Regulating Retinoic Acid Availability during Development and Regeneration: The Role of the CYP26 Enzymes

Abstract
1. Introduction
2. Retinoic Acid Synthesis and Receptor Signalling
3. Embryonic Defects Arising from Dysregulated RA Signalling
4. Retinoic Acid Degradation: The CYP26 Enzymes
5. CYP26 Structure, Biochemistry and Function
6. CYP26 in Human Genetic Disease
7. Function of CYP26 Enzymes during Development
7.1. Deletion of All Three Cyp26 Genes/Por
7.2. Loss-of Function Cyp26a1 Models
7.3. Loss-of Function Cyp26b1 Models
8. Forebrain
9. Hindbrain Patterning
10. Neuromesodermal Progenitors (NMPs) and Axis Extension
11. Skeletal Tissues: Craniofacial, Axis and Limb
12. Pharyngeal and Cardiovascular Systems
13. Other Systems
14. Regeneration
15. Paradoxical Feedback Loops in RA Signalling
16. Conclusions
Acknowledgments
Conflicts of Interest
References
- Mey, J.; McCaffery, P. Retinoic Acid Signaling in the Nervous System of Adult Vertebrates. Neuroscientist 2004, 10, 409–421. [Google Scholar] [CrossRef] [PubMed]
- McCaffery, P.; Zhang, J.; Crandall, J.E. Retinoic acid signaling and function in the adult hippocampus. J. Neurobiol. 2006, 66, 780–791. [Google Scholar] [CrossRef]
- Ransom, J.; Morgan, P.J.; McCaffery, P.J.; Stoney, P.N. The rhythm of retinoids in the brain. J. Neurochem. 2014, 129, 366–376. [Google Scholar] [CrossRef]
- Kiser, P.D.; Palczewski, K. Retinoids and Retinal Diseases. Annu. Rev. Vis. Sci. 2016, 2, 197–234. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, L.d.M.; Teixeira, E.F.M.; Sato, M.N. Impact of Retinoic Acid on Immune Cells and Inflammatory Diseases. Mediat. Infllam. 2018, 2018, 17. [Google Scholar] [CrossRef] [PubMed]
- Bonet, M.L.; Ribot, J.; Palou, A. Lipid metabolism in mammalian tissues and its control by retinoic acid. Biochim. Biophys. Acta 2012, 1821, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.A.; Kondo, N.; Okabe, T.; Takeshita, N.; Pilchak, D.M.; Koyama, E.; Ochiai, T.; Jensen, D.; Chu, M.-L.; Kane, M.A.; et al. Retinoic acid receptors are required for skeletal growth, matrix homeostasis and growth plate function in postnatal mouse. Dev. Biol. 2009, 328, 315–327. [Google Scholar] [CrossRef]
- Conaway, H.H.; Henning, P.; Lerner, U.H. Vitamin A Metabolism, Action, and Role in Skeletal Homeostasis. Endocr. Rev. 2013, 34, 766–797. [Google Scholar] [CrossRef]
- Lammer, E.J.; Chen, D.T.; Hoar, R.M.; Agnish, N.D.; Benke, P.J.; Braun, J.T.; Curry, C.J.; Fernhoff, P.M.; Grix, A.W.; Lott, I.T., Jr.; et al. Retinoic acid embryopathy. N. Engl. J. Med. 1985, 313, 837–841. [Google Scholar] [CrossRef]
- Niederreither, K.; Dolle, P. Retinoic acid in development: Towards an integrated view. Nat. Rev. Genet. 2008, 9, 541–553. [Google Scholar] [CrossRef]
- Rhinn, M.; Dollé, P. Retinoic acid signalling during development. Development 2012, 139, 843–858. [Google Scholar] [CrossRef] [PubMed]
- Stefanovic, S.; Zaffran, S. Mechanisms of retinoic acid signaling during cardiogenesis. Mech. Dev. 2017, 143, 9–19. [Google Scholar] [CrossRef]
- Zaffran, S.; Niederreither, K. Retinoic Acid Signaling and Heart Development. The Retinoids: Biology, Biochemistry, and Disease; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2015. [Google Scholar]
- Zaffran, S.; Robrini, E.N.; Bertrand, N. Retinoids and Cardiac Development. J. Dev. Biol. 2014, 2, 50–71. [Google Scholar] [CrossRef]
- Kam, R.K.T.; Deng, Y.; Chen, Y.; Zhao, H. Retinoic acid synthesis and functions in early embryonic development. Cell Biosci. 2012, 2, 11. [Google Scholar] [CrossRef]
- Shannon, S.R.; Moise, A.R.; Trainor, P.A. New insights and changing paradigms in the regulation of vitamin A metabolism in development. Wiley Interdiscip. Rev. Dev. Biol. 2017, 6, e264. [Google Scholar] [CrossRef] [PubMed]
- Simões-costa, M.S.; Azambuja, A.P.; Xavier-Neto, J. The search for non-chordate retinoic acid signaling: Lessons from chordates. J. Exp. Zool. Part Mol. Dev. Evol. 2008, 310, 54–72. [Google Scholar] [CrossRef] [PubMed]
- Pasutto, F.; Sticht, H.; Hammersen, G.; Gillessen-Kaesbach, G.; FitzPatrick, D.R.; Nürnberg, G.; Brasch, F.; Schirmer-Zimmermann, H.; Tolmie, J.L.; Chitayat, D.; et al. Mutations in STRA6 Cause a Broad Spectrum of Malformations Including Anophthalmia, Congenital Heart Defects, Diaphragmatic Hernia, Alveolar Capillary Dysplasia, Lung Hypoplasia, and Mental Retardation. Am. J. Hum. Genet. 2007, 80, 550–560. [Google Scholar] [CrossRef]
- Deltour, L.; Foglio, M.H.; Duester, G. Metabolic Deficiencies in Alcohol Dehydrogenase Adh1,Adh3, and Adh4 Null Mutant Mice: Overlapping roles of adh1 and adh4 in ethanol clearance and metabolism of retinol to retinoic acid. J. Biol. Chem. 1999, 274, 16796–16801. [Google Scholar] [CrossRef]
- Molotkov, A.; Fan, X.; Duester, G. Excessive vitamin A toxicity in mice genetically deficient in either alcohol dehydrogenase Adh1 or Adh3. Eur. J. Biochem. 2002, 269, 2607–2612. [Google Scholar] [CrossRef]
- Molotkov, A.; Fan, X.; Deltour, L.; Foglio, M.H.; Martras, S.; Farrés, J.; Parés, X.; Duester, G. Stimulation of retinoic acid production and growth by ubiquitously expressed alcohol dehydrogenase Adh3. Proc. Natl. Acad. Sci. USA 2002, 99, 5337–5342. [Google Scholar] [CrossRef]
- Molotkov, A.; Deltour, L.; Foglio, M.H.; Cuenca, A.E.; Duester, G. Distinct Retinoid Metabolic Functions for Alcohol Dehydrogenase Genes Adh1 and Adh4 in Protection against Vitamin A Toxicity or Deficiency Revealed in Double Null Mutant Mice. J. Biol. Chem. 2002, 277, 13804–13811. [Google Scholar] [CrossRef] [PubMed]
- Deltour, L.; Foglio, M.H.; Duester, G. Impaired retinol utilization in Adh4 alcohol dehydrogenase mutant mice. Dev. Genet. 1999, 25, 1–10. [Google Scholar] [CrossRef]
- Kumar, S.; Sandell, L.L.; Trainor, P.A.; Koentgen, F.; Duester, G. Alcohol and aldehyde dehydrogenases: Retinoid metabolic effects in mouse knockout models. Biochim. Biophys. Acta 2012, 1821, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Chambers, D.; Wilson, L.; Maden, M.; Lumsden, A. RALDH-independent generation of retinoic acid during vertebrate embryogenesis by CYP1B1. Development 2007, 134, 1369–1383. [Google Scholar] [CrossRef]
- Dragin, N.; Shi, Z.; Madan, R.; Karp, C.L.; Sartor, M.A.; Chen, C.; Gonzalez, F.J.; Nebert, D.W. Phenotype of the Cyp1a1/1a2/1b1(−/−) Triple-Knockout Mouse. Mol. Pharmacol. 2008, 73, 1844–1856. [Google Scholar] [CrossRef]
- Hollander, D.A.; Sarfarazi, M.; Stoilov, I.; Wood, I.S.; Fredrick, D.R.; Alvarado, J.A. Genotype and Phenotype Correlations in Congenital Glaucoma: CYP1B1 Mutations, Goniodysgenesis, and Clinical Characteristics. Am. J. Ophthalmol. 2006, 142, 993–1004. [Google Scholar] [CrossRef]
- Vincent, A.; Billingsley, G.; Priston, M.; Williams-Lyn, D.; Sutherland, J.; Glaser, T.; Oliver, E.; Walte, M.A.; Heathcote, G.; Levin, A.; et al. Phenotypic heterogeneity of CYP1B1: Mutations in a patient with Peters’ anomaly. J. Med Genet. 2001, 38, 324–326. [Google Scholar] [CrossRef]
- Tanwar, M.; Dada, T.; Dada, R. Axenfeld-Rieger Syndrome Associated with Congenital Glaucoma and Cytochrome P4501B1 Gene Mutations. Case Rep. Med. 2010, 2010, 6. [Google Scholar] [CrossRef]
- Millá, E.; Mañù, B.; Duch, S.; Hernan, I.; Borràs, E.; Planas, E.; de Sousa Dias, M.; Carballo, M.; Gamundi, M.J. Survey of familial glaucoma shows a high incidence of cytochrome P450, family 1, subfamily B, polypeptide 1 (CYP1B1) mutations in non-consanguineous congenital forms in a Spanish population. Mol. Vis. 2013, 19, 1707–1722. [Google Scholar]
- Maeda, A.; Maeda, T.; Sun, W.; Zhang, H.; Baehr, W.; Palczewski, K. Redundant and unique roles of retinol dehydrogenases in the mouse retina. Proc. Natl. Acad. Sci. USA 2007, 104, 19565–19570. [Google Scholar] [CrossRef]
- Maeda, A.; Maeda, T.; Golczak, M.; Palczewski, K. Retinopathy in Mice Induced by Disrupted All-trans-retinal Clearance. J. Biol. Chem. 2008, 283, 26684–26693. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, H.; Moosajee, M. Retinol dehydrogenase 12 (RDH12): Role in vision, retinal disease and future perspectives. Exp. Eye Res. 2019, 188, 107793. [Google Scholar] [CrossRef] [PubMed]
- Lhor, M.; Salesse, C. Retinol dehydrogenases: Membrane-bound enzymes for the visual function. Biochem. Cell Biol. 2014, 92, 510–523. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Cui, X.; Gu, Q.; Chen, Y.; Zhou, J.; Kuang, Y.; Wang, Z.; Xu, X. Retinol dehydrogenase 13 protects the mouse retina from acute light damage. Mol. Vis. 2012, 18, 1021–1030. [Google Scholar]
- Cui, X.; Ma, B.; Wang, Y.; Chen, Y.; Shen, C.; Kuang, Y.; Fei, J.; Lu, L.; Wang, Z. Rdh13 deficiency weakens carbon tetrachloride-induced liver injury by regulating Spot14 and Cyp2e1 expression levels. Front. Med. 2019, 13, 104–111. [Google Scholar] [CrossRef]
- Cui, X.; Dang, S.; Wang, Y.; Chen, Y.; Zhou, J.; Shen, C.; Kuang, Y.; Fei, J.; Lu, L.; Wang, Z. Retinol dehydrogenase 13 deficiency diminishes carbon tetrachloride-induced liver fibrosis in mice. Toxicol. Lett. 2017, 265, 17–22. [Google Scholar] [CrossRef]
- Wu, L.; Belyaeva, O.V.; Adams, M.K.; Klyuyeva, A.; Lee, S.-A.; Goggans, K.R.; Kesterson, R.A.; Popov, K.M.; Kedishvili, N.Y. Mice lacking the epidermal retinol dehydrogenases SDR16C5 and SDR16C6 display accelerated hair growth and enlarged meibomian glands. J. Biol. Chem. 2019, 294, 17060–17074. [Google Scholar] [CrossRef]
- Farjo, K.M.; Moiseyev, G.; Nikolaeva, O.; Sandell, L.L.; Trainor, P.A.; Ma, J.-X. RDH10 is the primary enzyme responsible for the first step of embryonic Vitamin A metabolism and retinoic acid synthesis. Dev. Biol. 2011, 357, 347–355. [Google Scholar]
- Rhinn, M.; Schuhbaur, B.; Niederreither, K.; Dollé, P. Involvement of retinol dehydrogenase 10 in embryonic patterning and rescue of its loss of function by maternal retinaldehyde treatment. Proc. Natl. Acad. Sci. USA 2011, 108, 16687–16692. [Google Scholar] [CrossRef]
- Sandell, L.L.; Sanderson, B.W.; Moiseyev, G.; Johnson, T.; Mushegian, A.; Young, K.; Rey, J.-P.; Ma, J.-X.; Staehling-Hampton, K.; Trainor, P.A. RDH10 is essential for synthesis of embryonic retinoic acid and is required for limb, craniofacial, and organ development. Genes Dev. 2007, 21, 1113–1124. [Google Scholar] [CrossRef]
- Sandell, L.L.; Lynn, M.L.; Inman, K.E.; McDowell, W.; Trainor, P.A. RDH10 Oxidation of Vitamin A Is a Critical Control. Step in Synthesis of Retinoic Acid during Mouse Embryogenesis. PLoS ONE 2012, 7, e30698. [Google Scholar]
- Strate, I.; Min, T.H.; Iliev, D.; Pera, E.M. Retinol dehydrogenase 10 is a feedback regulator of retinoic acid signalling during axis formation and patterning of the central nervous system. Development 2009, 136, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Hernandez, R.E.; Waxman, J.S.; Yelon, D.; Moens, C.B. Dhrs3a regulates retinoic acid biosynthesis through a feedback inhibition mechanism. Dev. Biol. 2010, 338, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Billings, S.E.; Pierzchalski, K.; Tjaden, N.E.B.; Pang, X.-Y.; Trainor, P.A.; Kane, M.A.; Moise, A.R. The retinaldehyde reductase DHRS3 is essential for preventing the formation of excess retinoic acid during embryonic development. FASWB J. 2013, 27, 4877–4889. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.K.; Belyaeva, O.V.; Wu, L.; Kedishvili, N.Y. The Retinaldehyde Reductase Activity of DHRS3 Is Reciprocally Activated by Retinol Dehydrogenase 10 to Control. Retinoid Homeostasis. J. Biol. Chem. 2014, 289, 14868–14880. [Google Scholar] [CrossRef]
- Belyaeva, O.V.; Adams, M.K.; Wu, L.; Kedishvili, N.Y. The Antagonistically Bifunctional Retinoid Oxidoreductase Complex. Is Required for Maintenance of All-trans-retinoic Acid Homeostasis. J. Biol. Chem. 2017, 292, 5884–5897. [Google Scholar] [CrossRef] [PubMed]
- Ang, H.L.; Duester, G. Stimulation of premature retinoic acid synthesis in Xenopus embryos following premature expression of aldehyde dehydrogenase ALDH1. FEBS J. 1999, 260, 227–234. [Google Scholar] [CrossRef]
- Duester, G. Involvement of alcohol dehydrogenase, short-chain dehydrogenase/reductase, aldehyde dehydrogenase, and cytochrome P450 in the control of retinoid signaling by activation of retinoic acid synthesis. Biochemistry 1996, 35, 12221–12227. [Google Scholar] [CrossRef]
- Mic, F.A.; Haselbeck, R.J.; Cuenca, A.E.; Duester, G. Novel retinoic acid generating activities in the neural tube and heart identified by conditional rescue of Raldh2 null mutant mice. Development 2002, 129, 2271–2282. [Google Scholar]
- Li, H.; Wagner, E.; McCaffery, P.; Smith, D.; Andreadis, A.; Dräger, U.C. A retinoic acid synthesizing enzyme in ventral retina and telencephalon of the embryonic mouse. Mech. Dev. 2000, 95, 283–289. [Google Scholar] [CrossRef]
- Mic, F.A.; Molotkov, A.; Fan, X.; Cuenca, A.E.; Duester, G. RALDH3, a retinaldehyde dehydrogenase that generates retinoic acid, is expressed in the ventral retina, otic vesicle and olfactory pit during mouse development. Mech. Dev. 2000, 97, 227–230. [Google Scholar] [CrossRef]
- Niederreither, K.; McCaffery, P.; Dräger, U.C.; Chambon, P.; Dollé, P. Restricted expression and retinoic acid-induced downregulation of the retinaldehyde dehydrogenase type 2 (RALDH-2) gene during mouse development. Mech. Dev. 1997, 62, 67–78. [Google Scholar] [CrossRef]
- Chatzi, C.; Brade, T.; Duester, G. Retinoic Acid Functions as a Key GABAergic Differentiation Signal. in the Basal Ganglia. PLoS Biol. 2011, 9, e1000609. [Google Scholar] [CrossRef] [PubMed]
- Dupe, V.; Matt, N.; Garnier, J.M.; Chambon, P.; Mark, M.; Ghyselinck, N.B. A newborn lethal defect due to inactivation of retinaldehyde dehydrogenase type 3 is prevented by maternal retinoic acid treatment. Proc. Natl. Acad. Sci. USA 2003, 100, 14036–14041. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Molotkov, A.; Manabe, S.I.; Donmoyer, C.M.; Deltour, L.; Foglio, M.H.; Cuenca, A.E.; Blaner, W.S.; Lipton, S.A.; Duester, G. Targeted Disruption of Aldh1a1 (Raldh1) Provides Evidence for a Complex. Mechanism of Retinoic Acid Synthesis in the Developing Retina. Mol. Cell. Biol. 2003, 23, 4637–4648. [Google Scholar] [PubMed]
- Matt, N.; Dupé, V.; Garnier, J.-M.; Dennefeld, C.; Chambon, P.; Mark, M.; Ghyselinck, N.B. Retinoic acid-dependent eye morphogenesis is orchestrated by neural crest cells. Development 2005, 132, 4789–4800. [Google Scholar] [CrossRef] [PubMed]
- Begemann, G.; Schilling, T.F.; Rauch, G.J.; Geisler, R.; Ingham, P.W. The zebrafish neckless mutation reveals a requirement for raldh2 in mesodermal signals that pattern the hindbrain. Development 2001, 128, 3081–3094. [Google Scholar]
- Hochgreb, T.; Linhares, V.L.; Menezes, D.C.; Sampaio, A.C.; Yan, C.Y.I.; Cardoso, W.V.; Rosentha, N.; Xavier-Neto, J. A caudorostral wave of RALDH2 conveys anteroposterior information to the cardiac field. Development 2003, 130, 5363–5374. [Google Scholar] [CrossRef]
- Niederreither, K.; Subbarayan, V.; Dolle, P.; Chambon, P. Embryonic retinoic acid synthesis is essential for early mouse post-implantation development. Nat. Genet. 1999, 21, 444–448. [Google Scholar] [CrossRef]
- Niederreither, K.; Vermot, J.; Schuhbaur, B.; Chambon, P.; Dollé, P. Embryonic retinoic acid synthesis is required for forelimb growth and anteroposterior patterning in the mouse. Development 2002, 129, 3563–3574. [Google Scholar]
- Niederreither, K.; Vermot, J.; Schuhbaur, B.; Chambon, P.; Dolle, P. Retinoic acid synthesis and hindbrain patterning in the mouse embryo. Development 2000, 127, 75–85. [Google Scholar]
- Niederreither, K.; Vermot, J.; Roux, I.L.; Schuhbaur, B.; Chambon, P.; Dollé, P. The regional pattern of retinoic acid synthesis by RALDH2 is essential for the development of posterior pharyngeal arches and the enteric nervous system. Development 2003, 130, 2525–2534. [Google Scholar] [CrossRef]
- Niederreither, K.; Vermot, J.; Messaddeq, N.; Schuhbaur, B.; Chambon, P.; Dolle, P. Embryonic retinoic acid synthesis is essential for heart morphogenesis in the mouse. Development 2001, 128, 1019–1031. [Google Scholar] [PubMed]
- Ryckebusch, L.; Wang, Z.; Bertrand, N.; Lin, S.C.; Chi, X.; Schwartz, R.; Zaffran, S.; Niederreither, K. Retinoic acid deficiency alters second heart field formation. Proc. Natl. Acad. Sci. USA 2008, 105, 2913–2918. [Google Scholar] [CrossRef] [PubMed]
- Vermot, J.; Llamas, J.G.; Fraulob, V.; Niederreither, K.; Chambon, P.; Dolle, P. Retinoic Acid Controls the Bilateral Symmetry of Somite Formation in the Mouse Embryo. Science 2005, 308, 563–566. [Google Scholar] [CrossRef] [PubMed]
- Vermot, J.; Pourquie, O. Retinoic acid coordinates somitogenesis and left-right patterning in vertebrate embryos. Nature 2005, 435, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Vermot, J.; Niederreither, K.; Garnier, J.-M.; Chambon, P.; Dollé, P. Decreased embryonic retinoic acid synthesis results in a DiGeorge syndrome phenotype in newborn mice. Proc. Natl. Acad. Sci. USA 2003, 100, 1763–1768. [Google Scholar] [CrossRef]
- Ryckebusch, L.; Bertrand, N.; Mesbah, K.; Bajolle, F.; Niederreither, K.; Kelly, R.G.; Zaffran, S. Decreased Levels of Embryonic Retinoic Acid Synthesis Accelerate Recovery From Arterial Growth Delay in a Mouse Model of DiGeorge Syndrome. Circ. Res. 2010, 106, 686–694. [Google Scholar] [CrossRef]
- Chambon, P. A decade of molecular biology of retinoic acid receptors. FASEB J. 1996, 10, 940–954. [Google Scholar] [CrossRef]
- Dollé, P. Developmental expression of retinoic acid receptors (RARs). Nucl. Recept. Signal. 2009, 7, nrs-07006. [Google Scholar] [CrossRef]
- Mark, M.; Ghyselinck, N.B.; Chambon, P. Function of retinoic acid receptors during embryonic development. Nucl. Recept. Signal. 2009, 7, nrs-07002. [Google Scholar] [CrossRef] [PubMed]
- Lyn, S.; Giguère, V. Localization of CRABP-I and CRABP-II mRNA in the early mouse embryo by whole-mount in situ hybridization: Implications for teratogenesis and neural development. Dev. Dyn. 1994, 199, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Cai, A.Q.; Radtke, K.; Linville, A.; Lander, A.D.; Nie, Q.; Schilling, T.F. Cellular retinoic acid-binding proteins are essential for hindbrain patterning and signal robustness in zebrafish. Development 2012, 139, 2150–2155. [Google Scholar] [CrossRef] [PubMed]
- Mic, F.A.; Molotkov, A.; Benbrook, D.M.; Duester, G. Retinoid activation of retinoic acid receptor but not retinoid X receptor is sufficient to rescue lethal defect in retinoic acid synthesis. Proc. Natl. Acad. Sci. USA 2003, 100, 7135–7140. [Google Scholar] [CrossRef]
- Kastner, P.; Mark, M.; Ghyselinck, N.; Krezel, W.; Dupe, V.; Grondona, J.M.; Chambon, P. Genetic evidence that the retinoid signal is transduced by heterodimeric RXR/RAR functional units during mouse development. Development 1997, 124, 313–326. [Google Scholar]
- Ghyselinck, N.B.; