patients with Pierre Robin sequence and literature review: 22q11.2 deletion is less common than other chromosomal anomalies. Am. J. Med. Genet. A 2016, 170, 870–880. [Google Scholar] [CrossRef] [PubMed]
- Bi, W.; Huang, W.; Whitworth, D.J.; Deng, J.M.; Zhang, Z.; Behringer, R.R.; de Crombrugghe, B. Haploinsufficiency of Sox9 results in defective cartilage primordia and premature skeletal mineralization. Proc. Natl. Acad. Sci. USA 2001, 98, 6698–6703. [Google Scholar] [CrossRef] [PubMed]
- Mori-Akiyama, Y.; Akiyama, H.; Rowitch, D.H.; de Crombrugghe, B. Sox9 is required for determination of the chondrogenic cell lineage in the cranial neural crest. Proc. Natl. Acad. Sci. USA 2003, 100, 9360–9365. [Google Scholar] [CrossRef] [PubMed]
- Ricks, J.E.; Ryder, V.M.; Bridgewater, L.C.; Schaalje, B.; Seegmiller, R.E. Altered mandibular development precedes the time of palate closure in mice homozygous for disproportionate micromelia: An oral clefting model supporting the Pierre-Robin sequence. Teratology 2002, 65, 116–120. [Google Scholar] [CrossRef]
- Clarke, L.; Hepworth, W.B.; Carey, J.C.; Seegmiller, R.E. Chondrodystrophic mice with coincidental agnathia: Evidence for the tongue obstruction hypothesis in cleft palate. Teratology 1988, 38, 565–570. [Google Scholar] [CrossRef]
- Oka, K.; Oka, S.; Sasaki, T.; Ito, Y.; Bringas, P.; Nonaka, K.; Chai, Y. The role of TGF-beta signaling in regulating chondrogenesis and osteogenesis during mandibular development. Dev. Biol. 2007, 303, 391–404. [Google Scholar] [CrossRef]
- Yuan, G.; Zhan, Y.; Gou, X.; Chen, Y.; Yang, G. TGF-β signaling inhibits canonical BMP signaling pathway during palate development. Cell Tissue Res. 2018, 371, 283–291. [Google Scholar] [CrossRef]
- Matzuk, M.M.; Kumar, T.R.; Bradley, A. Different phenotypes for mice deficient in either activins or activin receptor type II. Nature 1995, 374, 356–360. [Google Scholar] [CrossRef] [PubMed]
- Dudas, M.; Sridurongrit, S.; Nagy, A.; Okazaki, K.; Kaartinen, V. Craniofacial defects in mice lacking BMP type I receptor Alk2 in neural crest cells. Mech. Dev. 2004, 121, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, Z.; Chen, Y.; Zhang, Y. Conditional deletion of Bmp2 in cranial neural crest cells recapitulates Pierre Robin sequence in mice. Cell Tissue Res. 2019, 376, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Kouskoura, T.; El Fersioui, Y.; Angelini, M.; Graf, D.; Katsaros, C.; Chiquet, M. Dislocated tongue muscle attachment and cleft palate formation. J. Dent. Res. 2016, 95, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Liu, C.; Iwata, J.; Gu, S.; Suzuki, A.; Sun, C.; He, W.; Shu, R.; Li, L.; Chai, Y.; et al. Mice with Tak1 deficiency in neural crest lineage exhibit cleft palate associated with abnormal tongue development. J. Biol. Chem. 2013, 288, 10440–10450. [Google Scholar] [CrossRef] [PubMed]
- Dixon, J.; Jones, N.C.; Sandell, L.L.; Jayasinghe, S.M.; Crane, J.; Rey, J.-P.; Dixon, M.J.; Trainor, P.A. Tcof1/Treacle is required for neural crest cell formation and proliferation deficiencies that cause craniofacial abnormalities. Proc. Natl. Acad. Sci. USA 2006, 103, 13403–13408. [Google Scholar] [CrossRef] [PubMed]
- Sakai, D.; Trainor, P.A. Treacher Collins syndrome: Unmasking the role of Tcof1/treacle. Int. J. Biochem. Cell Biol. 2009, 41, 1229–1232. [Google Scholar] [CrossRef] [PubMed]
- Dash, S.; Bhatt, S.; Falcon, K.T.; Sandell, L.L.; Trainor, P.A. Med23 Regulates Sox9 Expression during Craniofacial Development. J. Dent. Res. 2020, 1–9, Epub ahead of print. [Google Scholar] [CrossRef]
- Watanabe, H.; Kimata, K.; Line, S.; Strong, D.; Gao, L.Y.; Kozak, C.A.; Yamada, Y. Mouse cartilage matrix deficiency (cmd) caused by a 7 bp deletion in the aggrecan gene. Nat. Genet. 1994, 7, 154–157. [Google Scholar] [CrossRef]
- Kouskoura, T.; Kozlova, A.; Alexiou, M.; Blumer, S.; Zouvelou, V.; Katsaros, C.; Chiquet, M.; Mitsiadis, T.A.; Graf, D. The etiology of cleft palate formation in BMP7-deficient mice. PLoS ONE 2013, 8, e59463. [Google Scholar] [CrossRef]
- Li, Y.; Lacerda, D.A.; Warman, M.L.; Beier, D.R.; Yoshioka, H.; Ninomiya, Y.; Oxford, J.T.; Morris, N.P.; Andrikopoulos, K.; Ramirez, F.; et al. A fibrillar collagen gene, Col11a1, is essential for skeletal morphogenesis. Cell 1995, 80, 423–430. [Google Scholar] [CrossRef]
- Kurihara, Y.; Kurihara, H.; Suzuki, H.; Kodama, T.; Maemura, K.; Nagai, R.; Oda, H.; Kuwaki, T.; Cao, W.H.; Kamada, N. Elevated blood pressure and craniofacial abnormalities in mice deficient in endothelin-1. Nature 1994, 368, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, P.J.; Chin, J.R.; Shum, L.; Slavkin, H.C.; Shuler, C.F.; Derynck, R.; Werb, Z. Epidermal growth factor receptor function is necessary for normal craniofacial development and palate closure. Nat. Genet. 1999, 22, 69–73. [Google Scholar] [CrossRef]
- Gendron-Maguire, M.; Mallo, M.; Zhang, M.; Gridley, T. Hoxa-2 mutant mice exhibit homeotic transformation of skeletal elements derived from cranial neural crest. Cell 1993, 75, 1317–1331. [Google Scholar] [CrossRef]
- Dash, S.; Bhatt, S.; Sandell, L.L.; Seidel, C.W.; Ahn, Y.; Krumlauf, R.E.; Trainor, P.A. The Mediator subunit, Med23 Is required for embryonic survival and regulation of canonical WNT signaling during cranial ganglia development. Front. Physiol. 2020, 11, 1284. [Google Scholar] [CrossRef]
- Satokata, I.; Maas, R. Msx1 deficient mice exhibit cleft palate and abnormalities of craniofacial and tooth development. Nat. Genet. 1994, 6, 348–356. [Google Scholar] [CrossRef]
- Stewart, K.; Uetani, N.; Hendriks, W.; Tremblay, M.L.; Bouchard, M. Inactivation of LAR family phosphatase genes Ptprs and Ptprf causes craniofacial malformations resembling Pierre-Robin sequence. Development 2013, 140, 3413–3422. [Google Scholar] [CrossRef]
- Britanova, O.; Depew, M.J.; Schwark, M.; Thomas, B.L.; Miletich, I.; Sharpe, P.; Tarabykin, V. Satb2 haploinsufficiency phenocopies 2q32-q33 deletions, whereas loss suggests a fundamental role in the coordination of jaw development. Am. J. Hum. Genet. 2006, 79, 668–678. [Google Scholar] [CrossRef]
- Murray, S.A.; Oram, K.F.; Gridley, T. Multiple functions of Snail family genes during palate development in mice. Development 2007, 134, 1789–1797. [Google Scholar] [CrossRef]
- Huang, H.; Yang, X.; Bao, M.; Cao, H.; Miao, X.; Zhang, X.; Gan, L.; Qiu, M.; Zhang, Z. Ablation of the Sox11 gene results in clefting of the secondary palate resembling the Pierre Robin sequence. J. Biol. Chem. 2016, 291, 7107–7118. [Google Scholar] [CrossRef]
- Jerome, L.A.; Papaioannou, V.E. DiGeorge syndrome phenotype in mice mutant for the T-box gene, Tbx1. Nat. Genet. 2001, 27, 286–291. [Google Scholar] [CrossRef] [PubMed]
- Dixon, J.; Brakebusch, C.; Fässler, R.; Dixon, M.J. Increased levels of apoptosis in the prefusion neural folds underlie the craniofacial disorder, Treacher Collins syndrome. Hum. Mol. Genet. 2000, 9, 1473–1480. [Google Scholar] [CrossRef] [PubMed]
