Genetics Underlying the Interactions between Neural Crest Cells and Eye Development

Abstract

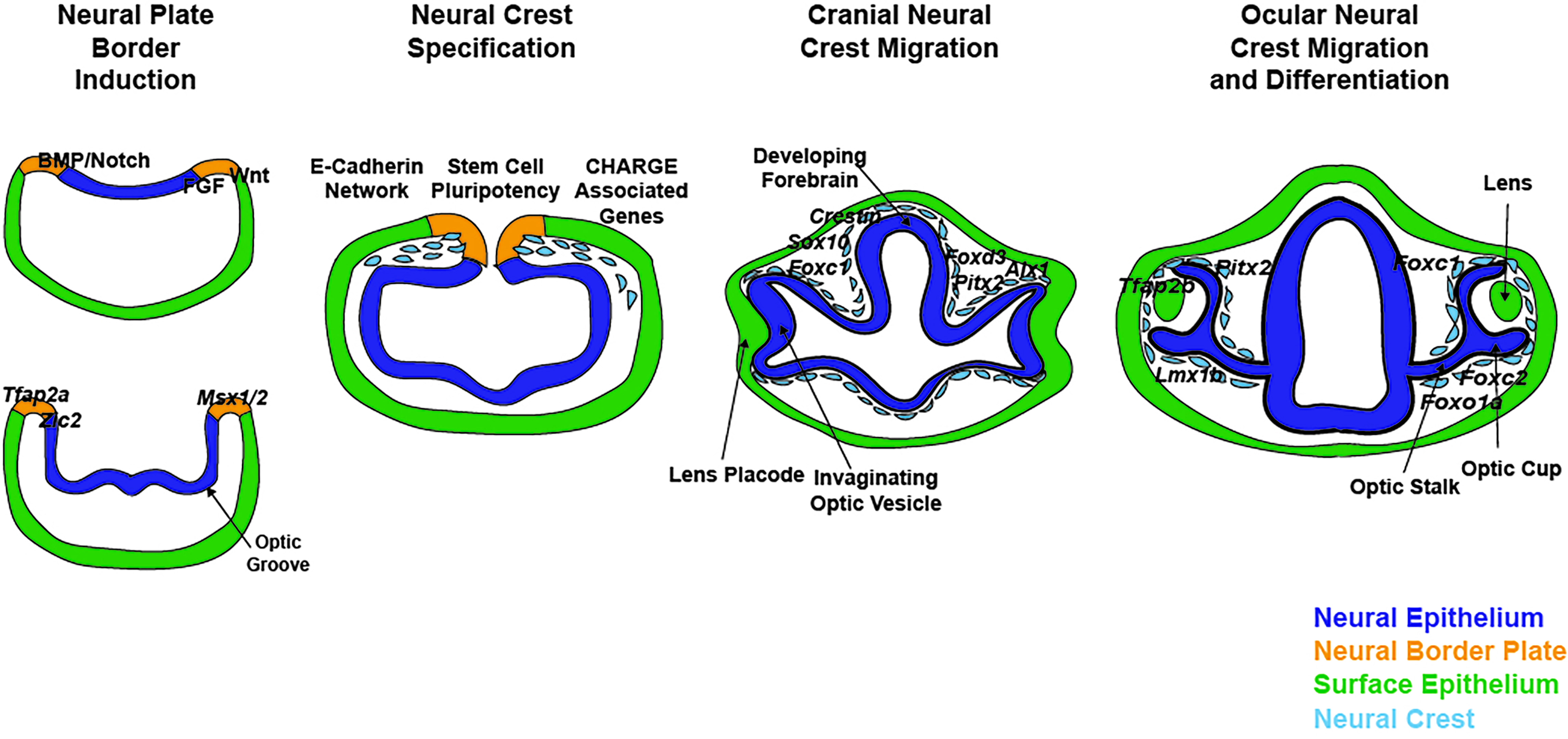{kind=link}
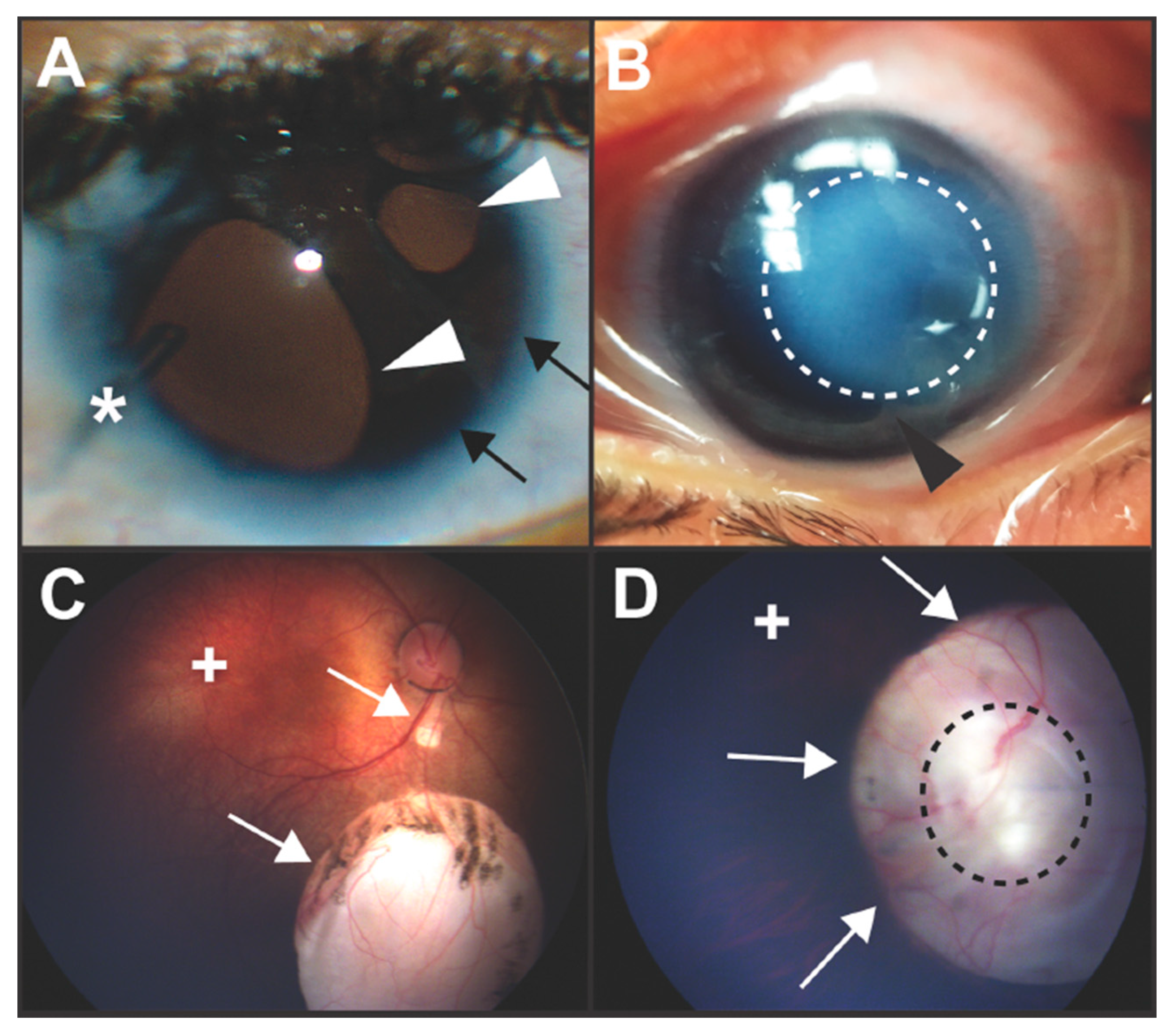{kind=link}
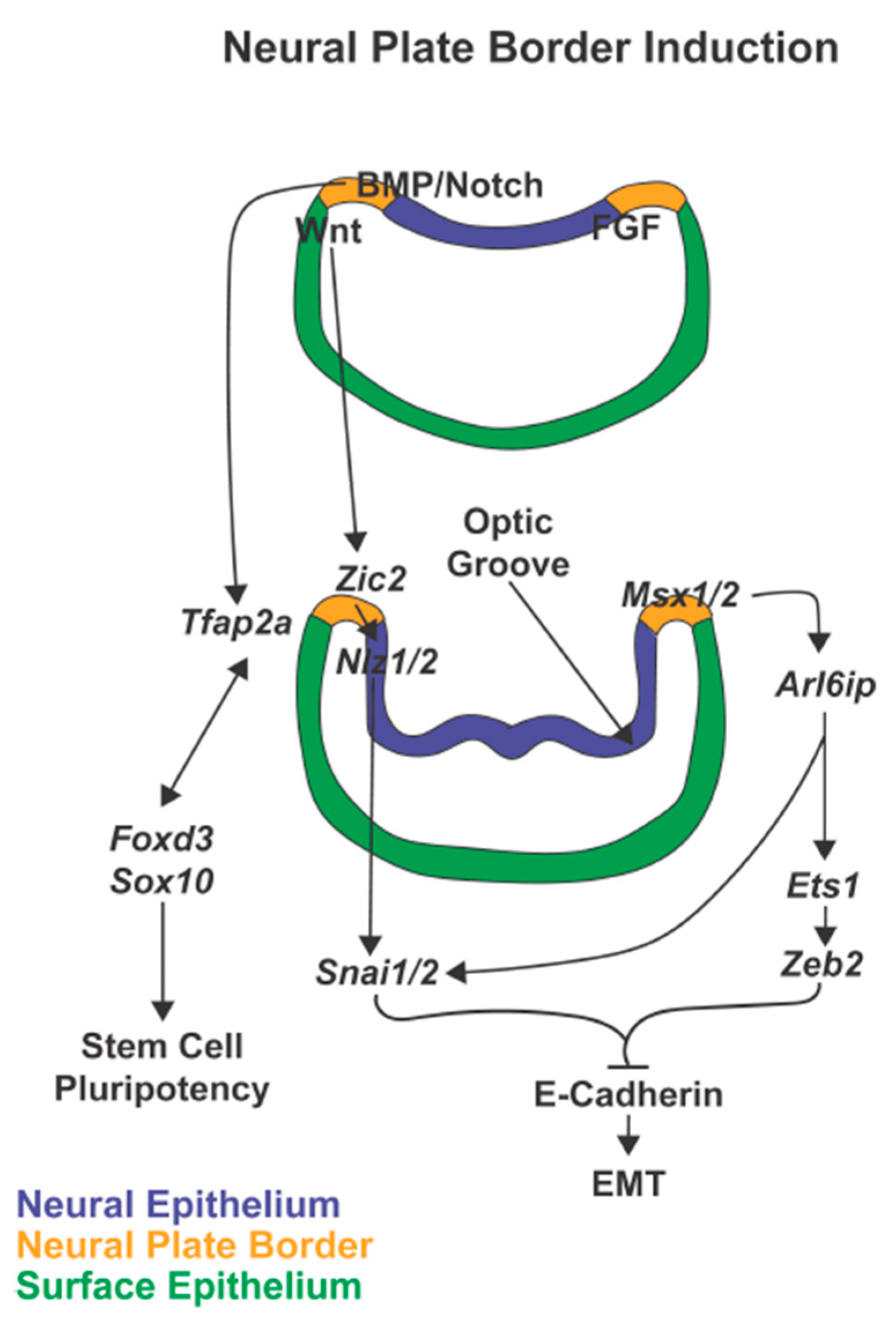{kind=link}
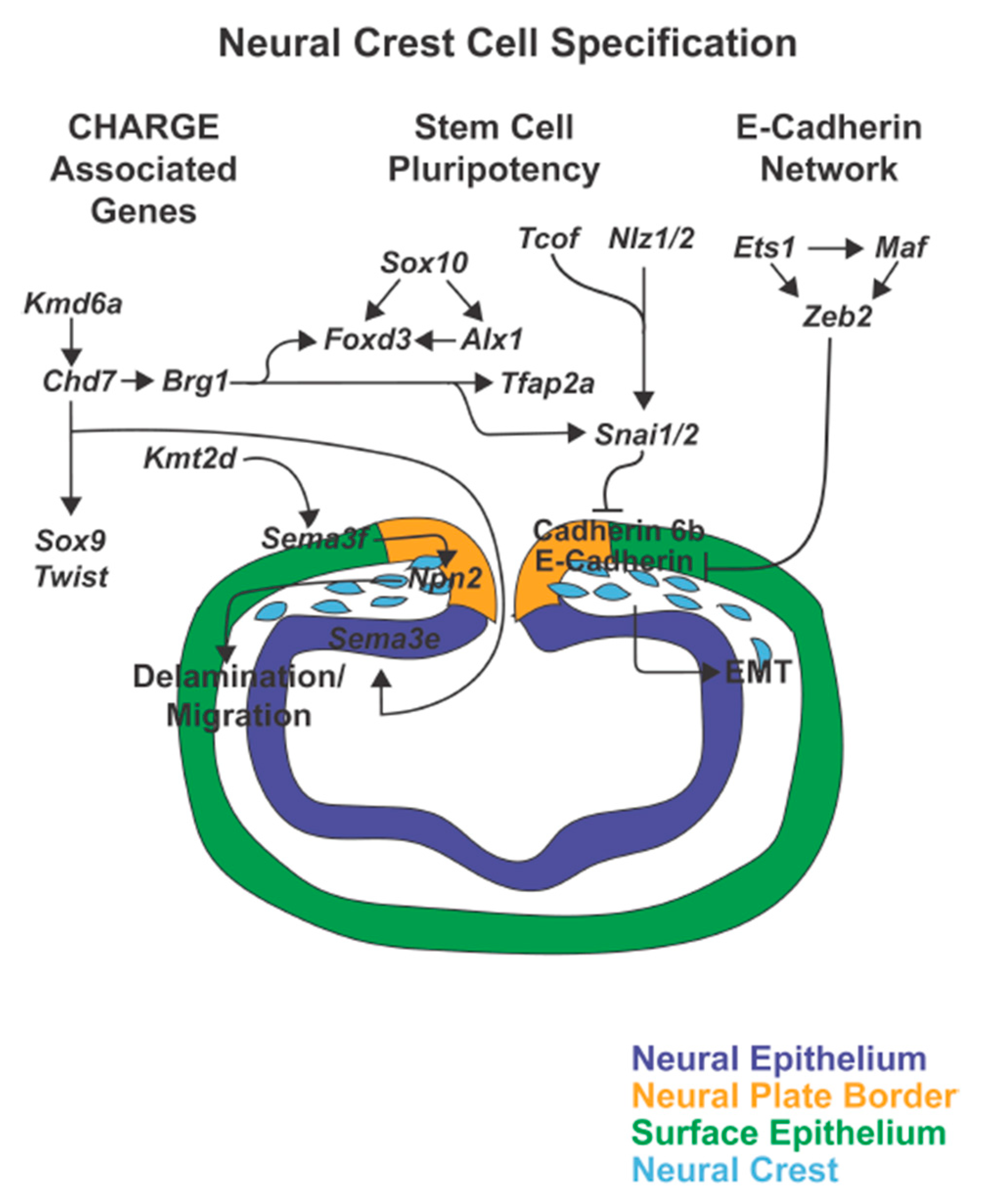{kind=link}
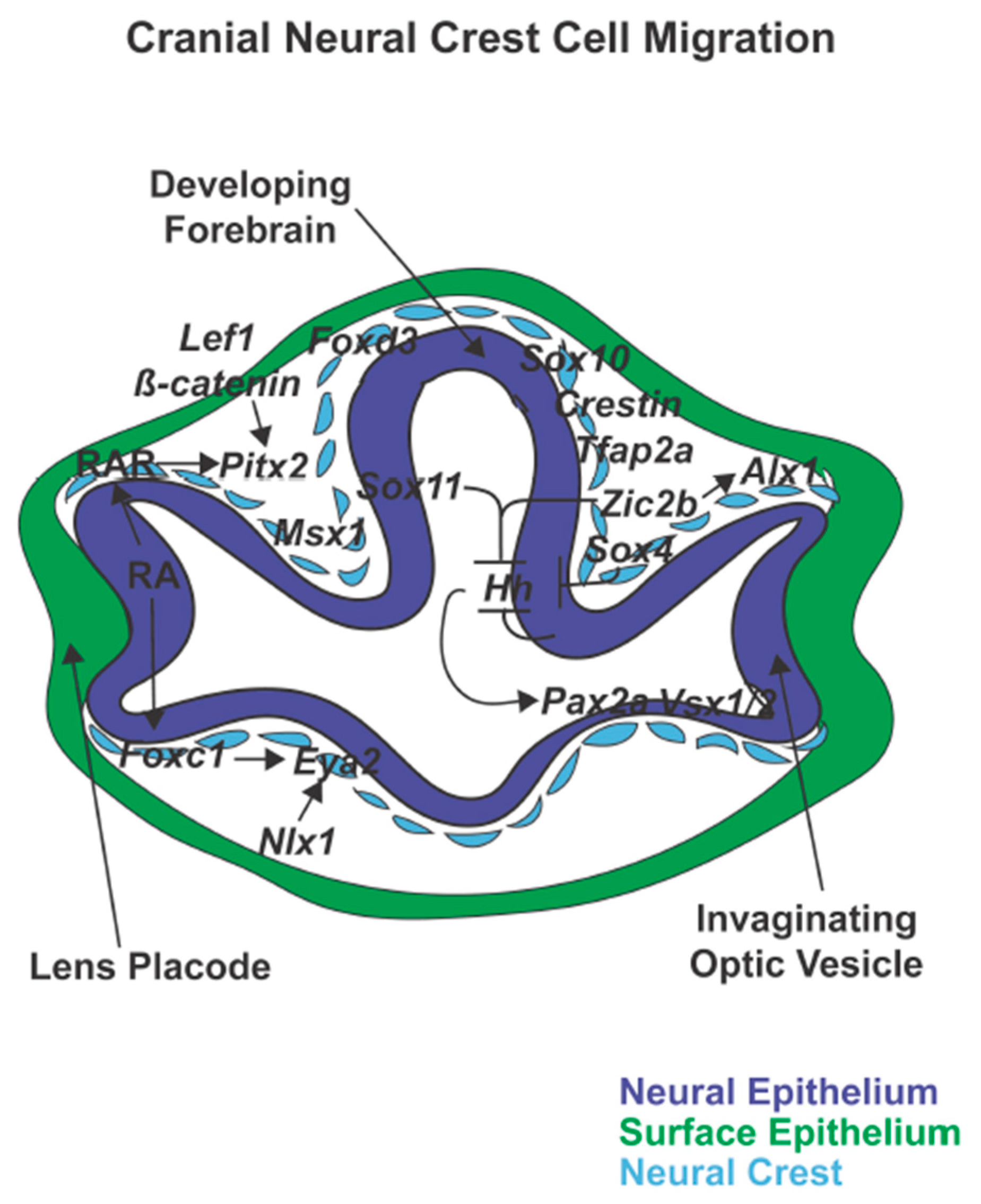{kind=link}
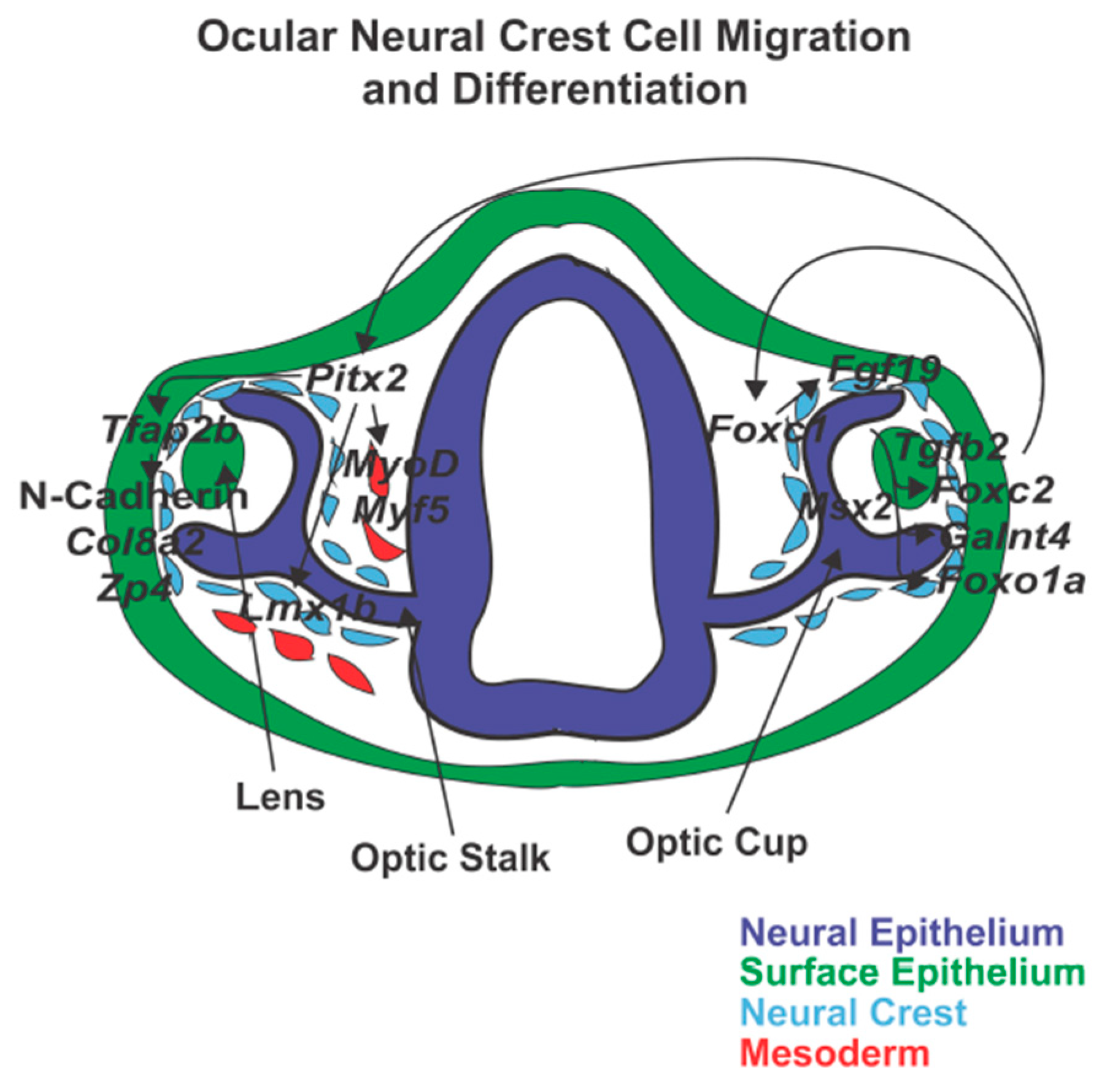{kind=link}
1. Introduction
2. Overview of Neural Crest Signaling Modules
3. Neural Plate Border Genes
3.1. Msx Gene Family
3.2. Zic Gene Family
3.3. TFAP2 Genes
4. CHARGE Syndrome Associated Genes
4.1. E-Cadherin in Neural Crest Cell EMT
4.2. Maintaining Embryonic Stem Cell Pluripotency and Cell Lineage-Specificity
5. Cranial Neural Crest Cell Migration Gene Regulation
6. Summary/Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hay, E. Development of the vertebrate cornea. Int. Rev. Cytol. 1979, 63, 263–322. [Google Scholar]
- Beebe, D.C.; Coats, J.M. The lens organizes the anterior segment: Specification of neural crest cell differentiation in the avian eye. Dev. Biol. 2000, 220, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Cvekl, A.; Tamm, E.R. Anterior eye development and ocular mesenchyme: New insights from mouse models and human disease. BioEssays 2004, 26, 374–386. [Google Scholar] [CrossRef] [PubMed]
- Gage, P.J.; Rhoades, W.; Prucka, S.K.; Hjalt, T. Fate maps of neural crest and mesoderm in the mammalian eye. Invest. Ophthalmol. Vis. Sci. 2005, 46, 4200–4208. [Google Scholar] [CrossRef] [PubMed]
- Kish, P.E.; Bohnsack, B.L.; Gallina, D.; Kasprick, D.S.; Kahana, A. The eye as an organizer of craniofacial development. Genesis 2011, 49, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.L.; Bohnsack, B.L. Neural crest derivatives in ocular development: Discerning the eye of the storm. Birth Defects Res. C Embryo Today 2015, 105, 87–95. [Google Scholar] [CrossRef]
- Cook, C.S.; Sulik, K.K. Keratolenticular dysgenesis (Peters’ anomaly) as a result of acute embryonic insult during gastrulation. J. Pediatr. Ophthalmol. Strabismus 1988, 25, 60–66. [Google Scholar]
- Ozeki, H.; Shirai, S.; Ikeda, K.; Ogura, Y. Anomalies associated with Axenfeld-Rieger syndrome. Graefes Arch. Clin. Exp. Ophthalmol. 1999, 237, 730–734. [Google Scholar] [CrossRef]
- Strungaru, M.H.; Dinu, I.; Walter, M.A. Genotype-phenotype correlations in Axenfeld-Rieger malformation and glaucoma patients with FOXC1 and PITX2 mutations. Invest. Ophthalmol. Vis. Sci. 2007, 48, 228–237. [Google Scholar] [CrossRef]
- Chawla, B.; Schley, E.; Williams, A.L.; Bohnsack, B.L. Retinoic acid and pitx2 regulate early neural crest survival and migration in craniofacial and ocular development. Birth Defects Res. B Dev. Reprod. Toxicol. 2016, 107, 126–135. [Google Scholar] [CrossRef]
- Eason, J.; Williams, A.L.; Chawla, B.; Apsey, C.; Bohnsack, B.L. Differences in neural crest sensitivity to ethanol account for the infrequency of anterior segment defects in the eye compared with craniofacial anomalies in a zebrafish model of fetal alcohol syndrome. Birth Defects Res. 2017, 109, 1212–1227. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.P.; Taylor, A.E.; Sowden, J.C.; Ragge, N.K.; Russell-Eggitt, I.; Rahi, J.S.; Gilbert, C.E.; Surveillance of Eye Anomalies (SEA-UK) Special Interest Group. Anophthalmos, microphthalmos, and typical coloboma in the United Kingdom: A prospective study of incidence and risk. Invest. Ophthalmol. Vis. Sci. 2011, 52, 558–564. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.P.; Taylor, A.E.; Sowden, J.C.; Ragge, N.; Russell-Eggitt, I.; Rahi, J.S.; Gilbert, C.E.; Surveillance of Eye Anomalies Special Interest Group. Anophthalmos, microphthalmos, and coloboma in the United Kingdom: Clinical features, results of investigations, and early management. Ophthalmology 2012, 119, 362–368. [Google Scholar] [CrossRef] [PubMed]
- Bryan, C.D.; Casey, M.A.; Pfeiffer, R.L.; Jones, B.W.; Kwan, K.M. Optic cup morphogenesis requires neural crest-mediated basement membrane assembly. Development 2020, 147, dev181420. [Google Scholar] [CrossRef] [PubMed]
- Betancur, P.; Bronner-Fraser, M.; Sauka-Spengler, T. Assembling neural crest regulatory circuits into a gene regulatory network. Annu. Rev. Cell Dev. Biol. 2010, 26, 581–603. [Google Scholar] [CrossRef]
- Mayor, R.; Theveneau, E. The neural crest. Development 2013, 140, 2247–2251. [Google Scholar] [CrossRef]
- Simões-Costa, M.; Bronner, M.E. Establishing neural crest identity: A gene regulatory recipe. Development 2015, 142, 2420257. [Google Scholar] [CrossRef]
- Ozair, M.Z.; Kintner, C.; Brivanlou, A.H. Neural induction and early patterning in vertebrates. Wiley Interdiscip. Rev. Dev. Biol. 2013, 2, 479–498. [Google Scholar] [CrossRef]
- Groves, A.K.; LaBonne, C. Setting appropriate boundaries: Fate, patterning and competence at the neural plate border. Dev. Biol. 2014, 389, 2–12. [Google Scholar] [CrossRef]
- Schille, C.; Schambony, A. Signaling pathways and tissue interactions in neural plate border formation. Neurogenesis (Austin) 2017, 4, e1292783. [Google Scholar] [CrossRef]
- Sauka-Spengler, T.; Bronner-Fraser, M. Development and evolution of the migratory neural crest: A gene regulatory perspective. Curr. Opin. Genet. Dev. 2006, 16, 360–366. [Google Scholar] [CrossRef] [PubMed]
- Creuzet, S.; Vincent, C.; Couly, G. Neural crest derivatives in ocular and periocular structures. Int. J. Dev. Biol. 2005, 49, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Theveneau, E.; Mayor, R. Neural crest delamination and migration: From epithelium-to-mesenchyme transition to collective cell migration. Dev. Biol. 2012, 366, 34–54. [Google Scholar] [CrossRef] [PubMed]
- Gammill, L.S.; Bronner-Fraser, M. Neural crest specification: Migrating into genomics. Nat. Rev. Neurosci. 2003, 4, 795–805. [Google Scholar] [CrossRef]
- Adler, R.; Canto-Soler, M.V. Molecular mechanisms of optic vesicle development: Complexities, ambiguities, and controversies. Dev. Biol. 2007, 305, 1–13. [Google Scholar] [CrossRef]
- Fuhrmann, S. Eye morphogenesis and patterning of the optic vesicle. Curr. Top. Dev. Biol. 2010, 93, 61–84. [Google Scholar]
- Graw, J. Eye development. Curr. Top. Dev. Biol. 2010, 90, 343–386. [Google Scholar]
- Kulesa, P.M.; Bailey, C.M.; Kasemeier-Kulesa, J.C.; McLennan, R. Cranial neural crest migration: New rules for an old road. Dev. Biol. 2010, 344, 543–554. [Google Scholar] [CrossRef]
