Perspective: Controlling Epidermal Terminal Differentiation with Transcriptional Bursting and RNA Bodies


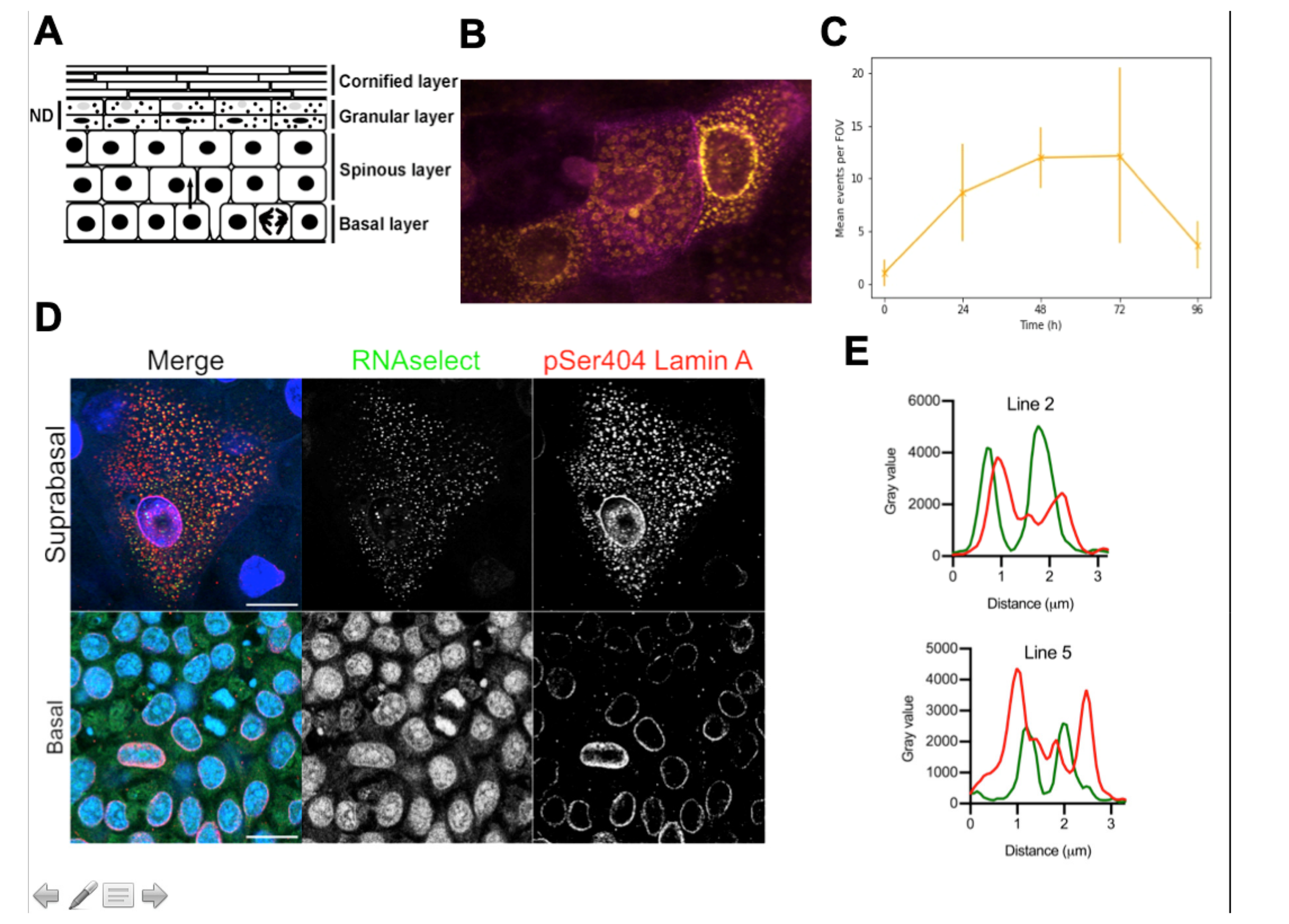{kind=link}
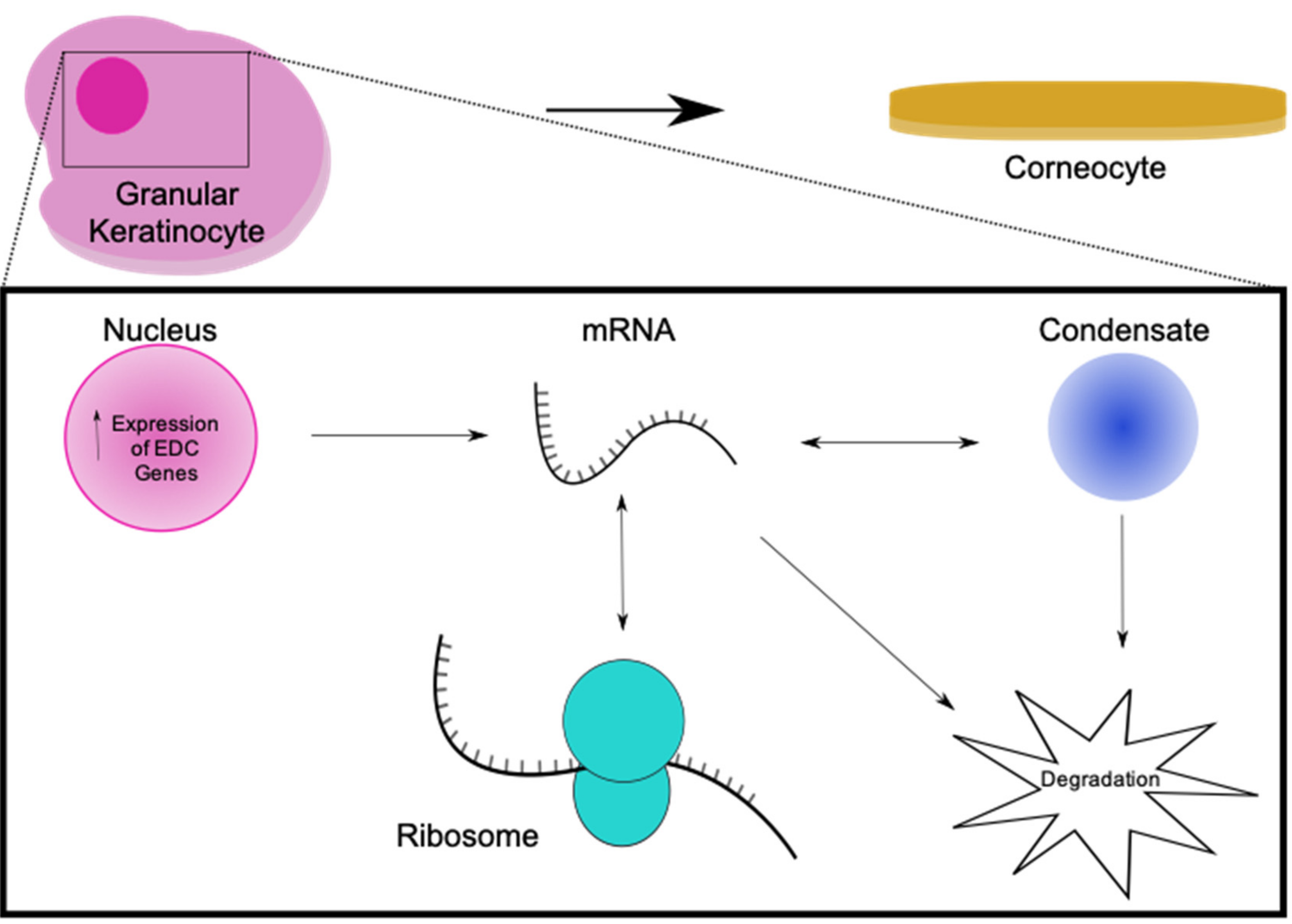{kind=link}
Abstract
1. Epidermal Terminal Differentiation Requires Increased Transcription during a Process of Nuclear Degradation
2. What Is Transcriptional Bursting—A Directed Process or just Noise?
3. Models of Bursting Behaviour and Their Relevance to Epidermal Differentiation
4. Modulators of Bursting Behaviour and Their Links to Epidermal Differentiation
5. Stochastic Transcription in Mammalian Cells
6. Biomolecular Condensates and Their Potential Roles in the Epidermis
7. Are Condensates and Transcription Linked?
8. Evidence for Transcriptional Bursting and Ribonuclear Protein Granules in Epidermal Terminal Differentiation
Author Contributions
Funding
Conflicts of Interest
References
- Candi, E.; Schmidt, R.; Melino, G. The cornified envelope: A model of cell death in the skin. Nat. Rev. Mol. Cell Biol. 2005, 6, 328–340. [Google Scholar] [CrossRef] [PubMed]
- Kypriotou, M.; Huber, M.; Hohl, D. The human epidermal differentiation complex: Cornified envelope precursors, S100 proteins and the ‘fused genes’ family. Exp. Dermatol. 2012, 21, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zieman, A.; Coulombe, P.A. Skin Keratins. Methods Enzym. 2016, 568, 303–350. [Google Scholar]
- Naeem, A.; Zhu, Y.; Di, W.L.; Marmiroli, S.; O’Shaughnessy, R.F.L. AKT1-mediated Lamin A/C degradation is required for nuclear degradation and normal epidermal terminal differentiation. Cell Death Differ. 2015, 22, 2123–2132. [Google Scholar] [CrossRef] [PubMed]
- Rogerson, C.; Wotherspoon, D.; O’Shaughnessy, R.F.L. BioRxiv preprint Akt1-associated actomyosin remodelling is required for nuclear lamina dispersal and nuclear shrinkage in epidermal terminal differentiation. bioRxiv 2020, 32, 1–32. [Google Scholar]
- Aho, S.; Harding, C.R.; Lee, J.-M.; Meldrum, H.; Bosko, C.A. Regulatory Role for the Profilaggrin N-Terminal Domain in Epidermal Homeostasis. J. Investig. Dermatol. 2012, 132, 2376–2385. [Google Scholar] [CrossRef] [PubMed]
- Elias, M.S.; Wright, S.C.; Nicholson, W.V.; Morrison, K.D.; Prescott, A.R.; Have, S.T.; Whitfield, P.D.; Lamond, A.I.; Brown, S.J. Functional and proteomic analysis of a full thickness filaggrin-deficient skin organoid model. Wellcome Open Res. 2019, 4, 134. [Google Scholar] [CrossRef]
- Bertrand, E.; Chartrand, P.; Schaefer, M.; Shenoy, S.M.; Singer, R.H.; Long, R.M. Localization of ASH1 mRNA Particles in Living Yeast. Mol. Cell 1998, 2, 437–445. [Google Scholar] [CrossRef]
- Femino, A.M. Visualization of Single RNA Transcripts in Situ. Science 1998, 280, 585–590. [Google Scholar] [CrossRef]
- Chubb, J.R.; Trcek, T.; Shenoy, S.M.; Singer, R.H. Transcriptional Pulsing of a Developmental Gene. Curr. Biol. 2006, 16, 1018–1025. [Google Scholar] [CrossRef]
- Golding, I.; Paulsson, J.; Zawilski, S.M.; Cox, E.C. Real-Time Kinetics of Gene Activity in Individual Bacteria. Cell 2005, 123, 1025–1036. [Google Scholar] [CrossRef] [PubMed]
- Raj, A.; Peskin, C.S.; Tranchina, D.; Vargas, D.Y.; Tyagi, S. Stochastic mRNA Synthesis in Mammalian Cells. PLoS Biol. 2006, 4, e309. [Google Scholar] [CrossRef] [PubMed]
- Elowitz, M.B.; Levine, A.J.; Siggia, E.D.; Swain, P.S. Stochastic Gene Expression in a Single Cell. Science 2002, 297, 1183–1186. [Google Scholar] [CrossRef] [PubMed]
- Paulsson, J. Summing up the noise in gene networks. Nat. Cell Biol. 2004, 427, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Darzacq, X.; Singer, R.H. The Dynamic Range of Transcription. Mol. Cell 2008, 30, 545–546. [Google Scholar] [CrossRef]
- Dieci, G.; Sentenac, A. Detours and shortcuts to transcription reinitiation. Trends Biochem. Sci. 2003, 28, 202–209. [Google Scholar] [CrossRef]
- Li, B.; Carey, M.; Workman, J.L. The Role of Chromatin during Transcription. Cell 2007, 128, 707–719. [Google Scholar] [CrossRef]
- Saunders, A.; Core, L.J.; Lis, J.T. Breaking barriers to transcription elongation. Nat. Rev. Mol. Cell Biol. 2006, 7, 557–567. [Google Scholar] [CrossRef]
- Struhl, K. Chromatin Structure and RNA Polymerase II Connection: Implications for Transcription. Cell 1996, 84, 179–182. [Google Scholar] [CrossRef]
- Thomas, M.C.; Chiang, C.-M. The General Transcription Machinery and General Cofactors. Crit. Rev. Biochem. Mol. Biol. 2006, 41, 105–178. [Google Scholar] [CrossRef]