- Geister, K.A.; Timms, A.E.; Beier, D.R. Optimizing genomic methods for mapping and identification of candidate variants in ENU mutagenesis screens using inbred mice. G3-Genes Genom. Genet. 2018, 8, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Herron, B.J.; Lu, W.; Rao, C.; Liu, S.; Peters, H.; Bronson, R.T.; Justice, M.J.; McDonald, J.D.; Beier, D.R. Efficient generation and mapping of recessive developmental mutations using ENU mutagenesis. Nat. Genet. 2002, 30, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Schubert, J.; Jahn, H.; Berginski, M. Experimental aspects of the pathogenesis of Robin sequence. Cleft Palate Craniofac. J. 2005, 42, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Wolf, Z.T.; Leslie, E.J.; Arzi, B.; Jayashankar, K.; Karmi, N.; Jia, Z.; Rowland, D.J.; Young, A.; Safra, N.; Sliskovic, S.; et al. A LINE-1 insertion in DLX6 is responsible for cleft palate and mandibular abnormalities in a canine model of Pierre Robin sequence. PLoS Genet. 2014, 10, e1004257. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Q.; Chiquet, B.T.; Devault, L.; Warman, M.L.; Nakamura, Y.; Swindell, E.C.; Hecht, J.T. Craniofacial abnormalities result from knock down of nonsyndromic clefting gene, crispld2, in zebrafish. Genesis 2012, 50, 871–881. [Google Scholar] [CrossRef]
- Ghassibe-Sabbagh, M.; Desmyter, L.; Langenberg, T.; Claes, F.; Boute, O.; Bayet, B.; Pellerin, P.; Hermans, K.; Backx, L.; Mansilla, M.A.; et al. FAF1, a gene that is disrupted in cleft palate and has conserved function in zebrafish. Am. J. Hum. Genet. 2011, 88, 150–161. [Google Scholar] [CrossRef][Green Version]
- Noack Watt, K.E.; Achilleos, A.; Neben, C.L.; Merrill, A.E.; Trainor, P.A. The roles of RNA polymerase I and III subunits Pozlr1c and Polr1d in craniofacial development and in zebrafish models of Treacher Collins syndrome. PLoS Genet. 2016, 12, e1006187. [Google Scholar] [CrossRef]
- Lau, M.C.C.; Kwong, E.M.L.; Lai, K.P.; Li, J.-W.; Ho, J.C.H.; Chan, T.-F.; Wong, C.K.C.; Jiang, Y.-J.; Tse, W.K.F. Pathogenesis of POLR1C-dependent Type 3 Treacher Collins Syndrome revealed by a zebrafish model. Biochim. Biophys. Acta 2016, 1862, 1147–1158. [Google Scholar] [CrossRef]
- Weiner, A.M.J.; Scampoli, N.L.; Calcaterra, N.B. Fishing the molecular bases of Treacher Collins syndrome. PLoS ONE 2012, 7, e29574. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Gene Name | Syndrome(s) | MIM Phenotype Number |
|---|---|---|---|
| AMER1 | Apc membrane recruitment protein 1 | Osteopathia striata with cranial sclerosis | 300373 |
| AP3D1 | Adaptor related protein complex 3 subunit delta 1 | Hermansky–Pudlak syndrome 10 | 617050 |
| BMP2 | Bone morphogenetic protein 2 | Short stature, facial dysmorphism, and skeletal anomalies with or without cardiac anomalies | 617877 |
| COG1 | Component of oligomeric golgi complex 1 | Congenital disorder of glycosylation, type IIg | 611209 |
| COL11A1 | Collagen, type XI, alpha-1 | Stickler syndrome, type II; Marshall syndrome | 604841; 154780 |
| COL11A2 | Collagen, type XI, alpha-2 | Otospondylomegaepiphyseal dysplasia, autosomal dominant; Otospondylomegaepiphyseal dysplasia, autosomal recessive | 184840; 215150 |
| COL2A1 | Collagen, type II, alpha-1 | Stickler syndrome, type I | 108300 |
| DHODH | Dihydroorotate dehydrogenase | Miller syndrome | 263750 |
| EDN1 | Endothelin 1 | Auriculocondylar syndrome 3 | 615706 |
| EFTUD2 | Elongation factor Tu guanosine triphosphate binding domain containing 2 | Mandibulofacial dysostosis, Guion–Almeida type | 610536 |
| EIF4A3 | Eukaryotic translation initiation factor 4a3 | Robin sequence with cleft mandible and limb anomalies | 268305 |
| MAP3K7 | Mitogen-activated protein kinase kinase kinase 7 | Frontometaphyseal dysplasia 2 | 617137 |
| MYMK | Myomaker, myoblast fusion factor | Carey–Fineman–Ziter syndrome | 254940 |
| PDHA1 | Pyruvate dehydrogenase E1 subunit alpha 1 | Pyruvate dehydrogenase E1-alpha deficiency | 312170 |
| PGAP3 | Post-glycophosphatidylinositol attachment to proteins phospholipase 3 | Hyperphosphatasia with mental retardation syndrome 4 | 615716 |
| PGM1 | Phosphoglucomutase 1 | Congenital disorder of glycosylation, type It | 614921 |
| PIGA | Phosphatidylinositol glycan anchor biosynthesis class A | Multiple congenital anomalies–hypotonia–seizures syndrome 2 | 300868 |
| POLR1C | RNA polymerase I and III subunit C | Treacher Collins syndrome 3 | 248390 |
| POLR1D | RNA polymerase I and III subunit D | Treacher Collins syndrome 2 | 613717 |
| RBM10 | RNA-binding motif protein 10 | TARP syndrome | 311900 |
| SATB2 | Special AT-rich sequence-binding protein 2 | Glass syndrome | 612313 |
| SLC10A7 | Solute carrier family 10 member 7 | Short stature, amelogenesis imperfecta, and skeletal dysplasia with scoliosis | 618363 |
| SLC26A2 | Solute carrier family 26 member 2 | Diastrophic dysplasia | 222600 |
| SNRPB | Small nuclear ribonucleoprotein polypeptides B and B1 | Cerebrocostomandibular syndrome | 117650 |
| SOX9 | Sry-box 9 | Campomelic dysplasia | 114290 |
| SF3B4 | Splicing factor 3b subunit 4 | Nager syndrome | 154400 |
| TBX1 | T-box transcription factor 1 | Velocardiofacial syndrome | 192430 |
| TCOF1 | Treacle ribosome biogenesis factor 1 | Treacher Collins syndrome 1 | 154500 |
| TGDS | Thymidine diphosphate-glucose 4,6-dehydratase | Catel–Manzke syndrome | 616145 |
| Animal Model | Species | Gene | Mutation | Phenotypes | References | |||
|---|---|---|---|---|---|---|---|---|
| Jaw | Tongue | Palate | Others | |||||
| Acancmd/cmd | Mouse | Acan | Intragenic deletion in Acan | Micrognathia or agnathia | Underdeveloped | Cleft palate | Short-limbed chondrodystrophy | [80,91] |
| Acvr2atm1Zuk | Mouse | Acvr2a | Acvr2a null | Micrognathia, defects in Meckel′s cartilage | None reported | Cleft palate | None reported | [83] |
| Acvr1fl/fl; Wnt1-Cre | Mouse | Acvr1 | Wnt1-Cre conditional knockout of Acvr1 | Micrognathia | None reported | Cleft palate | Enlarged frontal fontanels, incomplete zygomatic arches, squamosal bones lack the retrotympanic process; smaller temporal squama | [84] |
| Bmp2fl/fl; Wnt1-Cre;R26RmTmG | Mouse | Bmp2 | Wnt1-Cre conditional knockout of Bmp2 | Micrognathia | Malformed tongue | Cleft palate | A reduced size of craniofacial bones | [85] |
| Bmp7Δ/Δ | Mouse | Bmp7 | Bmp7 null | Impaired Meckel’s cartilage development; lack of a mandibular symphysis and mandibular mental spine formation | Misplaced origin of genioglossus muscle | Cleft palate | Alteration of oral cavity morphology | [86,92] |
| Col11a1cho/cho | Mouse | Col11a1 | Intragenic deletion in Col11a1 | Micrognathia or agnathia | Underdeveloped | Cleft palate | Short-limbed chondrodystrophy | [80,93] |
| Col2a1Dmm | Mouse | Col2a1 | Disproportionate micromelia (Dmm) semi-dominant mutation | Mandibular growth retardation, coupled with relative macroglossia in E14 | Relative tongue size to Meckel’s cartilage length significantly greater at E14.75 compared to control | Cleft palate | Mild dwarfism three weeks after birth in heterozygotes | [79] |
| Edn1−/− | Mouse | Edn1 | Edn1 null | Short and deformed mandibular bones | Most of tongue missing | Cleft palate | Thin anterior neck and hypoplastic auricles, aberrant zygomatic andtemporal bones, absent auditory ossicles and tympanic ring | [74,94] |
| Egfr−/− | Mouse | Egfr | Targeted intragenic deletion in Egfr | Under-developed lower jaw | None reported | Cleft palate | Narrow, elongated snouts | [95] |
| Erk2fl/fl; Wnt1-Cre | Mouse | Erk2 | Wnt1-Cre conditional knockout of Erk2 | Micrognathia and mandibular asymmetry | Malformed tongue | Cleft palate, failed palate elevation | None reported | [62] |
| pMes-Fgf10; Wnt1-Cre | Mouse | Fgf10 | Wnt1-Cre conditional transgene of Fgf10 | None reported | Heightened tongue | Failed palate elevation | None reported | [87] |
| Hoxa2D1 | Mouse | Hoxa2 | Hoxa2 null | Duplicated Meckel′s cartilage | None reported | Cleft palate | External ear defects, duplication of the ossification centers of the bones of the middle ear | [96] |
| Med23fl/fl; Wnt1-Cre | Mouse | Med23 | Wnt1-Cre conditional knockout of Med23 | Micrognathia, hypoplastic Meckel’s cartilage | Glossoptosis | Cleft palate | Cleidocranial dysplasia: Agenesis of nasal cartilage and bones, abnormal development of the tympanic ring and skull bones | [90,97] |
| Msx1−/− | Mouse | Msx1 | Msx1 null | Shortened mandible and maxilla | None reported | Cleft palate | Failure of tooth induction; Abnormalities of the nasal, frontal and parietal bones, and of the malleus in the middle ear; cyanosis | [98] |
| Prdm16cGT | Mouse | Prdm16 | Prdm16 null | Micrognathia, smaller Meckel′s cartilage | Abnormal positioning and morphology of the tongue | Cleft palate | Respiratory failure and abdominal distention, reduced ossification of the frontal and parietal bones, nasal cartilage appears shortened, abnormal retinal folds; hypoplasia of choroid plexi, salivary glands, lungs, cardiac ventricules | [39] |
| Prdm16csp1 | Mouse | Prdm16 | Intronic splice mutation in Prdm16 | Micrognathia, smaller Meckel′s cartilage | Abnormal positioning and morphology of the tongue | Cleft palate | Respiratory failure and abdominal distention, reduced ossification of the frontal and parietal bones, nasal cartilage appears shortened, abnormal retinal folds; hypoplasia of choroid plexi, salivary glands, lungs, cardiac ventricules | [38] |
| Ptprs−/−; Ptprf−/− | Mouse | Ptprs, Ptprf | Ptprs;Ptprf double-knockout | Micrognathia | Microglossia/glossoptosis | Cleft palate | Dysmorphic cranial bone and cartilage | [99] |
| Satb2tm1(cre)Vit | Mouse | Satb2 | Satb2 null | Micrognathia | Microglossia | Cleft palate | Microcephaly, nasocapsular and premaxillary hypoplasia; fully penetrant incisor adontia | [100] |
| Snai1/2-dko | Mouse | Snai1/Snai2 | Neural-crest-specific Snai1 deletion on a Snai2−/− genetic back-ground | Micrognathia, fused mandible and a failure of Meckel′s cartilage to extend the mandible | None reported | Cleft palate | Enlarged parietal foramen in skull vault | [101] |
| Sox9+/− | Mouse | Sox9 | Heterozygous knockout of Sox9 | Micrognathia | Bifurcated tongue | Cleft palate | Hypoplasia of cartilaginous skeletal elements | [77] |
| Sox9fl/+; Wnt1-Cre | Mouse | Sox9 | Heterozygous Wnt1-Cre conditional knockout of Sox9 | Micrognathia | None reported | Cleft palate | Mildly hypoplastic craniofacial skeleton | [78] |
| Sox9fl/+; Wnt1-Cre2 | Mouse | Sox9 | Heterozygous Wnt1-Cre conditional knockout of Sox9 | Micrognathia | None reported | Cleft palate in 50% of mutant embryos | None reported | [41] |
| Sox9 mEC1.45del/del | Mouse | Sox9 | Knockout of Sox9 enhancer mEC1.45 | Altered mandibular morphology | None reported | None reported | Reduction in weight gain | [41] |