- Canto-Soler, M.V.; Adler, R. Optic cup and lens development requires Pax6 expression in the early optic vesicle during a narrow time window. Dev. Biol. 2006, 294, 119–132. [Google Scholar] [CrossRef]
- Williams, A.L.; Eason, J.; Chawla, B.; Bohnsack, B.L. Cyp1b1 regulates ocular fissure closure through a retinoic acid-independent pathway. Invest. Ophthalmol. Vis. Sci. 2017, 58, 1084–1097. [Google Scholar] [CrossRef]
- Johnston, M.C.; Noden, D.M.; Hazelton, R.D.; Coulombre, J.L.; Coulombre, A. Origins of avian ocular and periocular tissues. Exp. Eye Res. 1979, 29, 27–43. [Google Scholar] [CrossRef]
- Takamiya, M.; Stegmaier, J.; Kobitski, A.Y.; Schott, B.; Weger, B.D.; Margariti, D.; Cereceda Delgado, A.R.; Gourain, V.; Scherr, T.; Yang, L.; et al. Pax6 organizes the anterior eye segment by guiding two distinct neural crest waves. PLoS Genet. 2020, 16, e1008774. [Google Scholar] [CrossRef] [PubMed]
- Akula, M.; Park, J.W.; West-Mays, J.A. Relationship between neural crest cell specification and rare ocular diseases. J. Neurosci. Res. 2019, 97, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.L.; Bohnsack, B.L. What’s retinoic acid got to do with it? Retinoic acid regulation of the neural crest in craniofacial and ocular development. Genesis 2019, 57, e23308. [Google Scholar] [CrossRef] [PubMed]
- Harris, M.L.; Erickson, C.A. Lineage specification in neural crest cell pathfinding. Dev. Dyn. 2007, 236, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Krispin, S.; Nitzan, E.; Kassem, Y.; Kalcheim, C. Evidence for a dynamic spatiotemporal fate map and early fate restrictions of premigratory avian neural crest. Development 2010, 137, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Nitzan, E.; Kalcheim, C. Neural crest and somitic mesoderm as paradigms to investigate cell fate decisions during development. Dev. Growth Differ. 2013, 55, 60–78. [Google Scholar] [CrossRef]
- Lukoseviciute, M.; Gavriouchkina, D.; Williams, R.M.; Hochgreb-Hagele, T.; Senanayake, U.; Chong-Morrison, V.; Thongjuea, S.; Repapi, E.; Mead, A.; Sauka-Spengler, T. From pioneer to repressor: Bimodal foxd3 activity dynamically remodels neural crest regulatory landscape in vivo. Dev. Cell. 2018, 47, 608–628. [Google Scholar] [CrossRef]
- Knecht, A.K.; Bronner-Fraser, M. Induction of the neural crest: A multigene process. Nat. Rev. 2002, 3, 453–461. [Google Scholar] [CrossRef]
- Streit, A.; Berliner, A.J.; Papanayotou, C.; Sirulnik, A.; Stern, C.D. Initiation of neural induction by FGF signaling before gastrulation. Nature 2000, 406, 74–78. [Google Scholar] [CrossRef]
- Milet, C.; Monsoro-Burq, A.H. Neural crest induction at the neural plate border in vertebrates. Dev. Biol. 2012, 366, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Stuhlmiller, T.J.; García-Castro, M.I. Current perspectives on the signaling pathways directing neural crest induction. Cell Mol. Life Sci. 2012, 69, 3715–3737. [Google Scholar] [CrossRef] [PubMed]
- Pla, P.; Monsoro-Burq, A.H. The neural border: Induction, specification and maturation of the territory that generates neural crest cells. Dev. Biol. 2018, 444, S36–S46. [Google Scholar] [CrossRef] [PubMed]
- Meulemans, D.; Bronner-Fraser, M. Gene-regulatory interactions in neural crest evolution and development. Dev. Cell. 2004, 7, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Khudyakov, J.; Bronner-Fraser, M. Comprehensive spatiotemporal analysis of early chick neural crest network genes. Dev. Dyn. 2009, 238, 716–723. [Google Scholar] [CrossRef] [PubMed]
- Prasad, M.S.; Sauka-Spengler, T.; LaBonne, C. Induction of the neural crest state: Control of stem cell attributes by gene regulatory, post-transcriptional and epigenetic interactions. Dev. Biol. 2012, 366, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Gheldof, A.; Berx, G. Cadherins and epithelial-to-mesenchymal transition. Prog. Mol. Biol. Transl. Sci. 2013, 116, 317–336. [Google Scholar]
- Schulz, Y.; Wehner, P.; Opitz, L.; Salinas-Riester, G.; Bongers, E.M.; van Ravenswaaij-Arts, C.M.; Wincent, J.; Schoumans, J.; Kohlhase, J.; Borchers, A.; et al. CHD7, the gene mutated in CHARGE syndrome, regulates genes involved in neural crest cell guidance. Hum. Genet. 2014, 133, 997–1009. [Google Scholar] [CrossRef]
- Dady, A.; Duband, J.L. Cadherin interplay during neural crest segregation from the non-neural ectoderm and neural tube in the early chick embryo. Dev. Dyn. 2017, 246, 550–565. [Google Scholar] [CrossRef]
- Bérubé-Simard, F.A.; Pilon, N. Molecular dissection of CHARGE syndrome highlights the vulnerability of neural crest cells to problems with alternative splicing and other transcription-related processes. Transcription 2019, 10, 21–28. [Google Scholar] [CrossRef]
- Scarpa, E.; Szabó, A.; Bibonne, A.; Theveneau, E.; Parsons, M.; Mayor, R. Cadherin switch during EMT in neural crest cells leads to contact inhibition of locomotion via repolarization of forces. Dev. Cell 2015, 34, 421–434. [Google Scholar] [CrossRef] [PubMed]
- Okuno, H.; Mihara, F.R.; Ohta, S.; Fukuda, K.; Kurosawa, K.; Akamatsu, W.; Sanosaka, T.; Kohyama, J.; Hayashi, K.; Nakajima, K.; et al. CHARGE syndrome modeling using patient-iPSCs reveals defective migration of neural crest cells harboring CHD7 mutations. Elife 2017, 6, e21114. [Google Scholar] [CrossRef] [PubMed]
- Kunisada, T.; Tezulka, K.-I.; Aoki, H.; Motohashi, T. The stemness of neural crest cells and their derivatives. Birth. Defects Res. C Embryo. Today 2014, 102, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Shellard, A.; Mayor, R. Integrating chemical and mechanical signals in neural crest cell migration. Curr. Opin. Genet. Dev. 2019, 57, 16–24. [Google Scholar] [CrossRef]
- Davidson, D. The function and evolution of Msx genes: Pointers and paradoxes. Trends Genet. 1995, 11, 405–411. [Google Scholar] [CrossRef]
- Alappat, S.; Zhang, Z.Y.; Chen, Y.P. Msx homeobox gene family and craniofacial development. Cell Res. 2003, 13, 429–442. [Google Scholar] [CrossRef]
- Catron, K.M.; Wang, H.; Hu, G.; Shen, M.M.; Abate-Shen, C. Comparison of Msx-1 and Msx-2 suggests a molecular basis for functional redundancy. Mech. Dev. 1996, 55, 185–199. [Google Scholar] [CrossRef]
- Tribulo, C.; Aybar, M.J.; Nguyen, V.H.; Mullins, M.C.; Mayor, R. Regulation of Msx genes by a Bmp gradient is essential for neural crest specification. Development 2003, 130, 6441–6452. [Google Scholar] [CrossRef]
- Ishii, M.; Han, J.; Yen, H.Y.; Sucov, H.M.; Chai, Y.; Maxson, R.E.J. Combined deficiencies of Msx1 and Msx2 cause impaired patterning and survival of the cranial neural crest. Development 2005, 132, 4937–4950. [Google Scholar] [CrossRef]
- Khadka, D.; Luo, T.; Sargent, T.D. Msx1 and Msx2 have shared essential functions in neural crest but may be dispensable in epidermis and axis formation in Xenopus. Int. J. Dev. Biol. 2006, 50, 499–502. [Google Scholar] [CrossRef]
- Phillips, B.T.; Kwon, H.J.; Melton, C.; Houghtaling, P.; Fritz, A.; Riley, B.B. Zebrafish msxB, msxC, and msxE function together to refine the neural-nonneural border and regulate cranial placodes and neural crest development. Dev. Biol. 2006, 294, 376–390. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lincecum, J.M.; Fannon, A.; Song, K.; Wang, Y.; Sassoon, D.A. Msh homeobox genes regulate cadherin-mediated cell adhesion and cell-cell sorting. J. Cell Biochem. 1998, 70, 22–28. [Google Scholar] [CrossRef]
- Monaghan, A.P.; Davidson, D.R.; Sime, C.; Graham, E.; Baldock, R.; Bhattacharya, S.S.; Hill, R.E. The Msh-like homeobox genes define domains in the developing vertebrate eye. Development 1991, 112, 1053–1061. [Google Scholar] [PubMed]
- Suzuki, H.R.; Padanilam, B.J.; Vitale, E.; Ramirez, F.; Solursh, M. Repeating developmental expression of G-Hox7, a novel homeobox-containing gene in the chicken. Dev. Biol. 1991, 148, 375–388. [Google Scholar] [CrossRef]
- Foerst-Potts, L.; Sadler, T.W. Disruption of Msx-1 and Msx-2 reveals roles for these genes in craniofacial, eye, and axial development. Dev. Dyn. 1997, 209, 70–84. [Google Scholar] [CrossRef]
- Aruga, J.; Hatayama, M. Comparative genomics of the zic family genes. Adv. Exp. Med. Biol. 2018, 1046, 3–26. [Google Scholar]
- Diamond, K.E.M.; Barratt, K.S.; Arkell, R.M. Overview of rodent zic genes. Adv. Exp. Med. Biol. 2018, 1046, 179–207. [Google Scholar]
- Winata, C.L.; Korzh, V. Zebrafish zic genes mediate developmental signaling. Adv. Exp. Med. Biol. 2018, 1046, 157–177. [Google Scholar]
- Nakata, K.; Nagai, T.; Aruga, J.; Mikoshiba, K. Xenopus Zic3, a primary regulator both in neural and neural crest development. Proc. Natl. Acad. Sci. USA 1997, 94, 11980–11985. [Google Scholar] [CrossRef]
- Nakata, K.; Nagai, T.; Aruga, J.; Mikoshiba, K. Xenopus Zic family and its role in neural and neural crest development. Mech. Dev. 1998, 75, 43–51. [Google Scholar] [CrossRef]
- Marchal, L.; Luxardi, G.; Thomé, V.; Kodjabachian, L. BMP inhibition initiates neural induction via FGF signaling and Zic genes. Proc. Natl. Acad. Sci. USA 2009, 106, 17437–17442. [Google Scholar] [CrossRef] [PubMed]
- Aruga, J. The role of Zic genes in neural development. Mol. Cell Neurosci. 2004, 26, 205–221. [Google Scholar] [CrossRef] [PubMed]
- Maurus, D.; Harris, W.A. Zic-associated holoprosencephaly: Zebrafish Zic1 controls midline formation and forebrain patterning by regulating Nodal, hedgehog, and retinoic acid signaling. Genes Dev. 2009, 23, 1461–1473. [Google Scholar] [CrossRef] [PubMed]
- McMahon, A.R.; Merzdorf, C.S. Expression of the zic1, zic2, zic3, and zic4 genes in early chick embryos. BMC Res. Notes 2010, 3, 167. [Google Scholar] [CrossRef] [PubMed]
- Aruga, J.; Millen, K.J. ZIC1 function in normal cerebellar development and human developmental pathology. Adv. Exp. Med. Biol. 2018, 1046, 249–268. [Google Scholar] [PubMed]
- Nyholm, M.K.; Abdelilah-Seyfried, S.; Grinblat, Y. A novel genetic mechanism regulates dorsolateral hinge-point formation during zebrafish cranial neurulation. J. Cell Sci. 2009, 122, 2137–2148. [Google Scholar] [CrossRef]
- Teslaa, J.J.; Keller, A.N.; Nyholm, M.K.; Grinblat, Y. Zebrafish zic2a and zic2b regulate neural crest and craniofacial development. Dev. Biol. 2013, 380, 73–86. [Google Scholar] [CrossRef]
- Elms, P.; Siggers, P.; Napper, D.; Greenfield, A.; Arkell, R. Zic2 is required for neural crest formation and hindbrain patterning during mouse development. Dev. Biol. 2003, 264, 391–406. [Google Scholar] [CrossRef]
- Sedykh, I.; Yoon, B.; Roberson, L.; Moskvin, O.; Dewey, C.N.; Grinblat, Y. Zebrafish zic2 controls formation of periocular neural crest and choroid fissure morphogenesis. Dev. Biol. 2017, 429, 92–104. [Google Scholar] [CrossRef]
- Sanek, N.A.; Grinblat, Y. A novel role for zebrafish zic2a during forebrain development. Dev. Biol. 2008, 317, 325–335. [Google Scholar] [CrossRef][Green Version]
- Pillai-Kastoori, L.; Wen, W.; Wilson, S.G.; Strachan, E.; Lo-Castro, A.; Fichera, M.; Musumeci, S.A.; Lehmann, O.J.; Morris, A.C. Sox11 is required to maintain proper levels of hedgehog signaling during vertebrate ocular morphogenesis. PLoS Genet. 2014, 10, e1004491. [Google Scholar] [CrossRef] [PubMed]
- Wen, W.; Pillai-Kastoori, L.; Wilson, S.G.; Morris, A.C. Sox4 regulates choroid fissure closure by limiting Hedgehog signaling during ocular morphogenesis. Dev. Biol. 2015, 399, 139–153. [Google Scholar] [CrossRef]
- Kuwajima, T.; Soares, C.A.; Sitko, A.A.; Lefebvre, V.; Mason, C. SoxC transcription factors promote contralateral retinal ganglion cell differentiation and axon guidance in the mouse visual system. Neuron 2017, 93, 1110–1125. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, T.L.; Javier, A.L.; Campeau, S.A.; Knight, R.D.; Schilling, T.F. Tfap2 transcription factors in zebrafish neural crest development and ectodermal evolution. J. Exp. Zool. B Mol. Dev. Evol. 2007, 308, 679–691. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Cornell, R.A. Redundant activities of Tfap2a and Tfap2c are required for neural crest induction and development of other non-neural ectoderm derivatives in zebrafish embryos. Dev. Biol. 2007, 304, 338–354. [Google Scholar] [CrossRef] [PubMed]
- Luo, T.; Zhang, Y.; Khadka, D.; Rangarajan, J.; Cho, K.W.Y.; Sargent, T.D. Regulatory targets for transcription factor AP2 in Xenopus embryos. Dev. Growth Differ. 2005, 47, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Luo, T.; Sargent, T.D. Expression of TFAP2beta and TFAP2gamma genes in Xenopus laevis. Gene Exp. Patterns 2006, 6, 589–595. [Google Scholar] [CrossRef]
- Van Otterloo, E.; Li, H.; Jones, K.L.; Williams, T. AP-2α and AP-2ß cooperatively orchestrate homeobox gene expression during branchial arch patterning. Development 2018, 145, dev157438. [Google Scholar] [CrossRef]
- Milunsky, J.M.; Maher, T.M.; Zhao, G.; Roberts, A.E.; Stalker, H.J.; Zori, R.T.; Burch, M.N.; Clemens, M.; Mulliken, J.B.; Smith, R.; et al. TFAP2A mutations result in branchio-oculo-facial syndrome. Am. J. Hum. Genet. 2008, 82, 1171–1177. [Google Scholar] [CrossRef]