- Kaern, M.; Elston, T.C.; Blake, W.J.; Collins, J.J. Stochasticity in gene expression: From theories to phenotypes. Nat. Rev. Genet. 2005, 6, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Kurakin, A. Self-organization vs Watchmaker: Stochastic gene expression and cell differentiation. Dev. Genes Evol. 2004, 215, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Maamar, H.; Raj, A.; Dubnau, D. Noise in Gene Expression Determines Cell Fate in Bacillus subtilis. Science 2007, 317, 526–529. [Google Scholar] [CrossRef] [PubMed]
- Maamar, H.; Dubnau, D. Bistability inthe Bacillus subtilisK-state (competence) system requires a positive feedback loop. Mol. Microbiol. 2005, 56, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Boettiger, A.N.; Levine, M. Synchronous and Stochastic Patterns of Gene Activation in the Drosophila Embryo. Science 2009, 325, 471–473. [Google Scholar] [CrossRef] [PubMed]
- Wernet, M.F.; Mazzoni, E.O.; Çelik, A.; Duncan, D.M.; Duncan, I.; Desplan, C. Stochastic spineless expression creates the retinal mosaic for colour vision. Nat. Cell Biol. 2006, 440, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Balázsi, G.; Van Oudenaarden, A.; Collins, J.J. Cellular Decision Making and Biological Noise: From Microbes to Mammals. Cell 2011, 144, 910–925. [Google Scholar] [CrossRef]
- Abranches, E.; Guedes, A.M.V.; Moravec, M.; Maamar, H.; Svoboda, P.; Raj, A.; Henrique, D. Stochastic NANOG fluctuations allow mouse embryonic stem cells to explore pluripotency. Development 2014, 141, 2770–2779. [Google Scholar] [CrossRef]
- Enver, T.; Pera, M.; Peterson, C.; Andrews, P.W. Stem Cell States, Fates, and the Rules of Attraction. Cell Stem Cell 2009, 4, 387–397. [Google Scholar] [CrossRef]
- Losick, R.; Desplan, C. Stochasticity and Cell Fate. Science 2008, 320, 65–68. [Google Scholar] [CrossRef]
- Mohammed, H.; Hernando-Herraez, I.; Savino, A.; Scialdone, A.; Macaulay, I.; Mulas, C.; Chandra, T.; Voet, T.; Dean, W.; Nichols, J.; et al. Single-Cell Landscape of Transcriptional Heterogeneity and Cell Fate Decisions during Mouse Early Gastrulation. Cell Rep. 2017, 20, 1215–1228. [Google Scholar] [CrossRef] [PubMed]
- Olsson, A.; Venkatasubramanian, M.; Chaudhri, V.K.; Aronow, B.J.; Salomonis, N.; Singh, H.; Leighton Grimeset, H. Single-cell analysis of mixed-lineage states leading to a binary cell fate choice. Nature 2016, 537, 698–702. [Google Scholar] [CrossRef] [PubMed]
- Raj, A.; Van Oudenaarden, A. Nature, Nurture, or Chance: Stochastic Gene Expression and Its Consequences. Cell 2008, 135, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Cacchiarelli, D.; Grimsby, J.; Pokharel, P.; Li, S.; Morse, M.A.; Lennon, N.J.; Livak, K.J.; Mikkelsen, T.; Rinn, J.L. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 2014, 32, 381–386. [Google Scholar] [CrossRef]
- Marinov, G.K.; Williams, B.A.; McCue, K.; Schroth, G.P.; Gertz, J.; Myers, R.M.; Wold, B. From single-cell to cell-pool transcriptomes: Stochasticity in gene expression and RNA splicing. Genome Res. 2014, 24, 496–510. [Google Scholar] [CrossRef]
- Peccoud, J.; Ycart, B. Markovian Modeling of Gene-Product Synthesis. Theor. Popul. Biol. 1995, 48, 222–234. [Google Scholar] [CrossRef]
- Armstrong, R.; Wen, W.; Meinkoth, J.; Taylor, S.; Montminy, M. A refractory phase in cyclic AMP-responsive transcription requires down regulation of protein kinase A. Mol. Cell. Biol. 1995, 15, 1826–1832. [Google Scholar] [CrossRef]
- Harper, C.V.; Finkenstädt, B.; Woodcock, D.J.; Friedrichsen, S.; Semprini, S.; Ashall, L.; Spiller, D.G.; Mullins, J.J.; Rand, D.A.; Davis, J.R.E.; et al. Dynamic Analysis of Stochastic Transcription Cycles. PLoS Biol. 2011, 9, e1000607. [Google Scholar] [CrossRef]
- Suter, D.M.; Molina, N.; Gatfield, D.; Schneider, K.; Schibler, U.; Naef, F. Mammalian Genes Are Transcribed with Widely Different Bursting Kinetics. Science 2011, 332, 472–474. [Google Scholar] [CrossRef]
- Tunnacliffe, E.; Chubb, J.R. What Is a Transcriptional Burst? Trends Genet. 2020, 36, 288–297. [Google Scholar] [CrossRef]
- Urban, E.A.; Robert, J.J., Jr. Buffering and Amplifying Transcriptional Noise during Cell Fate Specification. Front. Genet. 2018, 9, 591. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Hu, Z. Sensory Epithelial Cells Acquire Features of Prosensory Cells via Epithelial to Mesenchymal Transition. Stem Cells Dev. 2011, 21, 1812–1821. [Google Scholar] [CrossRef] [PubMed]
- Caveney, P.M.; Norred, S.E.; Chin, C.W.; Boreyko, J.B.; Razooky, B.S.; Retterer, S.T.; Collier, C.P.; Simpson, M.L. Resource Sharing Controls Gene Expression Bursting. ACS Synth. Biol. 2016, 6, 334–343. [Google Scholar] [CrossRef]
- Bartman, C.R.; Hsu, S.C.; Hsiung, C.C.-S.; Raj, A.; Blobel, G.A. Enhancer Regulation of Transcriptional Bursting Parameters Revealed by Forced Chromatin Looping. Mol. Cell 2016, 62, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Limi, S.; Senecal, A.; Coleman, R.; Lopez-Jones, M.; Guo, P.; Polumbo, C.; Singer, R.H.; Skoultchi, A.I.; Cvekl, A. Transcriptional burst fraction and size dynamics during lens fiber cell differentiation and detailed insights into the denucleation process. J. Biol. Chem. 2018, 293, 13176–13190. [Google Scholar] [CrossRef]
- Corrigan, A.M.; Tunnacliffe, E.; Cannon, D.; Chubb, J.R. A continuum model of transcriptional bursting. eLife 2016, 5, e13051. [Google Scholar] [CrossRef]
- Itzkovitz, S.S.; Van Oudenaarden, A. Validating transcripts with probes and imaging technology. Nat. Methods 2011, 8, S12–S19. [Google Scholar] [CrossRef]
- Itzkovitz, S.S.; Lyubimova, A.; Blat, I.C.; Maynard, M.; Van Es, J.; Lees, J.A.; Jacks, T.E.; Clevers, H.; Van Oudenaarden, A. Single-molecule transcript counting of stem-cell markers in the mouse intestine. Nat. Cell Biol. 2012, 14, 106–114. [Google Scholar] [CrossRef]
- Padovan-Merhar, O.; Nair, G.P.; Biaesch, A.G.; Mayer, A.; Scarfone, S.; Foley, S.W.; Wu, A.R.; Churchman, L.S.; Singh, A.; Raj, A. Single Mammalian Cells Compensate for Differences in Cellular Volume and DNA Copy Number through Independent Global Transcriptional Mechanisms. Mol. Cell 2015, 58, 339–352. [Google Scholar] [CrossRef]
- Finnegan, A.; Cho, R.J.; Luu, A.; Harirchian, P.; Lee, J.; Cheng, J.B.; Song, J.S. Single-Cell Transcriptomics Reveals Spatial and Temporal Turnover of Keratinocyte Differentiation Regulators. Front. Genet. 2019, 10. [Google Scholar] [CrossRef]
- Joost, S.; Zeisel, A.; Jacob, T.; Sun, X.; La Manno, G.; Lönnerberg, P.; Linnarsson, S.; Kasper, M. Single-Cell Transcriptomics Reveals that Differentiation and Spatial Signatures Shape Epidermal and Hair Follicle Heterogeneity. Cell Syst. 2016, 3, 221–237. [Google Scholar] [CrossRef] [PubMed]
- Tunnacliffe, E.; Corrigan, A.M.; Chubb, J.R. Promoter-mediated diversification of transcriptional bursting dynamics following gene duplication. Proc. Natl. Acad. Sci. USA 2018, 115, 8364–8369. [Google Scholar] [CrossRef] [PubMed]
- Fukaya, T.; Lim, B.; Levine, M.S. Enhancer Control of Transcriptional Bursting. Cell 2016, 166, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Hnisz, D.; Shrinivas, K.; Young, R.A.; Chakraborty, A.K.; Sharp, P.A. A Phase Separation Model for Transcriptional Control. Cell 2017, 169, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Larsson, A.J.M.; Johnsson, P.; Hagemann-Jensen, M.; Hartmanis, L.; Faridani, O.R.; Reinius, B.; Segerstolpe, Å.; Rivera, C.M.; Ren, B.; Sandberg, R. Genomic encoding of transcriptional burst kinetics. Nat. Cell Biol. 2019, 565, 251–254. [Google Scholar] [CrossRef] [PubMed]
- Hendy, O.; Campbell, J.; Weissman, J.D.; Larson, D.R.; Singer, D.S. Differential context-specific impact of individual core promoter elements on transcriptional dynamics. Mol. Biol. Cell 2017, 28, 3360–3370. [Google Scholar] [CrossRef] [PubMed]
- Larson, D.R.; Fritzsch, C.; Sun, L.; Meng, X.; Lawrence, D.S.; Singer, R.H. Direct observation of frequency modulated transcription in single cells using light activation. eLife 2013, 2. [Google Scholar] [CrossRef]
- Dar, R.D.; Razooky, B.S.; Singh, A.; Trimeloni, T.V.; Mccollum, J.M.; Cox, C.D.; Simpson, M.L.; Weinberger, L.S. Transcriptional burst frequency and burst size are equally modulated across the human genome. Proc. Natl. Acad. Sci. USA 2012, 109, 17454–17459. [Google Scholar] [CrossRef]
- Nicolas, D.; Zoller, B.; Suter, D.M.; Naef, F. Modulation of transcriptional burst frequency by histone acetylation. Proc. Natl. Acad. Sci. USA 2018, 115, 7153–7158. [Google Scholar] [CrossRef]
- Chen, L.-F.; Lin, Y.T.; Gallegos, D.A.; Hazlett, M.F.; Gómez-Schiavon, M.; Yang, M.G.; Kalmeta, B.; Zhou, A.S.; Holtzman, L.; Gersbach, C.A.; et al. Enhancer Histone Acetylation Modulates Transcriptional Bursting Dynamics of Neuronal Activity-Inducible Genes. Cell Rep. 2019, 26, 1174–1188. [Google Scholar] [CrossRef]
- Künzig, F.W.H.; Baptista, M.A.P.; Krammer, T.; Hennig, T.; Lange, M.; Arampatzi, P.; Jürges, C.S.; Theis, F.J.; Saliba, A.-E.; Dölken, L. scSLAM-seq reveals core features of transcription dynamics in single cells. Nat. Cell Biol. 2019, 571, 419–423. [Google Scholar] [CrossRef]
- Brouwer, I.; Lenstra, T.L. Visualizing transcription: Key to understanding gene expression dynamics. Curr. Opin. Chem. Biol. 2019, 51, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Zoller, B.; Little, S.C.; Gregor, T. Diverse Spatial Expression Patterns Emerge from Unified Kinetics of Transcriptional Bursting. Cell 2018, 175, 835–847. [Google Scholar] [CrossRef]
- Li, C.; Cesbron, F.; Oehler, M.; Brunner, M.; Höfer, T. Frequency Modulation of Transcriptional Bursting Enables Sensitive and Rapid Gene Regulation. Cell Syst. 2018, 6, 409–423. [Google Scholar] [CrossRef] [PubMed]
- Stavreva, D.A.; Garcia, D.A.; Fettweis, G.; Gudla, P.R.; Zaki, G.F.; Soni, V.; McGowan, A.; Williams, G.; Huynh, A.; Palangat, M.; et al. Transcriptional Bursting and Co-bursting Regulation by Steroid Hormone Release Pattern and Transcription Factor Mobility. Mol. Cell 2019, 75, 1161–1177. [Google Scholar] [CrossRef]
- Ochiai, H.; Sugawara, T.; Sakuma, T.; Yamamoto, T. Stochastic promoter activation affects Nanog expression variability in mouse embryonic stem cells. Sci. Rep. 2015, 4, 7125. [Google Scholar] [CrossRef]
- Kalo, A.; Kanter, I.; Shraga, A.; Sheinberger, J.; Tzemach, H.; Kinor, N.; Singer, R.H.; Lionnet, T.; Shav-Talet, Y. Cellular levels of signaling factors are sensed by β-actin alleles to modulate transcriptional pulse intensity. Cell Rep. 2015, 11, 419–432. [Google Scholar] [CrossRef]
- Senecal, A.; Munsky, B.; Proux, F.; Ly, N.; Braye, F.E.; Zimmer, C.; Mueller, F.; Darzacq, X. Transcription Factors Modulate c-Fos Transcriptional Bursts. Cell Rep. 2014, 8, 75–83. [Google Scholar] [CrossRef]
- Corrigan, A.M.; Chubb, J.R. Regulation of transcriptional bursting by a naturally oscillating signal. Curr. Biol. 2014, 24, 205–211. [Google Scholar] [CrossRef]
- Molina, N.; Suter, D.M.; Cannavo, R.; Zoller, B.; Gotic, I.; Naef, F. Stimulus-induced modulation of transcriptional bursting in a single mammalian gene. Proc. Natl. Acad. Sci. USA 2013, 110, 20563–20568. [Google Scholar] [CrossRef]
- Muramoto, T.; Müller, I.; Thomas, G.; Melvin, A.; Chubb, J.R. Methylation of H3K4 Is Required for Inheritance of Active Transcriptional States. Curr. Biol. 2010, 20, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Anna, M.; Mara, M.; Artem, S.; Eleonora, C.; Gerry, M.; Lello, Z. Multi-omics profiling of calcium-induced human keratinocytes differentiation reveals modulation of unfolded protein response signaling pathways. Cell Cycle 2019, 18, 2124–2140. [Google Scholar]
- Bikle, D.D.; Xie, Z.; Tu, C.-L. Calcium regulation of keratinocyte differentiation. Expert Rev. Endocrinol. Metab. 2012, 7, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Symmons, O.; Chang, M.; Mellis, I.A.; Kalish, J.M.; Park, J.; Susztak, K.; Bartolomei, M.S.; Raj, A. Allele-specific RNA imaging shows that allelic imbalances can arise in tissues through transcriptional bursting. PLoS Genet. 2019, 15, e1007874. [Google Scholar] [CrossRef]
- Das, S.; Moon, H.C.; Singer, R.H.; Park, H.Y. A transgenic mouse for imaging activity-dependent dynamics of endogenous Arc mRNA in live neurons. Sci. Adv. 2018, 4, eaar3448. [Google Scholar] [CrossRef]
- Wennekamp, S.; Hiiragi, T. Stochastic processes in the development of pluripotency in vivo. Biotechnol. J. 2012, 7, 737–744. [Google Scholar] [CrossRef]
- Richard, A.; Boullu, L.; Herbach, U.; Bonnafoux, A.; Morin, V.; Vallin, E.; Guillemin, A.; Gao, N.P.; Gunawan, R.; Cosette, J.; et al. Single-Cell-Based Analysis Highlights a Surge in Cell-to-Cell Molecular Variability Preceding Irreversible Commitment in a Differentiation Process. PLoS Biol. 2016, 14, e1002585. [Google Scholar] [CrossRef]
- Meredith, M.M.; Zemmour, D.; Mathis, D.; Benoist, C. Aire controls gene expression in the thymic epithelium with ordered stochasticity. Nat. Immunol. 2015, 16, 942–949. [Google Scholar] [CrossRef]
- Limi, S.; Zhao, Y.; Guo, P.; Lopez-Jones, M.; Zheng, D.; Singer, R.H.; Skoultchi, A.I.; Cvekl, A. Bidirectional Analysis of Cryba4-Crybb1 Nascent Transcription and Nuclear Accumulation of Crybb3 mRNAs in Lens Fibers. Investig. Opthalmology Vis. Sci. 2019, 60, 234–244. [Google Scholar] [CrossRef]
- Rogerson, C.; Bergamaschi, D.; O’Shaughnessy, R.F.L. Uncovering mechanisms of nuclear degradation in keratinocytes: A paradigm for nuclear degradation in other tissues. Nucleus 2018, 9, 56–64. [Google Scholar] [CrossRef]
- Foreman, R.; Wollman, R. Mammalian gene expression variability is explained by underlying cell state. Mol. Syst. Biol. 2020, 16, e9146. [Google Scholar] [CrossRef] [PubMed]
- Dacheux, E.; Malys, N.; Meng, X.; Ramachandran, V.; Mendes, P.; McCarthy, J.E.G. Translation initiation events on structured eukaryotic mRNAs generate gene expression noise. Nucleic Acids Res. 2017, 45, 6981–6992. [Google Scholar] [CrossRef] [PubMed]
- Battich, N.; Stoeger, T.; Pelkmans, L. Control of Transcript Variability in Single Mammalian Cells. Cell 2015, 163, 1596–1610. [Google Scholar] [CrossRef] [PubMed]
- Janes, K.A. Cell-to-Cell Transcript Variability: Seeing Signal in the Noise. Cell 2015, 163, 1566–1568. [Google Scholar] [CrossRef]
- Wu, B.; Eliscovich, C.; Yoon, Y.J.; Singer, R.H. Translation dynamics of single mRNAs in live cells and neurons. Science 2016, 352, 1430–1435. [Google Scholar] [CrossRef]
- Quiroz, F.G.; Fiore, V.F.; Levorse, J.; Polak, L.; Wong, E.; Pasolli, H.A.; Fuchs, E. Liquid-liquid phase separation drives skin barrier formation. Science 2020, 367, eaax9554. [Google Scholar] [CrossRef]
- Banani, S.F.; Lee, H.O.; Hyman, A.; Rosen, M.K. Biomolecular condensates: Organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 2017, 18, 285–298. [Google Scholar] [CrossRef]
- Sawyer, I.A.; Sturgill, D.; Dundr, M. Membraneless nuclear organelles and the search for phases within phases. Wiley Interdiscip. Rev. RNA 2019, 10, e1514. [Google Scholar] [CrossRef]
- Sawyer, I.A.; Bartek, J.; Dundr, M. Phase separated microenvironments inside the cell nucleus are linked to disease and regulate epigenetic state, transcription and RNA processing. Semin. Cell Dev. Biol. 2019, 90, 94–103. [Google Scholar] [CrossRef]
- Dundr, M.; Hebert, M.D.; Karpova, T.S.; Stanek, D.; Xu, H.; Shpargel, K.B.; Meier, U.T.; Neugebauer, K.M.; Matera, A.G.; Misteli, T. In vivo kinetics of Cajal body components. J. Cell Biol. 2004, 164, 831–842. [Google Scholar] [CrossRef]
- Phair, R.D.; Misteli, T. High mobility of proteins in the mammalian cell nucleus. Nat. Cell Biol. 2000, 404, 604–609. [Google Scholar] [CrossRef] [PubMed]
- Weidtkamp-Peters, S.; Lenser, T.; Negorev, D.; Gerstner, N.; Hofmann, T.G.; Schwanitz, G.; Hoischen, C.; Maul, G.; Dittrich, P.; Hemmerich, P. Dynamics of component exchange at PML nuclear bodies. J. Cell Sci. 2008, 121, 2731–2743. [Google Scholar] [CrossRef]
- Brangwynne, C.P.; Eckmann, C.R.; Courson, D.S.; Rybarska, A.; Hoege, C.; Gharakhani, J.; Jülicher, F.; Hyman, A.A. Germline P Granules Are Liquid Droplets That Localize by Controlled Dissolution/Condensation. Science 2009, 324, 1729–1732. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-C.M.; Kappel, C.; Beaudouin, J.; Eils, R.; Spector, D.L. Live Cell Dynamics of Promyelocytic Leukemia Nuclear Bodies upon Entry into and Exit from Mitosis. Mol. Biol. Cell 2008, 19, 3147–3162. [Google Scholar] [CrossRef]
- Dellaire, G.; Ching, R.W.; Dehghani, H.; Ren, Y.; Bazett-Jones, D.P. The number of PML nuclear bodies increases in early S phase by a fission mechanism. J. Cell Sci. 2006, 119, 1026–1033. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Gao, Y.-S.; Tousson, A.; Shah, A.; Chen, T.-L.L.; Vertel, B.M.; Sztul, E. Nuclear Aggresomes Form by Fusion of PML-associated Aggregates. Mol. Biol. Cell 2005, 16, 4905–4917. [Google Scholar] [CrossRef] [PubMed]
- Platani, M.; Goldberg, I.; Swedlow, J.R.; Lamond, A.I. In Vivo Analysis of Cajal Body Movement, Separation, and Joining in Live Human Cells. J. Cell Biol. 2000, 151, 1561–1574. [Google Scholar] [CrossRef]
- Shaw, P.J.; Jordan, E.G. The Nucleolus. Annu. Rev. Cell Dev. Biol. 1995, 11, 93–121. [Google Scholar] [CrossRef]
- Berry, J.; Weber, S.C.; Vaidya, N.; Haataja, M.; Brangwynne, C.P. RNA transcription modulates phase transition-driven nuclear body assembly. Proc. Natl. Acad. Sci. USA 2015, 112, E5237–E5245. [Google Scholar] [CrossRef]
- Feric, M.; Vaidya, N.; Harmon, T.S.; Mitrea, D.M.; Zhu, L.; Richardson, T.M.; Kriwacki, R.W.; Pappu, R.V.; Brangwynne, C.P. Coexisting Liquid Phases Underlie Nucleolar Subcompartments. Cell 2016, 165, 1686–1697. [Google Scholar] [CrossRef]
- Kato, M.; Han, T.W.; Xie, S.; Shi, K.; Du, X.; Wu, L.C.; Mirzaei, H.; Goldsmith, E.J.; Longgood, J.; Pei, J.; et al. Cell-free Formation of RNA Granules: Low Complexity Sequence Domains Form Dynamic Fibers within Hydrogels. Cell 2012, 149, 753–767. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, J.R.; Matheny, T.; Jain, S.; Abrisch, R.; Parker, R. Distinct stages in stress granule assembly and disassembly. eLife 2016, 5, e18413. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Banjade, S.; Cheng, H.-C.; Kim, S.; Chen, B.; Guo, L.; Llaguno, M.C.; Hollingsworth, J.V.; King, D.S.; Banani, S.F.; et al. Phase transitions in the assembly of multivalent signalling proteins. Nat. Cell Biol. 2012, 483, 336–340. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Protter, D.S.W.; Rosen, M.K.; Parker, R. Formation and Maturation of Phase-Separated Liquid Droplets by RNA-Binding Proteins. Mol. Cell 2015, 60, 208–219. [Google Scholar] [CrossRef]
- Molliex, A.; Temirov, J.; Lee, J.; Coughlin, M.; Kanagaraj, A.P.; Kim, H.J.; Mittag, T.; Taylor, J.P. Phase Separation by Low Complexity Domains Promotes Stress Granule Assembly and Drives Pathological Fibrillization. Cell 2015, 163, 123–133. [Google Scholar] [CrossRef]
- Nott, T.J.; Petsalaki, E.; Farber, P.; Jervis, D.; Fussner, E.; Plochowietz, A.; Craggs, T.D.; Bazett-Jones, D.P.; Pawson, T.; Forman-Kay, J.D.; et al. Phase Transition of a Disordered Nuage Protein Generates Environmentally Responsive Membraneless Organelles. Mol. Cell 2015, 57, 936–947. [Google Scholar] [CrossRef]
- Wright, P.E.; Dyson, H.J. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 2015, 16, 18–29. [Google Scholar] [CrossRef]
- Murakami, T.; Qamar, S.; Lin, J.Q.; Schierle, G.S.K.; Rees, E.J.; Miyashita, A.; Costa, A.R.; Dodd, R.B.; Chan, F.T.; Michel, C.H.; et al. ALS/FTD Mutation-Induced Phase Transition of FUS Liquid Droplets and Reversible Hydrogels into Irreversible Hydrogels Impairs RNP Granule Function. Neuron 2015, 88, 678–690. [Google Scholar] [CrossRef]
- Patel, A.; Lee, H.O.; Jawerth, L.; Maharana, S.; Jahnel, M.; Hein, M.Y.; Stoynov, S.; Mahamid, J.; Saha, S.; Franzmann, T.M.; et al. A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell 2015, 162, 1066–1077. [Google Scholar] [CrossRef]
- McSwiggen, D.T.; Mir, M.; Darzacq, X.; Tjian, R. Evaluating phase separation in live cells: Diagnosis, caveats, and functional consequences. Genes Dev. 2019, 33, 1619–1634. [Google Scholar] [CrossRef]
- Ivanov, P.; Kedersha, N.; Anderson, P. Stress Granules and Processing Bodies in Translational Control. Cold Spring Harb. Perspect. Biol. 2020, 11, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Protter, D.S.W.; Parker, R.R. Principles and Properties of Stress Granules. Trends Cell Biol. 2016, 26, 668–679. [Google Scholar] [CrossRef] [PubMed]
- Youn, J.-Y.; Dyakov, B.J.; Zhang, J.; Knight, J.D.; Vernon, R.M.; Forman-Kay, J.D.; Gingras, A.-C. Properties of Stress Granule and P-Body Proteomes. Mol. Cell 2019, 76, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Aizer, A.; Kalo, A.; Kafri, P.; Shraga, A.; Ben-Yishay, R.; Jacob, A.; Kinor, N.; Shav-Tal, Y. Quantifying mRNA targeting to P-bodies in living human cells reveals their dual role in mRNA decay and storage. J. Cell Sci. 2014, 127, 4443–4456. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Wheeler, J.R.; Walters, R.W.; Agrawal, A.; Barsic, A.; Parker, R.R. ATPase-Modulated Stress Granules Contain a Diverse Proteome and Substructure. Cell 2016, 164, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Kedersha, N.L.; Gupta, M.; Li, W.; Miller, I.; Anderson, P. RNA-Binding Proteins Tia-1 and Tiar Link the Phosphorylation of Eif-2α to the Assembly of Mammalian Stress Granules. J. Cell Biol. 1999, 147, 1431–1442. [Google Scholar] [CrossRef]
- Kedersha, N.; Anderson, P. Stress granules: Sites of mRNA triage that regulate mRNA stability and translatability. Biochem. Soc. Trans. 2002, 30, 963–969. [Google Scholar] [CrossRef]
- Kedersha, N.; Stoecklin, G.; Ayodele, M.; Yacono, P.; Lykke-Andersen, J.; Fritzler, M.J.; Scheuner, D.; Kaufman, R.J.; Golan, D.E.; Anderson, P. Stress granules and processing bodies are dynamically linked sites of mRNP remodeling. J. Cell Biol. 2005, 169, 871–884. [Google Scholar] [CrossRef]
- Moon, S.L.; Morisaki, T.; Khong, A.; Lyon, K.; Parker, R.R.; Stasevich, T.J. Multicolour single-molecule tracking of mRNA interactions with RNP granules. Nat. Cell Biol. 2019, 21, 162–168. [Google Scholar] [CrossRef]
- An, H.; Tan, J.T.; Shelkovnikova, T.A. Stress granules regulate stress-induced paraspeckle assembly. J. Cell Biol. 2019, 218, 4127–4140. [Google Scholar] [CrossRef]
- Buchan, J.R. MRNP granules Assembly, function, and connections with disease. RNA Biol. 2014, 11, 1019–1030. [Google Scholar] [CrossRef] [PubMed]
- Decker, C.J.; Teixeira, D.; Parker, R. Edc3p and a glutamine/asparagine-rich domain of Lsm4p function in processing body assembly in Saccharomyces cerevisiae. J. Cell Biol. 2007, 179, 437–449. [Google Scholar] [CrossRef] [PubMed]
- Kedersha, N.; Panas, M.D.; Achorn, C.A.; Lyons, S.; Tisdale, S.; Hickman, T.; Thomas, M.; Lieberman, J.; McInerney, G.M.; Ivanov, P.; et al. G3BP–Caprin1–USP10 complexes mediate stress granule condensation and associate with 40S subunits. J. Cell Biol. 2016, 212, 845–860. [Google Scholar] [CrossRef] [PubMed]
- Klosin, A.; Oltsch, F.; Harmon, T.S.; Honigmann, A.; Jülicher, F.; Hyman, A.A.; Zechner, C. Phase separation provides a mechanism to reduce noise in cells. Science 2020, 367, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Kalinowski, A.; Dahl, K.N.; Buehler, M.J. Structure and stability of the lamin A tail domain and HGPS mutant. J. Struct. Biol. 2011, 175, 425–433. [Google Scholar] [CrossRef]
- Wilson, K.L.; Foisner, R. Lamin-binding Proteins. Cold Spring Harb. Perspect. Biol. 2010, 2, a000554. [Google Scholar] [CrossRef]
- Gutowska-Owsiak, D.; De La Serna, J.B.; Fritzsche, M.; Naeem, A.; Podobas, E.I.; Leeming, M.; Colin-York, H.; O’Shaughnessy, R.; Eggeling, C.; Ogg, G.S. Orchestrated control of filaggrin–actin scaffolds underpins cornification. Cell Death Dis. 2018, 9, 1–18. [Google Scholar] [CrossRef]
- Lechler, T. Arp2/3 complex function in the epidermis. Tissue Barriers 2014, 2, e944445. [Google Scholar] [CrossRef][Green Version]
- Akinduro, O.; Sully, K.; Patel, A.; Robinson, D.J.; Chikh, A.; McPhail, G.; Braun, K.M.; Philpott, M.P.; Harwood, C.A.; Byrne, C.; et al. Constitutive Autophagy and Nucleophagy during Epidermal Differentiation. J. Investig. Dermatol. 2016, 136, 1460–1470. [Google Scholar] [CrossRef]
- Bhaduri, A.; Ungewickell, A.; Boxer, L.D.; Lopez-Pajares, V.; Zarnegar, B.J.; Khavari, P.A. Network Analysis Identifies Mitochondrial Regulation of Epidermal Differentiation by MPZL3 and FDXR. Dev. Cell 2015, 35, 444–457. [Google Scholar] [CrossRef]
- Mahanty, S.; Dakappa, S.S.; Shariff, R.; Patel, S.; Swamy, M.M.; Majumdar, A.; Setty, S.R.G. Keratinocyte differentiation promotes ER stress-dependent lysosome biogenesis. Cell Death Dis. 2019, 10, 269–285. [Google Scholar] [CrossRef] [PubMed]
- Arimoto, K.; Fukuda, H.; Imajoh-Ohmi, S.; Saito, H.; Takekawa, M. Formation of stress granules inhibits apoptosis by suppressing stress-responsive MAPK pathways. Nat. Cell Biol. 2008, 10, 1324–1332. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.J.; Back, S.H.; Kim, V.; Ryu, I.; Jang, S.K. Sequestration of TRAF2 into Stress Granules Interrupts Tumor Necrosis Factor Signaling under Stress Conditions. Mol. Cell. Biol. 2005, 25, 2450–2462. [Google Scholar] [CrossRef] [PubMed]
- Takahara, T.; Maeda, T. Transient Sequestration of TORC1 into Stress Granules during Heat Stress. Mol. Cell 2012, 47, 242–252. [Google Scholar] [CrossRef] [PubMed]
- Thedieck, K.; Holzwarth, B.; Prentzell, M.T.; Boehlke, C.; Kl#xE4;sener, K.; Ruf, S.; Sonntag, A.G.; Maerz, L.; Grellscheid, S.-N.; Kremmer, E.; et al. Inhibition of mTORC1 by astrin and stress granules prevents apoptosis in cancer cells. Cell 2013, 154, 859–874. [Google Scholar] [CrossRef]
- Adams, D.R.; Ron, D.; Kiely, P.A. RACK1, A multifaceted scaffolding protein: Structure and function. Cell Commun. Signal. 2011, 9, 22. [Google Scholar] [CrossRef]
- Kovalenko, A.; Kim, J.C.; Kang, T.B.; Rajput, A.; Bogdanov, K.; Dittrich-Breiholz, O.; Kracht, M.; Brenner, O.; Wallach, D. Caspase-8 deficiency in epidermal keratinocytes triggers an inflammatory skin disease. J. Exp. Med. 2009, 206, 2161–2177. [Google Scholar] [CrossRef]
- Ding, X.; Bloch, W.; Iden, S.; Rüegg, M.A.; Hall, M.N.; Leptin, M.; Partridge, L.; Eming, S.A. mTORC1 and mTORC2 regulate skin morphogenesis and epidermal barrier formation. Nat. Commun. 2016, 7, 13226. [Google Scholar] [CrossRef]
- Naeem, A.S.; Tommasi, C.; Cole, C.; Brown, S.J.; Zhu, Y.; Way, B.; Willis-Owen, S.; Moffatt, M.; Cookson, W.O.; Harper, J.I.; et al. A mechanistic target of rapamycin complex 1/2 (mTORC1)/V-Akt murine thymoma viral oncogene homolog 1 (AKT1)/cathepsin H axis controls filaggrin expression and processing in skin, a novel mechanism for skin barrier disruption in patients with atopic dermat. J. Allergy Clin. Immunol. 2017, 139, 1228–1241. [Google Scholar] [CrossRef]
- Ding, X.; Willenborg, S.; Bloch, W.; Wickström, S.A.; Wagle, P.; Brodesser, S.; Roers, A.; Jais, A.; Brüning, J.C.; Hall, M.N.; et al. Epidermal mammalian target of rapamycin complex 2 controls lipid synthesis and filaggrin processing in epidermal barrier formation. J. Allergy Clin. Immunol. 2020, 145, 283–300. [Google Scholar] [CrossRef]
- Sully, K.; Akinduro, O.; Philpott, M.P.; Naeem, A.S.; Harwood, C.A.; Reeve, V.E.; O’Shaughnessy, R.; Byrneet, C. The mTOR inhibitor rapamycin opposes carcinogenic changes to epidermal Akt1/PKBα isoform signaling. Oncogene 2013, 32, 3254–3262. [Google Scholar] [CrossRef] [PubMed]
- Ling, Y.; Wong, C.; Li, K.; Chan, K.; Boukamp, P.; Liu, W.K. CCHCR1 interacts with EDC4, suggesting its localization in P-bodies. Exp. Cell Res. 2014, 327, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Tervaniemi, M.H.; Katayama, S.; Skoog, T.; Siitonen, H.A.; Vuola, J.; Nuutila, K.; Tammimies, K.; Suomela, S.; Kankuri, E.; Kere, J.; et al. Intracellular signalling pathways and cytoskeletal functions converge on the psoriasis candidate gene CCHCR1 expressed at P-bodies and centrosomes. BMC Genom. 2018, 19, 432. [Google Scholar] [CrossRef] [PubMed]
- Kubo, E.; Fatma, N.; Sharma, P.; Shinohara, T.; Chylack, L.T., Jr.; Akagi, Y.; Singh, D.P. Transactivation of Involucrin, A Marker of Differentiation in Keratinocytes, by Lens Epithelium-Derived Growth Factor (LEDGF). J. Mol. Biol. 2002, 320, 1053–1063. [Google Scholar] [CrossRef]
- Nishizawa, Y.; Usukura, J.; Singh, D.; Chylack, L.T.; Shinohara, T. Spatial and temporal dynamics of two alternatively spliced regulatory factors, lens epithelium-derived growth factor (ledgf/p75) and p52, in the nucleus. Cell Tissue Res. 2001, 305, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Strong, C.D.G.; Conlan, S.; Deming, C.B.; Cheng, J.; Sears, K.E.; Segre, J.A. A milieu of regulatory elements in the epidermal differentiation complex syntenic block: Implications for atopic dermatitis and psoriasis. Hum. Mol. Genet. 2010, 19, 1453–1460. [Google Scholar] [CrossRef]
- Joost, S.; Jacob, T.; Sun, X.; Annusver, K.; La Manno, G.; Sur, I.; Kasper, M. Single-Cell Transcriptomics of Traced Epidermal and Hair Follicle Stem Cells Reveals Rapid Adaptations during Wound Healing. Cell Rep. 2018, 25, 585–597. [Google Scholar] [CrossRef]
- Decker, C.J.; Parker, R.R. P-Bodies and Stress Granules: Possible Roles in the Control of Translation and mRNA Degradation. Cold Spring Harb. Perspect. Biol. 2012, 4, a012286. [Google Scholar] [CrossRef]
- Luo, Y.; Na, Z.; Slavoff, S.A. P-Bodies: Composition, Properties, and Functions. Biochemistry 2018, 57, 2424–2431. [Google Scholar] [CrossRef]
- Hubstenberger, A.; Courel, M.; Benard, M.; Souquère, S.; Ernoult-Lange, M.; Chouaib, R.; Yi, Z.; Morlot, J.-B.; Munier, A.; Fradet, M.; et al. P-Body Purification Reveals the Condensation of Repressed mRNA Regulons. Mol. Cell 2017, 68, 144–157. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wotherspoon, D.; Rogerson, C.; O’Shaughnessy, R.F.L. Perspective: Controlling Epidermal Terminal Differentiation with Transcriptional Bursting and RNA Bodies. J. Dev. Biol. 2020, 8, 29. https://doi.org/10.3390/jdb8040029
Wotherspoon D, Rogerson C, O’Shaughnessy RFL. Perspective: Controlling Epidermal Terminal Differentiation with Transcriptional Bursting and RNA Bodies. Journal of Developmental Biology. 2020; 8(4):29. https://doi.org/10.3390/jdb8040029
Chicago/Turabian StyleWotherspoon, Duncan, Clare Rogerson, and Ryan F.L. O’Shaughnessy. 2020. "Perspective: Controlling Epidermal Terminal Differentiation with Transcriptional Bursting and RNA Bodies" Journal of Developmental Biology 8, no. 4: 29. https://doi.org/10.3390/jdb8040029
APA StyleWotherspoon, D., Rogerson, C., & O’Shaughnessy, R. F. L. (2020). Perspective: Controlling Epidermal Terminal Differentiation with Transcriptional Bursting and RNA Bodies. Journal of Developmental Biology, 8(4), 29. https://doi.org/10.3390/jdb8040029