Transcriptional Trajectories in Mouse Limb Buds Reveal the Transition from Anterior-Posterior to Proximal-Distal Patterning at Early Limb Bud Stage


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Approval
2.2. Mouse Lines
2.3. 3′End Single-Cell RNA-seq
2.4. Single-Cell RNA-seq Data Analysis
2.4.1. Pre-Processing
2.4.2. Quality Control and Filtering
2.4.3. Normalization and Scaling
2.4.4. Dimensional Reduction
2.4.5. Clustering
2.4.6. Monocle3 Analysis
2.5. Single-Cell ATAC-seq Data Analysis
2.6. Whole-Mount In Situ Hybridization
2.7. Data Availability
3. Results
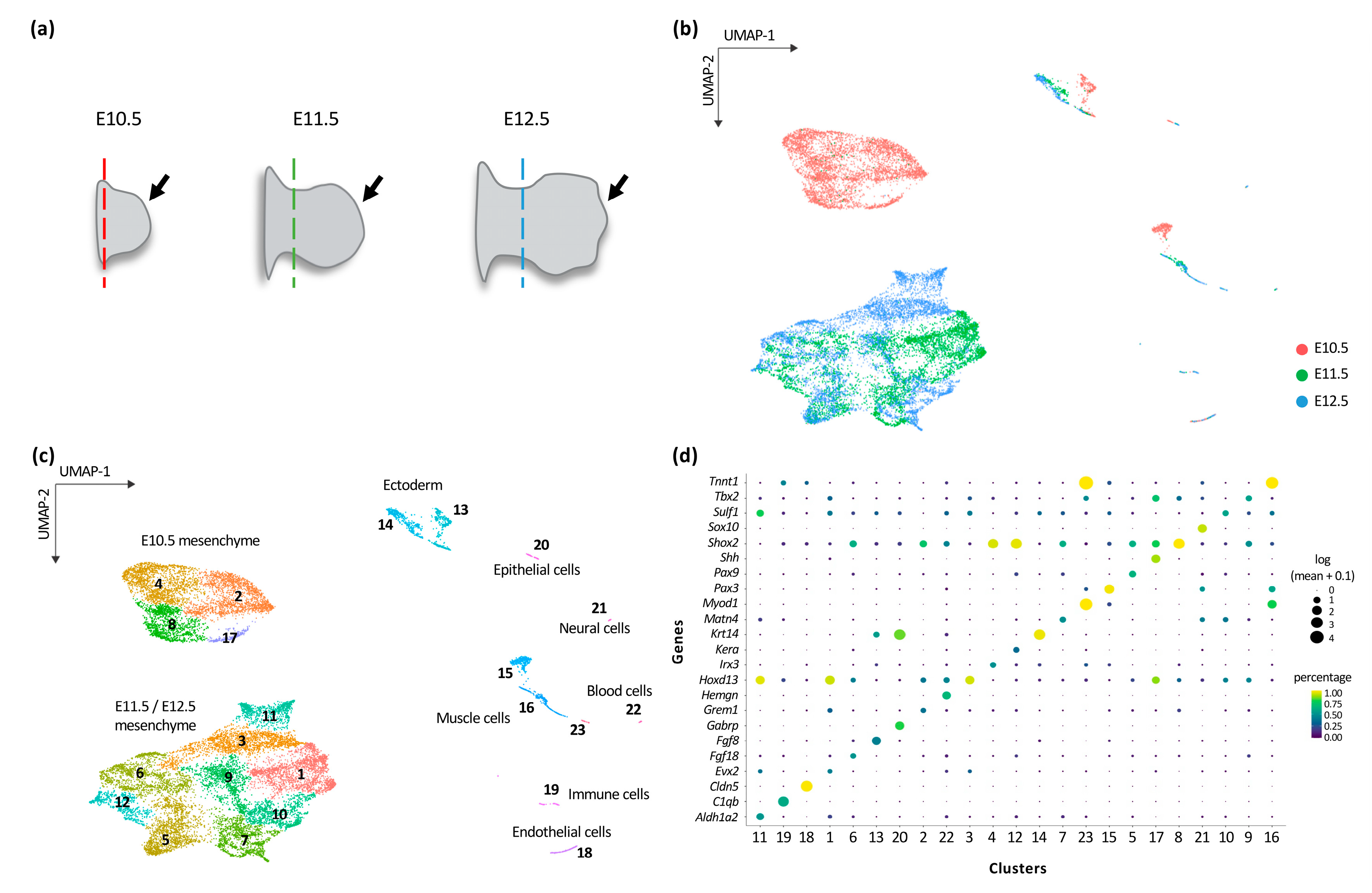
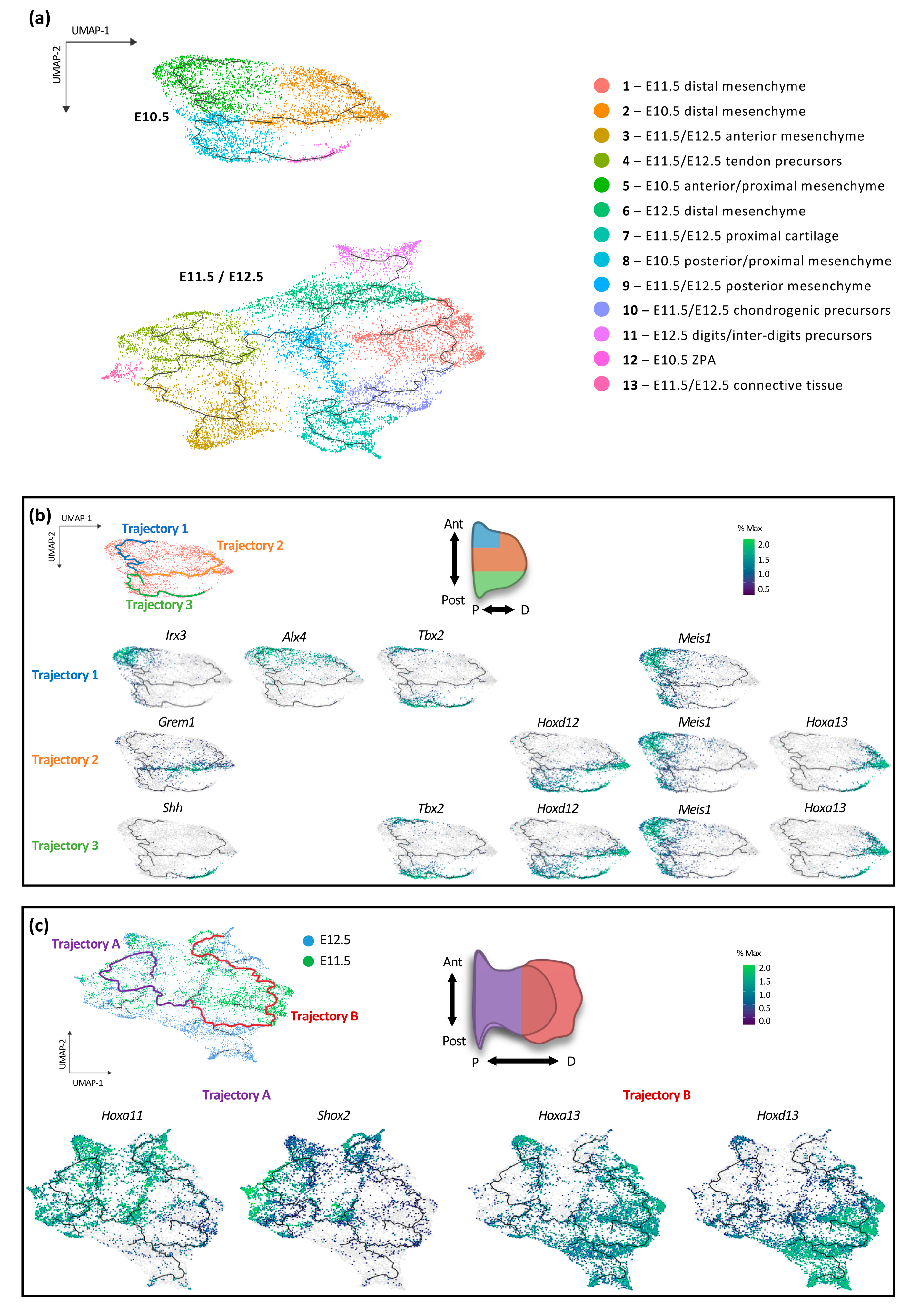
3.1. Transcriptional Trajectories in the Developing Forelimb Bud Reveal a Major Switch in the Developmental Program between E10.5 and E11.5