- Milunsky, J.M.; Maher, T.M.; Zhao, G.; Wang, Z.; Mulliken, J.B.; Chitayat, D.; Clemens, M.; Stalker, H.J.; Bauer, M.; Burch, M.; et al. Genotype-phenotype analysis of the branchio-oculo-facial syndrome. Am. J. Med. Genet. A 2011, 144A, 22–32. [Google Scholar] [CrossRef]
- Kwon, H.J.; Bhat, N.; Sweet, E.M.; Cornell, R.A.; Riley, B.B. Identification of early requirements for preplacodal ectoderm and sensory organ development. PLoS Genet. 2010, 6, e1001133. [Google Scholar] [CrossRef] [PubMed]
- Stewart, R.A.; Arduini, B.L.; Berghmans, S.; George, R.E.; Kanki, J.P.; Henion, P.D.; Look, A.T. Zebrafish foxd3 is selectively required for neural crest specification, migration and survival. Dev. Biol. 2006, 292, 174–188. [Google Scholar] [CrossRef] [PubMed]
- Dooley, C.M.; Wali, N.; Sealy, I.M.; White, R.J.; Stemple, D.L.; Collins, J.E.; Busch-Nentwich, E.M. The gene regulatory basis of genetic compensation during neural crest induction. PLoS Genet. 2019, 15, e1008213. [Google Scholar] [CrossRef] [PubMed]
- Gestri, G.; Osborne, R.J.; Wyatt, A.W.; Gerrelli, D.; Gribble, S.; Stewart, H.; Fryer, A.; Bunyan, D.J.; Prescott, K.; Collin, J.R.O.; et al. Reduced TFAP2A function causes variable optic fissure closure and retinal defects and sensitizes eye development to mutations in other morphogenetic regulators. Hum. Genet. 2009, 126, 791–803. [Google Scholar] [CrossRef] [PubMed]
- Bassett, E.A.; Pontoriero, G.F.; Feng, W.; Marquardt, T.; Fini, M.E.; Williams, T.; West-Mays, J.A. Conditional deletion of activating protein 2alpha (AP-2alpha) in the developing retina demonstrates non-cell-autonomous roles for AP-2alpha in optic cup development. Mol. Cell. Biol. 2007, 27, 7497–7510. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.M. Epigenetic developmental disorders: CHARGE syndrome, a case study. Curr. Genet. Med. Rep. 2015, 3, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Baralena, L.; Marcocci, C.; Tanda, M.L.; Manetti, L.; Dell’Unto, E.; Bartolomei, M.P.; Nardi, M.; Martino, E.; Pinchera, A. Cigarette smoking and treatment outcomes in Graves ophthalmopathy. Ann. Intern. Med. 1998, 129, 632–635. [Google Scholar] [CrossRef] [PubMed]
- Bergman, J.E.; Janssen, N.; Hoefsloot, L.H.; Jongmans, M.C.; Hofstra, R.M.; van Ravenswaaij-Arts, C.M. CHD7 mutations in CHARGE syndrome: The clinical implications of an expanding phenotype. J. Med. Genet. 2011, 48, 334–342. [Google Scholar] [CrossRef]
- Hsu, P.; Ma, A.; Wilson, M.; Williams, G.; Curotta, J.; Munns, C.F.; Mehr, S. CHARGE syndrome: A review. J. Paediatr. Child. Health 2014, 50, 504–511. [Google Scholar] [CrossRef]
- Bajpai, R.; Chen, D.A.; Rada-Iglesias, A.; Zhang, J.; Xiong, Y.; Helms, J.; Chang, C.-P.; Zhao, Y.; Swigut, T.; Wysocka, J. CHD7 cooperates with PBAF to control multipotent neural crest formation. Nature 2010, 463, 958–962. [Google Scholar] [CrossRef]
- Ufartes, R.; Schwenty-Lara, J.; Freese, L.; Neuhofer, C.; Möller, J.; Wehner, P.; van Ravenswaaij-Arts, C.M.A.; Wong, M.T.Y.; Schanze, I.; Tzschach, A.; et al. Sema3a plays a role in the pathogenesis of CHARGE syndrome. Hum. Mol. Genet. 2018, 27, 1343–1352. [Google Scholar] [CrossRef] [PubMed]
- Sperry, E.D.; Hurd, E.A.; Durham, M.A.; Reamer, E.N.; Stein, A.B.; Martin, D.M. The chromatin remodeling protein CHD7, mutated in CHARGE syndrome, is necessary for proper craniofacial and tracheal development. Dev. Dyn. 2014, 243, 1055–1066. [Google Scholar] [CrossRef] [PubMed]
- Gage, P.J.; Hurd, E.A.; Martin, D.M. Mouse models for the dissection of CHD7 functions in eye development and the molecular basis for ocular defects in CHARGE syndrome. Investig. Ophthalmol. Vis. Sci. 2015, 56, 7923–7930. [Google Scholar] [CrossRef] [PubMed]
- Shpargel, K.B.; Starmer, J.; Wang, C.; Ge, K.; Magnuson, T. UTX-guided neural crest function underlies craniofacial features of Kabuki syndrome. Proc. Natl. Acad. Sci. USA 2017, 114, E9046–E9055. [Google Scholar] [CrossRef]
- Schwenty-Lara, J.; Nehl, D.; Borchers, A. The histone methyltransferase KMT2D, mutated in Kabuki syndrome patients, is required for neural crest cell formation and migration. Hum. Mol. Genet. 2020, 29, 305–319. [Google Scholar] [CrossRef]
- Shpargel, K.B.; Mangini, C.L.; Xie, G.; Ge, K.; Magnuson, T. The KMT2D Kabuki syndrome histone methylase controls neural crest cell differentiation and facial morphology. Development 2020, 147, dev187997. [Google Scholar] [CrossRef]
- Cocciadiferro, D.; Augello, B.; De Nittis, P.; Zhang, J.; Mandriani, B.; Malerba, N.; Squeo, G.M.; Romano, A.; Piccinni, B.; Verri, T.; et al. Dissecting KMT2D missense mutations in Kabuki syndrome patients. Hum. Mol. Genet. 2018, 27, 3651–3668. [Google Scholar] [CrossRef]
- Shangguan, H.; Su, C.; Ouyang, Q.; Cao, B.; Wang, J.; Gong, C.; Chen, R. Kabuki syndrome: Novel pathogenic variants, new phenotypes and review of literature. Orphanet. J. Rare Dis. 2019, 14, 255. [Google Scholar] [CrossRef]
- Wang, Y.-R.; Xu, N.-X.; Wang, J.; Wang, X.-M. Kabuki syndrome: Review of the clinical features, diagnosis and epigenetic mechanisms. World J. Pediatr. 2019, 15, 528–535. [Google Scholar] [CrossRef]
- Schwarz, Q.; Vieira, J.M.; Howard, B.; Eickholt, B.J.; Ruhrberg, C. Neuropilin 1 and 2 control cranial gangliogenesis and axon guidance through neural crest cells. Development 2008, 135, 1605–1613. [Google Scholar] [CrossRef]
- Nakayama, H.; Bruneau, S.; Kochupurakkal, N.; Coma, S.; Briscoe, D.M.; Klagsbrun, M. Regulation of mTOR signaling by semaphorin 3F-neuropilin 2 interactions in vitro and in vivo. Sci. Rep. 2015, 5, 11789. [Google Scholar] [CrossRef] [PubMed]
- Van Nostrand, J.L.; Brady, C.A.; Jung, H.; Fuentes, D.R.; Kozak, M.M.; Johnson, T.M.; Lin, C.-Y.; Lin, C.-J.; Swiderski, D.L.; Vogel, H.; et al. Inappropriate p53 activation during development induces features of CHARGE syndrome. Nature 2014, 514, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, W.A.; Trainor, P.A. Neural crest cell evolution: How and when did a neural crest cell become a neural crest cell. Curr. Top. Dev. Biol. 2015, 111, 3–26. [Google Scholar] [PubMed]
- Higashi, Y.; Maruhashi, M.; Nelles, L.; Van de Putte, T.; Verschueren, K.; Miyoshi, T.; Yoshimoto, A.; Kondoh, H.; Huylebroeck, D. Generation of the floxed allele of the SIP1 (Smad-interacting protein 1) gene for Cre-mediated conditional knockout in the mouse. Genesis 2002, 33, 82–84. [Google Scholar] [CrossRef] [PubMed]
- Van de Putte, T.; Maruhashi, M.; Francis, A.; Nelles, L.; Kondoh, H.; Huylebroeck, D.; Higashi, Y. Mice lacking ZFHX1B, the gene that codes for Smad-interacting protein-1, reveal a role for multiple neural crest defects in the etiology of Hirschsprung disease-mental retardation syndrome. Am. J. Hum. Genet. 2003, 72, 465–470. [Google Scholar] [CrossRef] [PubMed]
- Pettersson, M.; Bessonova, M.; Gu, H.F.; Groop, L.C.; Jönsson, J.I. Characterization, chromosomal localization, and expression during hematopoietic differentiation of the gene encoding Arl6ip, ADP-ribosylation-like factor-6 interacting protin (ARL6). Genomics 2000, 68, 351–354. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Esmail, M.A.; Ansley, S.J.; Blacque, O.E.; Boroevich, K.; Ross, A.J.; Moore, S.J.; Badano, J.L.; May-Simera, H.; Compton, D.S.; et al. Mutations in a member of the Ras superfamily of small GTP-binding proteins causes Bardet-Biedel syndrome. Nat. Genet. 2004, 36, 989–993. [Google Scholar] [CrossRef]
- Kobayashi, T.; Hori, Y.; Ueda, N.; Kajiho, H.; Muraoka, S.; Shima, F.; Kataoka, T.; Kontani, K.; Katada, T. Biochemical characterization of missense mutations in the Arf/Arl-family small GTPase Arl6 causing Bardet-Biedl syndrome. Biochem. Biophys. Res. Commun. 2009, 381, 439–442. [Google Scholar] [CrossRef]
- Tu, C.-T.; Yang, T.-C.; Huang, H.-Y.; Tsai, H.-J. Zebrafish arl6ip1 is required for neural crest development during embryogenesis. PLoS ONE 2012, 7, e32899. [Google Scholar] [CrossRef]
- Barembaum, M.; Bronner, M.E. Identification and dissection of a key enhancer mediating cranial neural crest specific expression of transcription factor, Ets-1. Dev. Biol. 2013, 382, 567–575. [Google Scholar] [CrossRef][Green Version]
- Sieweke, M.H.; Tekotte, H.; Frampton, J.; Graf, T. MafB is an interaction partner and repressor of Ets-1 that inhibits erythroid differentiation. Cell 1996, 85, 49–60. [Google Scholar] [CrossRef]
- Javadiyan, S.; Craig, J.E.; Sharma, S.; Lower, K.M.; Casey, T.; Haan, E.; Souzeau, E.; Burdon, K.P. Novel missense mutation in the bZIP transcription factor, MAF, associated with congenital cataract, developmental delay, seizures, and hearing loss (Aymé-Gripp syndrome). BMC Med. Genet. 2017, 18, 52. [Google Scholar] [CrossRef] [PubMed]
- Niceta, M.; Barbuti, D.; Gupta, N.; Ruggiero, C.; Tizzano, E.F.; Graul-Neumann, L.; Barresi, S.; Nishimura, G.; Valenzuela, I.; López-Grondona, F.; et al. Skeletal abnormalities are common features in Aymé-Gripp syndrome. Clin. Genet. 2020, 97, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Lu, N.; Eberspaecher, H.; De Crombrugghe, B. A new long form of c-Maf cooperates with Sox9 to activate the type II collagen gene. J. Biol. Chem. 2002, 277, 50668–50675. [Google Scholar] [CrossRef]
- Rogers, C.D.; Phillips, J.L.; Bronner, M.E. Elk3 is essential for the progression from progenitor to definitive neural crest cell. Dev. Biol. 2013, 374, 225–263. [Google Scholar] [CrossRef]
- Delalande, J.-M.; Guyote, M.E.; Smith, C.M.; Shepherd, I.T. Zebrafish sip1a and sip1b are essential for normal axial and neural patterning. Dev. Dyn. 2008, 237, 1060–1069. [Google Scholar] [CrossRef]
- Mowat, D.R.; Wilson, M.J.; Goossens, M. Mowat-Wilson syndrome. J. Med. Genet. 2003, 40, 305–310. [Google Scholar] [CrossRef]
- Garavelli, L.; Mainardi, P.C. Mowat-Wilson syndrome. Orphanet. J. Rare Dis. 2007, 2, 42. [Google Scholar] [CrossRef]
- Garcia-Castro, M.I.; Marcelle, C.; Bronner-Fraser, M. Ectodermal Wnt function as a neural crest inducer. Science 2002, 297, 848–851. [Google Scholar]
- Chen, Y.; Gridley, T. The SNAI1 and SNAI2 proteins occupy their own and each other’s promoter during chondrogenesis. Biochem. Biophys. Res. Commun. 2013, 435, 356–360. [Google Scholar] [CrossRef]
- Plouhinec, J.-L.; Roche, D.D.; Pegoraro, C.; Figueiredo, A.L.; Maczkowiak, F.; Brunet, L.J.; Milet, C.; Vert, J.-P.; Pollet, N.; Harland, R.M.; et al. Pax3 and Zic1 trigger the early neural crest gene regulatory network by the direct activation of multiple key neural crest specifiers. Dev. Biol. 2014, 386, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Dixon, M.J. Treacher Collins syndrome. J. Med. Genet. 1995, 32, 806–808. [Google Scholar] [CrossRef]
- Valdez, B.C.; Henning, C.; So, R.B.; Dixon, J.; Dixon, M.J. The Treacher Collins syndrome (TCOF1) gene product is involved in ribosomal DNA gene transcription by interacting with upstream binding factor. Proc. Natl. Acad. Sci. USA 2004, 101, 10709–10714. [Google Scholar] [CrossRef] [PubMed]
- Dixon, J.; Jones, N.C.; Sandell, L.L.; Jayasinghe, S.M.; Crane, J.; Rey, J.P.; Dixon, M.J.; Trainor, P.A. Tcof1/Treacle is required for neural crest cell formation and proliferation deficiencies that cause craniofacial abnormalities. Proc. Natl. Acad. Sci. USA 2006, 103, 13403–13408. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.-S.; Saint-Jeannet, J.-P. Znf703, a novel target of Pax3 and Zic1, regulates hindbrain and neural crest development in Xenopus. Genesis 2017, 55, 10. [Google Scholar] [CrossRef]
- Janesick, A.; Tang, W.; Ampig, K.; Blumberg, B. Znf703 is a novel RA target in the neural plate border. Sci. Rep. 2019, 9, 8275. [Google Scholar] [CrossRef]
- Cano, A.; Pérez-Moreno, M.A.; Locascio, R.A.; Blanco, M.J.; del Barrio, M.G.; Portillo, F.; Nieto, M.A. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat. Cell Biol. 2000, 2, 76–83. [Google Scholar] [CrossRef]
- Taneyhill, L.A.; Coles, E.G.; Bronner-Fraser, M. Snail2 directly represses cadherin6B during epithelial-to-mesenchymal transitions of the neural crest. Development 2007, 134, 1481–1490. [Google Scholar] [CrossRef]
- Coles, E.G.; Taneyhill, L.A.; Bronner-Fraser, M. A critical role for Cadherin6B in regulating avian neural crest emigration. Dev. Biol. 2007, 312, 533–544. [Google Scholar] [CrossRef]
- Blanco, M.J.; Barrallo-Gimeno, A.; Acloque, H.; Reyes, A.E.; Tada, M.; Allende, M.L.; Mayor, R.; Nieto, M.A. Snail1a and Snail1b cooperate in the anterior migration of the axial mesendoderm in the zebrafish embryo. Development 2007, 134, 4073–4081. [Google Scholar] [CrossRef]
- Qiao, L.; Guo, H.; Zhang, T.; Jing, L.; Xiao, C.; Xiao, Y.; Luo, N.; Zhu, H.; Meng, W.; Xu, H.; et al. Snail modulates the assembly of fibronectin via α5 integrin for myocardial migration in zebrafish embryos. Sci. Rep. 2014, 26, 4470. [Google Scholar] [CrossRef] [PubMed]
- Le Douarin, N.M.; Dupin, E. The pluripotency of neural crest cells and their role in brain development. Curr. Top. Dev. Biol. 2016, 116, 659–678. [Google Scholar] [PubMed]
- Sarkar, A.; Hochedlinger, K. The sox family of transcription factors: Versatile regulators of stem and progenitor cell fate. Cell Stem. Cell 2013, 12, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Julian, L.M.; McDonald, A.C.; Stanford, W.L. Direct reprogramming with SOX factors: Masters of cell fate. Curr. Opin. Genet. Dev. 2017, 46, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Southard-Smith, E.M.; Kos, L.; Pavan, W.J. Sox10 mutation disrupts neural crest development in Dom Hirschsprung mouse model. Nat. Genet. 1998, 18, 60–64. [Google Scholar] [CrossRef] [PubMed]
- Wegner, M.; Stolt, C.C. From stem cells to neurons and glia: A Soxist’s view of neural development. Trends Neurosci. 2005, 28, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Dutton, K.A.; Pauliny, A.; Lopes, S.S.; Elworthy, S.; Carney, T.J.; Rauch, J.; Geisler, R.; Haffter, P.; Kelsh, R.N. Zebrafish colourless encodes sox10 and specifies non-ectomesenchymal neural crest fates. Development 2001, 128, 4113–4125. [Google Scholar] [PubMed]
- Matsuoka, T.; Ahlberg, P.; Kessaris, N.; Iannarelli, P.; Dennehy, U.; Richardson, W.D.; McMahon, A.P.; Koentges, G. Neural crest origins of the neck and shoulder. Nature 2005, 436, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Bohnsack, B.L.; Gallina, D.; Kahana, A. Phenothiourea sensitizes zebrafish cranial neural crest and extraocular muscle development to changes in retinoic acid and insulin-like growth factor signaling. PLoS ONE 2011, 6, e22991. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, M.; Kamel, G.; Shubinets, V.; Hickey, G.; Grimaldi, M.; Liao, E.C. Embryonic fate map of first pharyngeal arch structures in the sox10:kaede zebrafish transgenic model. J. Craniofac. Surg. 2012, 23, 1333–1337. [Google Scholar] [CrossRef] [PubMed]
- Read, A.P.; Newton, V.E. Waardenburg syndrome. J. Med. Genet. 1997, 34, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Pingault, V.; Ente, D.; Dastot-Le Moal, F.; Goossens, M.; Marlin, S.; Bondurand, N. Review and update of mutations causing Waardenburg syndrome. Hum. Mutat. 2010, 31, 391–406. [Google Scholar] [CrossRef] [PubMed]
- Britsch, S.; Goerich, D.E.; Riethmacher, D.; Peirano, R.I.; Rossner, M.; Nave, K.A. The transcription factor Sox10 is a key regulator of peripheral glial development. Genes Dev. 2001, 15, 66–78. [Google Scholar] [CrossRef] [PubMed]
- Truch, K.; Arter, J.; Turnescu, T.; Weider, M.; Hartwig, A.C.; Tamm, E.R.; Sock, E.; Wegner, M. Analysis of the human SOX10 mutation Q377X in mice and its implications for genotype-phenotype correlation in SOX10-related human disease. Hum. Mol. Genet. 2018, 27, 1078–1092. [Google Scholar] [CrossRef]
- Teng, L.; Mundell, N.A.; Frist, A.Y.; Wang, Q.; Labosky, P.A. Requirement for Foxd3 in the maintenance of neural crest progenitors. Development 2008, 135, 1615–1624. [Google Scholar] [CrossRef]
- Mundell, N.A.; Labosky, P.A. Neural crest stem cell multipotency requires Foxd3 to maintain neural potential and repress mesenchymal fates. Development 2011, 138, 641–652. [Google Scholar] [CrossRef]
- Schunter, J.A.; Löffler, D.; Wiesner, T.; Kovacs, P.; Badenhoop, K.; Aust, G.; Tönjes, A.; Müller, P.; Baber, R.; Simon, J.C.; et al. A novel FoxD3 variant is associated with vitiligo and elevated thyroid auto-antibodies. J. Clin. Endocrinol. Metab. 2015, 100, E1335–E1342. [Google Scholar] [CrossRef]
- Lee, H.-C.; Huang, H.-Y.; Lin, C.-Y.; Chen, Y.-H.; Tsai, H.-J. Foxd3 mediates zebrafish myf5 expression during early somitogenesis. Dev. Biol. 2006, 290, 359–372. [Google Scholar] [CrossRef]
- Curran, K.; Raible, D.W.; Lister, J.A. Foxd3 controls melanophore specification in the zebrafish neual crest by regulation of Mitf. Dev. Biol. 2009, 332, 408–417. [Google Scholar] [CrossRef]
- Wang, W.D.; Melville, D.B.; Montero-Balaguer, M.; Hatzopoulos, A.K.; Knapik, E.W. Tfap2a and Foxd3 regulate early steps in the development of the neural crest progenitor population. Dev. Biol. 2011, 360, 173–185. [Google Scholar] [CrossRef]
- Patel, S.; Rauf, A.; Khan, H.; Meher, B.R.; Hassan, S.S.U. A holistic review on the autoimmune disease vitiligo with emphasis on the causal factors. Biomed. Pharmacother. 2017, 92, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Kloss, B.A.; Reis, L.M.; Brémond-Gignac, D.; Glaser, T.; Semina, E.V. Analysis of FOXD3 sequence variation in human ocular disease. Mol. Vis. 2012, 18, 1740–1749. [Google Scholar] [PubMed]
- McGonnell, I.M.; Graham, A.; Richardson, J.; Fish, J.L.; Depew, M.J.; Dee, C.T.; Holland, P.W.H.; Takahashi, T. Evolution of the Alx homeobox gene family: Parallel retention and independent loss of the vertebrate Alx3 gene. Evol. Dev. 2011, 13, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Dee, C.T.; Szymoniuk, C.R.; Mills, P.E.D.; Takahashi, T. Defective neural crest migration revealed by a zebrafish model of Alx1-related frontonasasl dysplasia. Hum. Mol. Genet. 2013, 22, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Uz, E.; Alanay, Y.; Aktas, D.; Vargel, I.; Gucer, S.; Tuncbilek, G.; von Eggeling, F.; Yilmaz, E.; Deren, O.; Posorski, N.; et al. Disruption of ALX1 causes extreme microphthalmia and severe facial clefting: Expandig the spectrum of autosomal-recessive ALX-related frontonasasl dysplasia. Am. J. Hum. Genet. 2010, 86, 789–796. [Google Scholar] [CrossRef]
- Pini, J.; Kueper, J.; Hu, Y.D.; Kawasaki, K.; Yeung, P.; Tsimbal, C.; Yoon, B.; Carmichael, N.; Maas, R.L.; Cotney, J.; et al. ALX-1 related frontonasal dysplasia results from defective neural crest cell development and migration. EMBO Mol. Med. 2020, 12, e12013. [Google Scholar] [CrossRef]
- Twigg, S.R.; Versnel, S.L.; Nürnberg, G.; Lees, M.M.; Bhat, M.; Hammond, P.; Hennekam, R.C.; Hoogeboom, A.J.; Hurst, J.A.; Johnson, D.; et al. Frontorhiny, a distinctive presentation of frontonasal dysplasia caused by recessive mutations in the ALX3 homeobox gene. Am. J. Hum. Genet. 2009, 84, 698–705. [Google Scholar] [CrossRef]
- Bertola, D.R.; Rodrigues, M.G.; Quaio, C.R.; Kim, C.A.; Passos-Bueno, M.R. Vertical transmission of a frontonasal phenotype caused by a novel ALX4 mutation. Am. J. Med. Genet. A 2013, 161A, 600–604. [Google Scholar] [CrossRef]
- Trainor, P.A.; Tam, P.P. Cranial paraxial mesoderm and neural crest of the mouse embryo-codistribution in the craniofacial mesenchyme but distinct segreation in the branchial arches. Development 1995, 229, 14–29. [Google Scholar]
- Trainor, P.A.; Krumlauf, R. Patterning the cranial neural crest: Hindbrain segmentation and Hox gene plasticity. Nat. Rev. Neurosci. 2000, 1, 116–124. [Google Scholar] [CrossRef]
- Trainor, P.A. Specification of neural crest cell formation and migration in mouse embryos. Sem. Cell Dev. Biol. 2005, 16, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Bohnsack, B.L.; Kahana, A. Thyroid hormone and retinoic acid interact to regulate zebrafish craniofacial neural crest development. Dev. Biol. 2013, 373, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; An, M.; Arduini, B.L.; Henion, P.D. Specific pan-neural crest expression of zebrafish crestin throughout embryonic development. Dev. Dyn. 2001, 220, 169–174. [Google Scholar] [CrossRef]
- Van Der Meulen, K.L.; Vöcking, O.; Weaver, M.L.; Meshram, N.N.; Famulski, J.K. Spatiotemporal characterization of anterior segment mesenchyme heterogeneity during zebrafish ocular anterior segment development. Front. Cell Dev. Biol. 2020, 8, 379. [Google Scholar] [CrossRef] [PubMed]
- Gage, P.J.; Suh, H.; Camper, S.A. Dosage requirement of Pitx2 for development of multiple organs. Development 1999, 126, 4643–4651. [Google Scholar] [PubMed]
- Bohnsack, B.L.; Kasprick, D.; Kish, P.E.; Goldman, D.; Kahana, A. A zebrafish model of Axenfeld-Rieger Syndrome reveals that pitx2 regulation by retinoic acid is essential for ocular and craniofacial development. Invest. Ophthalmol. Vis. Sci. 2012, 53, 7–22. [Google Scholar] [CrossRef]
- Hendee, K.E.; Sorokina, E.A.; Muheisen, S.S.; Reis, L.M.; Tyler, R.C.; Markovic, V.; Cuturilo, G.; Link, B.A.; Semina, E.V. PITX2 deficiency and associated human disease: Insights from the zebrafish model. Hum. Mol. Genet. 2018, 27, 1675–1695. [Google Scholar] [CrossRef]
- Evans, A.L.; Gage, P.J. Expression of the homeobox gene Pitx2 in neural crest is required for optic stalk and ocular anterior segment development. Hum. Mol. Genet. 2005, 14, 3347–3359. [Google Scholar] [CrossRef]
- Bhate, M.; Martin, F.J. Unilateral inferior rectus hypoplasia in a child with Axenfeld-Rieger syndrome. J. AAPOS 2012, 16, 304–306. [Google Scholar] [CrossRef]
- Shah, B.M.; Dada, T.; Panda, T.; Tanwar, M.; Bhartiya, S.; Dada, R. Novel occurence of Axenfeld-Rieger syndrome in a patient with blepharophimosis ptosis epicanthus inversus syndrome. Indian J. Ophthalmol. 2014, 62, 358–360. [Google Scholar]
- Semina, E.V.; Reiter, R.; Leysens, N.J.; Alward, W.L.; Small, K.W.; Datson, N.A.; Siegel-Barelt, J.; Bierke-Nelson, D.; Bitou, P.; Zabel, B.U.; et al. Cloning and characterization of a novel bicoid-related homeobox transcription factor gene, RIEG, involved in Rieger syndrome. Nat. Genet. 1996, 14, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Kulak, S.C.; Kozlowski, K.; Semina, E.V.; Pearce, W.G.; Walter, M.A. Mutation in the RIEG1 gene in patients with iridogoniodysgenesis syndrome. Hum. Mol. Genet. 1998, 7, 1113–1117. [Google Scholar] [CrossRef] [PubMed]
- Saadi, I.; Semina, E.V.; Amendt, B.A.; Harris, D.J.; Murphy, K.P.; Murray, J.C.; Russo, A.F. Identification of a dominant negative homeodomain mutation in Rieger syndrome. J. Biol. Chem. 2001, 276, 23034–23041. [Google Scholar] [CrossRef] [PubMed]
- Saadi, I.; Toro, R.; Kuburas, A.; Semina, E.V.; Murray, J.C.; Russo, A.F. An unusual class of PITX2 mutations in Axenfeld-Rieger syndrome. Birth. Defects Res. A Clin. Mol. Teratol. 2006, 76, 175–181. [Google Scholar] [CrossRef]
- Zepeda, E.M.; Branham, K.; Moroi, S.E.; Bohnsack, B.L. Surgical outcomes of glaucoma associated with Axenfeld-Rieger syndrome. BMC Ophthalmol. 2020, 20, 172. [Google Scholar] [CrossRef]
- Arikawa, A.; Yoshida, S.; Yoshikawa, H.; Ishikawa, K.; Yamaji, Y.; Arita, R.-I.; Ueno, A.; Ishibashi, T. Case of novel PITX2 gene mutation associated with Peters’ anomaly and persistent hyperplastic primary vitreous. Eye (Lond.) 2009, 24, 391–393. [Google Scholar] [CrossRef][Green Version]
- Kumar, S.; Duester, G. Retinoic acid signaling in perioptic mesenchyme represses Wnt signaling via induction of Pitx2 and Dkk2. Dev. Biol. 2010, 340, 67–74. [Google Scholar] [CrossRef]
- Kitamura, K.; Miura, H.; Miyagawa-Tomita, S.; Yanazawa, M.; Katoh-Fukui, Y.; Suzuki, R.; Ohuchi, H.; Suehiro, A.; Motegi, Y.; Nakahara, Y.; et al. Mouse Pitx2 deficiency leads to anomalies of the ventral body wall, heart, extra- and periocular mesoderm and right pulmonary isomerism. Development 1999, 126, 5749–5758. [Google Scholar]
- Asai-Coakwell, M.; Backhouse, C.; Casey, R.J.; Gage, P.J.; Lehmann, O.J. Reduced human and murine corneal thickness in an Axenfeld-Rieger Syndrome subtype. Invest. Ophthalmol. Vis. Sci. 2006, 47, 4905–4909. [Google Scholar] [CrossRef]
- Matt, N.; Ghyselinck, N.B.; Wendling, O.; Chambon, P.; Mark, M. Retinoic acid-induced developmental defects are mediated by RARbeta/RXR heterodimers in the pharyngeal endoderm. Development 2003, 130, 2083–2093. [Google Scholar] [CrossRef]
- Molotkov, A.; Molotkova, N.; Duester, G. Retinoic acid guides eye morphogenetic movements via paracrine signaling but is unnecessary for retinal dorsoventral patterning. Development 2006, 133, 1901–1910. [Google Scholar] [CrossRef] [PubMed]
- Matt, N.; Ghyselinck, N.B.; Pellerin, I.; Dupe, V. Impairing retinoic acid signaling in the neural crest cells is sufficient to alter entire eye morphogenesis. Dev. Biol. 2008, 320, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Berry, F.B.; Lines, M.A.; Ooas, J.M.; Footz, T.; Underhill, D.A.; Gage, P.J.; Walter, M.A. Functional interactions between FOXC1 and PITX2 underlie the sensitifvity to FOXC! gene dose in Axenfeld=Rieger syndrome and anterior segment dysgenesis. Hum. Mol. Genet. 2006, 15, 905–919. [Google Scholar] [CrossRef] [PubMed]
- Gage, P.J.; Qian, M.; Wu, D.; Rosenberg, K.I. The canonical Wnt signaling antagonist DKK2 is an essential effector of PITX2 function during normal eye development. Dev. Biol. 2008, 317, 310–324. [Google Scholar] [CrossRef]
- Pressman, C.L.; Chen, H.; Johnson, R.L. LMX1B, a LIM homeodomain class transcription factor, is necessary for normal development of multiple tissues in the anterior segment of the murine eye. Genesis 2000, 26, 15–25. [Google Scholar] [CrossRef]
- Vollrath, D.; Jaramillo-Babb, V.L.; Clough, M.V.; McIntosh, I.; Scott, K.M.; Lichter, P.R.; Richards, J.E. Loss-of-function mutations in the LIM-homeodomain gene, LMX1B in nail-patella syndrome. Hum. Mol. Genet. 1998, 7, 1091–1098. [Google Scholar] [CrossRef]