| Sox11fl/fl; EIIa-Cre | Mouse | Sox11 | Sox11 null | Micrognathia | Displaced tongue position | Cleft palate with retardation to palatal shelf elevation | None reported | [102] |
| Tak1fl/fl; Wnt1-Cre | Mouse | Tak1 | Wnt1-Cre conditional knockout of Tak1 | Micrognathia | Malformed tongue | Cleft palate | Hypoplastic calvarial bones | [87] |
| Tbx1−/− | Mouse | Tbx1 | Tbx1 null | Micrognathia | None reported | Cleft palate | Hypoplasia of the thymus and parathyroid glands, cardiac outflow tract abnormalities, abnormal facial structures, abnormal vertebrae | [103] |
| Tcof1+/− | Mouse | Tcof1 | Heterozygous knockout of Tcof1 | Micrognathia/retrognathia | None reported | Cleft palate | Agenesis of the nasal passages, abnormal maxilla, exencephaly, anophthalmia | [88,104] |
| Tgdsbub/Tgdsbub | Mouse | Tgds | N-ethyl-N-nitrosourea-induced mutation | Micrognathia | None reported | Cleft palate | None reported | [105] |
| hpmd-line 171a | Mouse | Unknown | N-ethyl-N-nitrosou-rea-induced mutation | Hypoplastic mandible | None reported | Cleft palate | Split in xyphoid process, malformation of first brachial arch derivatives | [76,106] |
| A/WySn | Mouse | Unknown | Unknown | Retrognathia | None reported | Cleft palate | None reported | [107] |
| CP1 NSDTR | Dog | DLX6 | A long interspersed nuclear element-1 insertion in DLX6 | Relative micrognathia | None reported | Cleft palate | None reported | [108] |
| crispld2KD | Zebrafish | crispld2 | Morpholino knockdown of crispld2 | Loss of lower jaw structures | None reported | Malformations of the palate | Truncated body, shortened and curved tail with cardiac edema, clefting of the ethmoid plate | [109] |
| faf1KD | Zebrafish | faf1 | Morpholino knockdown of faf1 | Under-developed jaw | None reported | None reported | Smaller head; “open-mouth” phenotype | [110] |
| polr1c−/− | Zebrafish | polr1c | polr1c knockout (polr1chi1124Tg) generated by insertion mutagenesis | Hypoplastic mandible | None reported | Cleft palate, smaller ethmoid plate | Smaller heads, microphthalmia, pericardial edema | [111,112] |
| polr1d−/− | Zebrafish | polr1d | polr1d knockout (polr1dhi2393Tg) generated by insertion mutagenesis | Hypoplastic mandible | None reported | Smaller ethmoid plate | Smaller heads, microphthalmia, pericardial edema | [111] |
| tcof1KD | Zebrafish | tcof1 | Morpholino knockdown of tcof1 | Hypoplastic mandible | None reported | Smaller and dysmorphic ethmoid plate | Cranioskeletal hypoplasia in the frontal, premaxillary, and maxillary elements | [113] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Motch Perrine, S.M.; Wu, M.; Holmes, G.; Bjork, B.C.; Jabs, E.W.; Richtsmeier, J.T. Phenotypes, Developmental Basis, and Genetics of Pierre Robin Complex. J. Dev. Biol. 2020, 8, 30. https://doi.org/10.3390/jdb8040030
Motch Perrine SM, Wu M, Holmes G, Bjork BC, Jabs EW, Richtsmeier JT. Phenotypes, Developmental Basis, and Genetics of Pierre Robin Complex. Journal of Developmental Biology. 2020; 8(4):30. https://doi.org/10.3390/jdb8040030
Chicago/Turabian StyleMotch Perrine, Susan M., Meng Wu, Greg Holmes, Bryan C. Bjork, Ethylin Wang Jabs, and Joan T. Richtsmeier. 2020. "Phenotypes, Developmental Basis, and Genetics of Pierre Robin Complex" Journal of Developmental Biology 8, no. 4: 30. https://doi.org/10.3390/jdb8040030
APA StyleMotch Perrine, S. M., Wu, M., Holmes, G., Bjork, B. C., Jabs, E. W., & Richtsmeier, J. T. (2020). Phenotypes, Developmental Basis, and Genetics of Pierre Robin Complex. Journal of Developmental Biology, 8(4), 30. https://doi.org/10.3390/jdb8040030