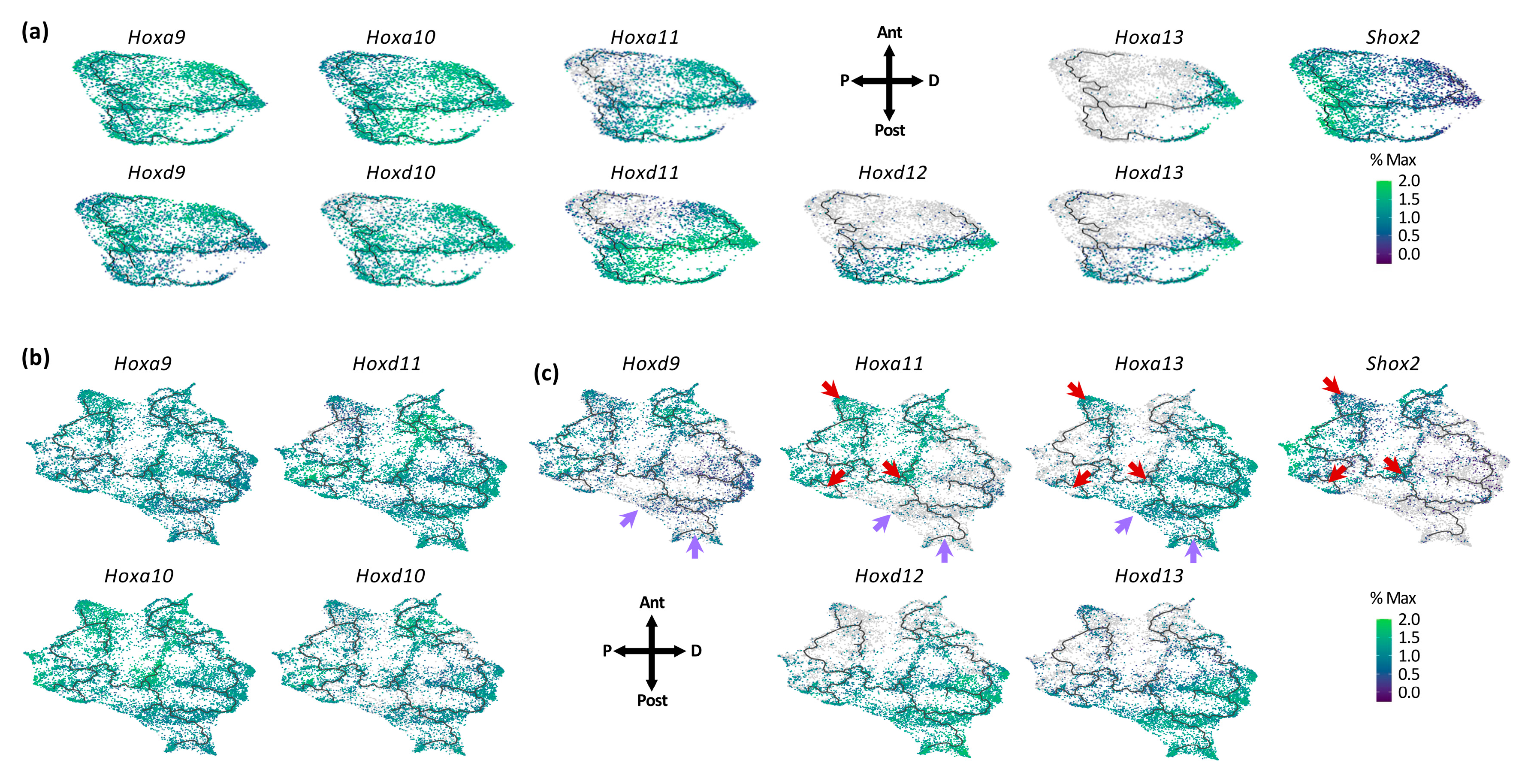
3.2. Individual Transcriptional Trajectories Are Characterized by a Specific Hox Code
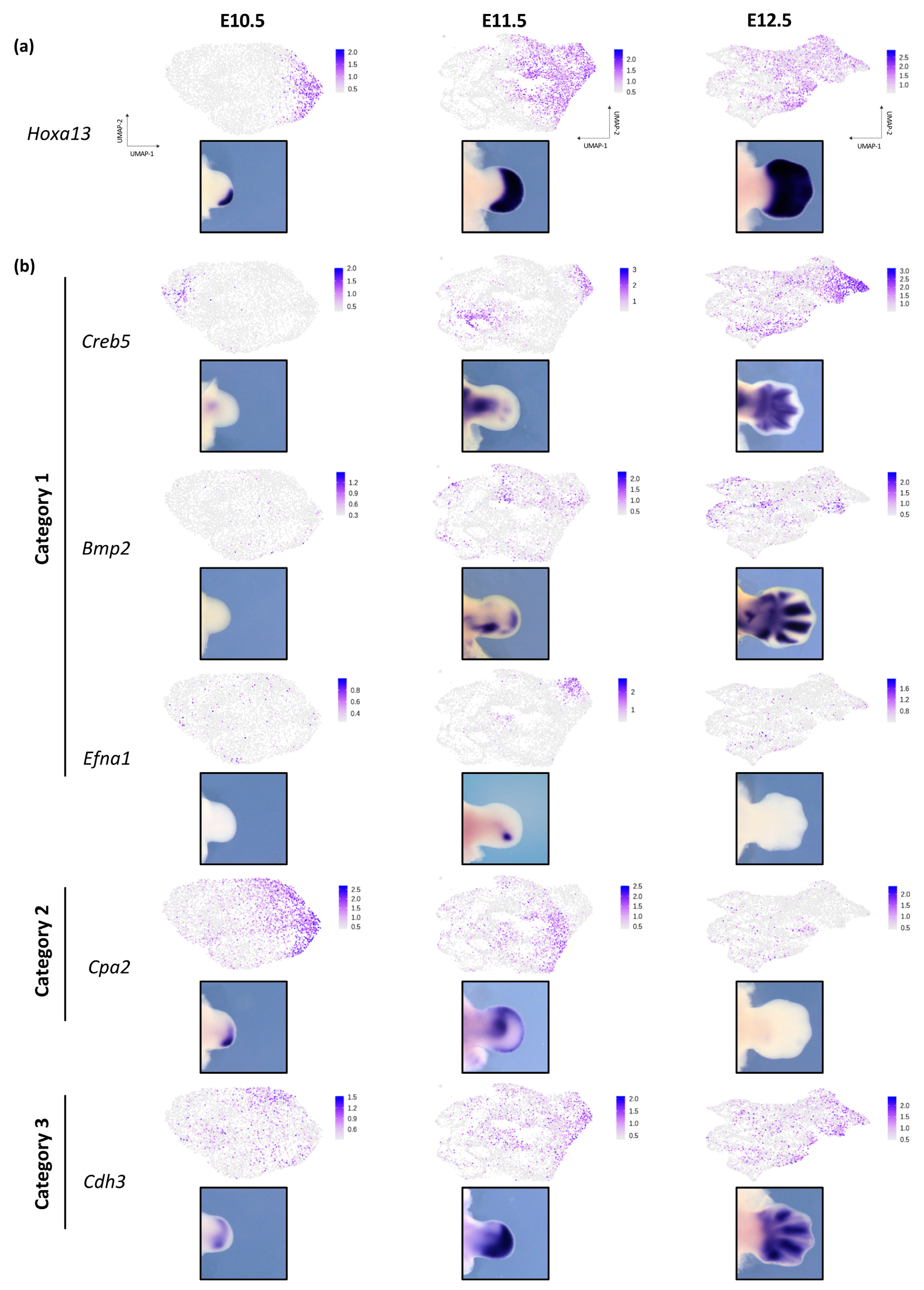
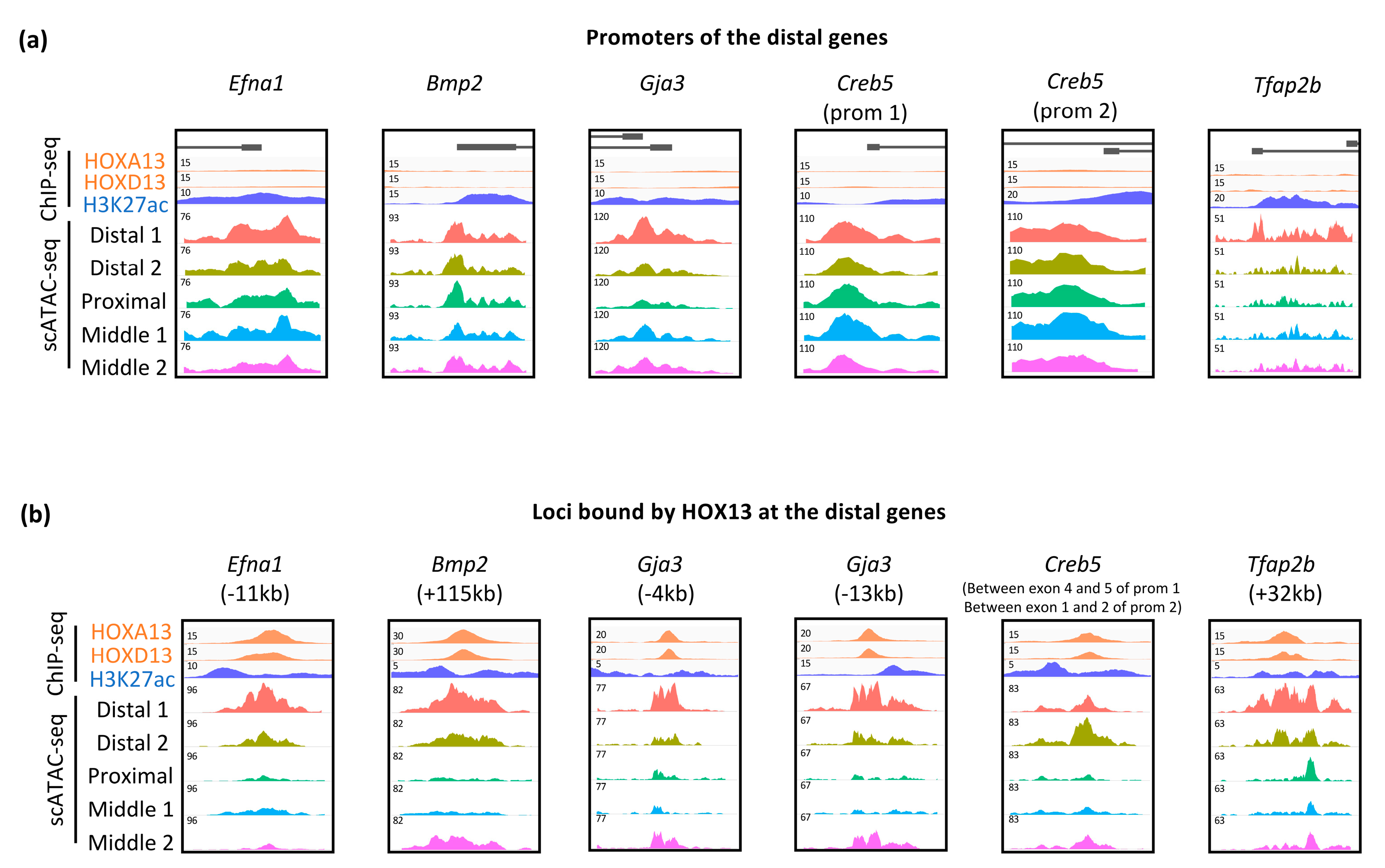
3.3. Dynamic Expression of Distal Genes Largely Relies on HOX13 Function
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lewandoski, M.; Sun, X.; Martin, G.R. Fgf8 signalling from the AER is essential for normal limb development. Nat. Genet. 2000, 26, 460–463. [Google Scholar] [CrossRef] [PubMed]
- Delgado, I.; López-Delgado, A.C.; Roselló-Díez, A.; Giovinazzo, G.; Cadenas, V.; Fernández-de-Manuel, L.; Sánchez-Cabo, F.; Anderson, M.J.; Lewandoski, M.; Torres, M. Proximo-distal positional information encoded by an Fgf-regulated gradient of homeodomain transcription factors in the vertebrate limb. Sci. Adv. 2020, 6, eaaz0742. [Google Scholar] [CrossRef]