- McMahon, C.; Gestri, G.; Wilson, S.W.; Link, B.A. Lmx1b is essentail for survival of periocular mesenchymal cells and influences Fgf-mediated retinal patterning in zebrafish. Dev. Biol. 2009, 332, 287–298. [Google Scholar] [CrossRef]
- Chen, L.; Martino, V.; Dombkowski, A.; Williams, T.; West-Mays, J.; Gage, P.J. AP-2ß is a downstream effector of PITX2 required to specify endothelium and establish angiogenic privilege during corneal development. Invest. Ophthalmol. Vis. Sci. 2016, 57, 1072–1081. [Google Scholar] [CrossRef]
- Martino, V.B.; Sabljic, T.; Deschamps, P.; Green, R.M.; Akula, M.; Peacock, E.; Ball, A.; Williams, T.; West-Mays, J.A. Conditional deletion of AP-2ß in mouse cranial neural crest results in anterior segment dysgenesis and early-onset glaucoma. Dis. Model Mech. 2016, 9, 849–961. [Google Scholar] [CrossRef]
- Bohnsack, B.L.; Gallina, D.; Thompson, H.; Kasprick, D.; Lucarelli, M.J.; Dootz, G.; Nelson, C.; McGonnell, I.M.; Kahana, A. Development of extraocular muscles require early signals from periocular neural crest and the developing eye. Arch. Ophthalmol. 2011, 129, 1030–1041. [Google Scholar] [CrossRef]
- Diehl, A.G.; Zareparst, S.; Qian, M.; Khanna, R.; Angeles, R.; Gage, P.J. Extraocular muscle morphogenesis and gene expression are regulated by Pitx 2 gene dose. Invest. Ophthalmol. Vis. Sci. 2006, 47, 1785–1793. [Google Scholar] [CrossRef] [PubMed]
- Zacharias, A.L.; Lewandoski, M.; Rudnicki, M.A.; Gage, P.J. Pitx2 is an upstream activator of extraocular myogenesis and survival. Dev. Biol. 2011, 349, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Langenberg, T.; Kahana, A.; Wszalek, J.A.; Halloran, M.C. The Eye Organizes Neural Crest Cell Migration. Dev. Dyn. 2008, 237, 1645–16521. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.S.; Zabealeta, A.; Kume, T.; Savinova, O.V.; Kidson, S.H.; Martin, J.E.; Nishimura, D.Y.; Alward, W.L.; Hogan, B.L.; John, S.W.M. Haploinsufficiency of the transcription factors FOXC1 and FOXC2 results in aberrant ocular development. Hum. Mol. Genet. 2000, 9, 1021–1032. [Google Scholar] [CrossRef]
- French, C.R.; Seshadri, S.; Destafano, A.L.; Fornage, M.; Arnold, C.R.; Gage, P.J.; Skarie, J.M.; Dobyns, W.B.; Millen, K.J.; Liu, T.; et al. Mutation of FOXC1 and PITX2 induces cerebral small-vessel disease. J. Clin. Investig. 2014, 124, 4877–4881. [Google Scholar] [CrossRef]
- Tumer, Z.; Bach-Holm, D. Axenfeld-Rieger syndrome and spectrum of Pitx2 and Foxc1 mutations. Eur. J. Hum. Genet. 2009, 17, 1527–1539. [Google Scholar] [CrossRef]
- Saleem, R.A.; Banerjee-Basu, S.; Murphy, T.C.; Baxevanis, A.D.; Walter, M.A. Essential structural and functional determinants within the forkhead domain of FOXC1. Nucleic Acids Res. 2004, 32, 4182–4193. [Google Scholar] [CrossRef]
- David, D.; Cardoso, J.; Marques, B.; Marques, R.; Silva, E.D.; Santos, H.; Boavida, M.G. Molecular characterization of a familial translocation implicates disruption of HDAC9 and possible position effect on TGFbeta2 in the pathogenesis of Peters’ anomaly. Genomics 2003, 81, 489–503. [Google Scholar] [CrossRef]
- Iwao, K.; Inatani, M.; Matsumoto, Y.; Ogata-Iwao, M.; Takihara, Y.; Irie, F.; Yamaguchi, Y.; Okinami, S.; Tanihara, H. Heparan sulfate deficiency leads to Peters anomaly in mice by disturbing neural crest TGF-β2 signaling. J. Clin. Investig. 2009, 119, 997–1008. [Google Scholar] [CrossRef]
- Silla, Z.T.; Naidoo, J.; Kidson, S.H.; Sommer, P. Signals from the lens and Foxc1 regulate the expression of key genes during the onset of corneal endothelial development. Exp. Cell Res. 2014, 322, 381–388. [Google Scholar] [CrossRef]
- Matt, N.; Dupe, V.; Garnier, J.-M.; Dennefeld, C.; Chambon, P.; Mark, M.; Ghyselinck, N.B. Retinoic acid-dependent eye morphogenesis is orchestrated by neural crest cells. Development 2005, 132, 4789–4800. [Google Scholar] [CrossRef] [PubMed]
- Sommer, P.; Napier, H.R.; Hogan, B.L.; Kidson, S.H. Identification of Tgfbeta1i4 as a downstream target of Foxc1. Dev. Growth Differ. 2006, 48, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Lupo, G.; Gestri, G.; O’Brien, M.; Denton, R.M.; Chandraratna, R.A.S.; Ley, S.V.; Harris, W.A.; Wilson, S.W. Retinoic acid receptor signaling regulates choroid fissure closure through independent mechanisms in the ventral optic cup and periocular mesenchyme. Proc. Natl. Acad. Sci. USA 2011, 108, 8698–8703. [Google Scholar] [CrossRef]
- Tamimi, Y.; Skarie, J.M.; Footz, T.; Berry, F.B.; Link, B.A.; Walter, M.A. FGF19 is a target for FOXC1 regulation in ciliary body-derived cells. Hum. Mol. Genet. 2006, 15, 3229–3240. [Google Scholar] [CrossRef]
- Berry, F.B.; Skarie, J.M.; Mirzayans, F.; Fortin, Y.; Hudson, T.J.; Raymond, V.; Link, B.A.; Walter, M.A. FOXC1 is required for cell viability and resistance to oxidative stress in the eye through the transcriptional regulation of FOXO1A. Hum. Mol. Genet. 2008, 17, 490–505. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.; Chen, L.; Liu, W.; Zhao, D.; Schultz, K.M.; Sasman, A.; Liu, T.; Zhang, H.F.; Gage, P.J.; Kume, T. Foxc1 and Foxc2 in the neural crest are required for ocular anterior segment development. Investig. Ophthalmol. Vis. Sci. 2017, 58, 1368–1377. [Google Scholar] [CrossRef] [PubMed]
- Schimmenti, L.A.; de la Cruz, J.; Lewis, R.A.; Karkera, J.D.; Manligas, G.S.; Ressler, E.; Muenke, M. Novel mutation in sonic hedgehog in non-syndrome colobomatous microphthalmia. Am. J. Med. Genet. 2003, 116A, 215–221. [Google Scholar] [CrossRef]
- Solebo, A.L.; Teoh, L.; Rahi, J. Epidemiology of blindness in children. Arch. Dis. Child. 2017, 102, 853–857. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weigele, J.; Bohnsack, B.L. Genetics Underlying the Interactions between Neural Crest Cells and Eye Development. J. Dev. Biol. 2020, 8, 26. https://doi.org/10.3390/jdb8040026
Weigele J, Bohnsack BL. Genetics Underlying the Interactions between Neural Crest Cells and Eye Development. Journal of Developmental Biology. 2020; 8(4):26. https://doi.org/10.3390/jdb8040026
Chicago/Turabian StyleWeigele, Jochen, and Brenda L. Bohnsack. 2020. "Genetics Underlying the Interactions between Neural Crest Cells and Eye Development" Journal of Developmental Biology 8, no. 4: 26. https://doi.org/10.3390/jdb8040026
APA StyleWeigele, J., & Bohnsack, B. L. (2020). Genetics Underlying the Interactions between Neural Crest Cells and Eye Development. Journal of Developmental Biology, 8(4), 26. https://doi.org/10.3390/jdb8040026