- Panman, L.; Zeller, R. Patterning the limb before and after SHH signalling. J. Anat. 2003, 202, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Zakany, J.; Duboule, D. The role of Hox genes during vertebrate limb development. Curr. Opin. Genet. Dev. 2007, 17, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Kmita, M.; Tarchini, B.; Zàkàny, J.; Logan, M.; Tabin, C.J.; Duboule, D. Early developmental arrest of mammalian limbs lacking HoxA/HoxD gene function. Nature 2005, 435, 1113–1116. [Google Scholar] [CrossRef]
- Tarchini, B.; Duboule, D. Control of Hoxd genes’ collinearity during early limb development. Dev. Cell 2006, 10, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Berlivet, S.; Paquette, D.; Dumouchel, A.; Langlais, D.; Dostie, J.; Kmita, M. Clustering of Tissue-Specific Sub-TADs Accompanies the Regulation of HoxA Genes in Developing Limbs. PLoS Genet. 2013, 9, e1004018. [Google Scholar] [CrossRef] [PubMed]
- Lonfat, N.; Montavon, T.; Darbellay, F.; Gitto, S.; Duboule, D. Convergent evolution of complex regulatory landscapes and pleiotropy at Hox loci. Science 2014, 346, 1004–1006. [Google Scholar] [CrossRef]
- Andrey, G.; Montavon, T.; Mascrez, B.; Gonzalez, F.; Noordermeer, D.; Leleu, M.; Trono, D.; Spitz, F.; Duboule, D. A switch between topological domains underlies HoxD genes collinearity in mouse limbs. Science 2013, 340, 1234167. [Google Scholar] [CrossRef]
- Beccari, L.; Yakushiji-Kaminatsui, N.; Woltering, J.M.; Necsulea, A.; Lonfat, N.; Rodríguez-Carballo, E.; Mascrez, B.; Yamamoto, S.; Kuroiwa, A.; Duboule, D. A role for HOX13 proteins in the regulatory switch between TADs at the HoxD locus. Genes Dev. 2016, 30, 1172–1186. [Google Scholar] [CrossRef]
- Sheth, R.; Barozzi, I.; Langlais, D.; Osterwalder, M.; Nemec, S.; Carlson, H.L.; Stadler, H.S.; Visel, A.; Drouin, J.; Kmita, M. Distal Limb Patterning Requires Modulation of cis-Regulatory Activities by HOX13. Cell Rep. 2016, 17, 2913–2926. [Google Scholar] [CrossRef] [PubMed]
- Scotti, M.; Kherdjemil, Y.; Roux, M.; Kmita, M. A Hoxa13:Cre mouse strain for conditional gene manipulation in developing limb, hindgut, and urogenital system. Genesis 2015, 53, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Muzumdar, M.D.; Tasic, B.; Miyamichi, K.; Li, L.; Luo, L. A global double-fluorescent Cre reporter mouse. Genesis 2007, 45, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotech. 2018, 36, 411–420. [Google Scholar] [CrossRef]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M., III; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive Integration of Single-Cell Data. Cell 2019, 177, 1888–1902.e21. [Google Scholar] [CrossRef]
- Aztekin, C.; Hiscock, T.W.; Marioni, J.C.; Gurdon, J.B.; Simons, B.D.; Jullien, J. Identification of a regeneration-organizing cell in the Xenopus tail. Science 2019, 364, 653. [Google Scholar] [CrossRef]
- McInnes, L.; Healy, J.; Melville, J. UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. arXiv 2020, arXiv:1802.03426. Available online: https://arxiv.org/abs/1802.03426 (accessed on 13 October 2020).
- Blondel, V.D.; Guillaume, J.-L.; Lambiotte, R.; Lefebvre, E. Fast unfolding of communities in large networks. J. Stat. Mech. Theory Exp. 2008, 2008, P10008. [Google Scholar] [CrossRef]
- Lancichinetti, A.; Fortunato, S. Community detection algorithms: A comparative analysis. Phys. Rev. E 2009, 80, 056117. [Google Scholar] [CrossRef]
- Jolliffe, I.T.; Cadima, J. Principal component analysis: A review and recent developments. Phil. Trans. R. Soc. A 2016, 374, 20150202. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Prot. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Cacchiarelli, D.; Grimsby, J.; Pokharel, P.; Li, S.; Morse, M.; Lennon, N.J.; Livak, K.J.; Mikkelsen, T.S.; Rinn, J.L. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotech. 2014, 32, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Mao, Q.; Tang, Y.; Wang, L.; Chawla, R.; Pliner, H.A.; Trapnell, C. Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods 2017, 14, 979–982. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Spielmann, M.; Qiu, X.; Huang, X.; Ibrahim, D.M.; Hill, A.J.; Zhang, F.; Mundlos, S.; Christiansen, L.; Steemers, F.J.; et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature 2019, 566, 496–502. [Google Scholar] [CrossRef] [PubMed]
- Traag, V.A.; Waltman, L.; van Eck, N.J. From Louvain to Leiden: Guaranteeing well-connected communities. Sci. Rep. 2019, 9, 5233. [Google Scholar] [CrossRef]
- Desanlis, I.; Kherdjemil, Y.; Mayran, A.; Bouklouch, Y.; Gentile, C.; Sheth, R.; Zeller, R.; Drouin, J.; Kmita, M. HOX13-dependent chromatin accessibility underlies the transition towards the digit development program. Nat. Commun. 2020, 11, 2491. [Google Scholar] [CrossRef]
- Cusanovich, D.A.; Daza, R.; Adey, A.; Pliner, H.A.; Christiansen, L.; Gunderson, K.L.; Steemers, F.J.; Trapnell, C.; Shendure, J. Multiplex single-cell profiling of chromatin accessibility by combinatorial cellular indexing. Science 2015, 348, 910. [Google Scholar] [CrossRef]
- Waltman, L.; van Eck, N.J. A smart local moving algorithm for large-scale modularity-based community detection. Eur. Phys. J. B 2013, 86, 471. [Google Scholar] [CrossRef]
- Warot, X.; Fromental-Ramain, C.; Fraulob, V.; Chambon, P.; Dollé, P. Gene dosage-dependent effects of the Hoxa-13 and Hoxd-13 mutations on morphogenesis of the terminal parts of the digestive and urogenital tracts. Development 1997, 124, 4781–4791. [Google Scholar]
- Dickinson, M.E.; Kobrin, M.S.; Silan, C.M.; Kingsley, D.M.; Justice, M.J.; Miller, D.A.; Ceci, J.D.; Lock, L.F.; Lee, A.; Buchberg, A.M.; et al. Chromosomal localization of seven members of the murine TGF-β superfamily suggests close linkage to several morphogenetic mutant loci. Genomics 1990, 6, 505–520. [Google Scholar] [CrossRef]
- Li, D.; Sakuma, R.; Vakili, N.A.; Mo, R.; Puviindran, V.; Deimling, S.; Zhang, X.; Hopyan, S.; Hui, C. Formation of Proximal and Anterior Limb Skeleton Requires Early Function of Irx3 and Irx5 and Is Negatively Regulated by Shh Signaling. Dev. Cell 2014, 29, 233–240. [Google Scholar] [CrossRef]
- Michos, O.; Panman, L.; Vintersten, K.; Beier, K.; Zeller, R.; Zuniga, A. Gremlin-mediated BMP antagonism induces the epithelial-mesenchymal feedback signaling controlling metanephric kidney and limb organogenesis. Development 2004, 131, 3401–3410. [Google Scholar] [CrossRef] [PubMed]
- Riddle, R.D.; Johnson, R.L.; Laufer, E.; Tabin, C. Sonic hedgehog mediates the polarizing activity of the ZPA. Cell 1993, 75, 1401–1416. [Google Scholar] [CrossRef]
- Kuijper, S.; Feitsma, H.; Sheth, R.; Korving, J.; Reijnen, M.; Meijlink, F. Function and regulation of Alx4 in limb development: Complex genetic interactions with Gli3 and Shh. Dev. Biol 2005, 285, 533–544. [Google Scholar] [CrossRef]
- Farin, H.F.; Lüdtke, T.H.-W.; Schmidt, M.K.; Placzko, S.; Schuster-Gossler, K.; Petry, M.; Christoffels, V.M.; Kispert, A. Tbx2 terminates shh/fgf signaling in the developing mouse limb bud by direct repression of gremlin1. PLoS Genet. 2013, 9, e1003467. [Google Scholar] [CrossRef] [PubMed]
- Knezevic, V.; De Santo, R.; Schughart, K.; Huffstadt, U.; Chiang, C.; Mahon, K.A.; Mackem, S. Hoxd-12 differentially affects preaxial and postaxial chondrogenic branches in the limb and regulates Sonic hedgehog in a positive feedback loop. Development 1997, 124, 4523. [Google Scholar]
- Mercader, N.; Selleri, L.; Criado, L.M.; Pallares, P.; Parras, C.; Cleary, M.L.; Torres, M. Ectopic Meis1 expression in the mouse limb bud alters P-D patterning in a Pbx1-independent manner. Int J. Dev. Biol. 2009, 53, 1483–1494. [Google Scholar] [CrossRef]
- Haack, H.; Gruss, P. The Establishment of Murine Hox-1 Expression Domains during Patterning of the Limb. Dev. Biol. 1993, 157, 410–422. [Google Scholar] [CrossRef]
- Lu, P.; Yu, Y.; Perdue, Y.; Werb, Z. The apical ectodermal ridge is a timer for generating distal limb progenitors. Development 2008, 135, 1395. [Google Scholar] [CrossRef]
- Cobb, J.; Dierich, A.; Huss-Garcia, Y.; Duboule, D. A mouse model for human short-stature syndromes identifies Shox2 as an upstream regulator of Runx2 during long-bone development. Proc. Natl. Acad. Sci. USA 2006, 103, 4511. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.P.; Witte, D.P.; Hsieh-Li, H.M.; Potter, S.S.; Capecchi, M.R. Absence of radius and ulna in mice lacking hoxa-11 and hoxd-11. Nature 1995, 375, 791–795. [Google Scholar] [CrossRef] [PubMed]
- Kherdjemil, Y.; Lalonde, R.L.; Sheth, R.; Dumouchel, A.; de Martino, G.; Pineault, K.M.; Wellik, D.M.; Stadler, H.S.; Akimenko, M.-A.; Kmita, M. Evolution of Hoxa11 regulation in vertebrates is linked to the pentadactyl state. Nature 2016, 539, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Woltering, J.M.; Noordermeer, D.; Leleu, M.; Duboule, D. Conservation and Divergence of Regulatory Strategies at Hox Loci and the Origin of Tetrapod Digits. PLoS Biol. 2014, 12, e1001773. [Google Scholar] [CrossRef]
- Sheth, R.; Bastida, M.F.; Kmita, M.; Ros, M. “Self-regulation,” a new facet of Hox genes’ function. Dev. Dyn. 2014, 243, 182–191. [Google Scholar] [CrossRef]
- Mayan, M.D.; Carpintero-Fernandez, P.; Gago-Fuentes, R.; Martinez-de-Ilarduya, O.; Wang, H.-Z.; Valiunas, V.; Brink, P.; Blanco, F.J. Human articular chondrocytes express multiple gap junction proteins: Differential expression of connexins in normal and osteoarthritic cartilage. Am. J. Pathol 2013, 182, 1337–1346. [Google Scholar] [CrossRef]
- Zhao, F.; Bosserhoff, A.-K.; Buettner, R.; Moser, M. A Heart-Hand Syndrome Gene: Tfap2b Plays a Critical Role in the Development and Remodeling of Mouse Ductus Arteriosus and Limb Patterning. PLoS ONE 2011, 6, e22908. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Desanlis, I.; Paul, R.; Kmita, M. Transcriptional Trajectories in Mouse Limb Buds Reveal the Transition from Anterior-Posterior to Proximal-Distal Patterning at Early Limb Bud Stage. J. Dev. Biol. 2020, 8, 31. https://doi.org/10.3390/jdb8040031
Desanlis I, Paul R, Kmita M. Transcriptional Trajectories in Mouse Limb Buds Reveal the Transition from Anterior-Posterior to Proximal-Distal Patterning at Early Limb Bud Stage. Journal of Developmental Biology. 2020; 8(4):31. https://doi.org/10.3390/jdb8040031
Chicago/Turabian StyleDesanlis, Ines, Rachel Paul, and Marie Kmita. 2020. "Transcriptional Trajectories in Mouse Limb Buds Reveal the Transition from Anterior-Posterior to Proximal-Distal Patterning at Early Limb Bud Stage" Journal of Developmental Biology 8, no. 4: 31. https://doi.org/10.3390/jdb8040031
APA StyleDesanlis, I., Paul, R., & Kmita, M. (2020). Transcriptional Trajectories in Mouse Limb Buds Reveal the Transition from Anterior-Posterior to Proximal-Distal Patterning at Early Limb Bud Stage. Journal of Developmental Biology, 8(4), 31. https://doi.org/10.3390/jdb8040031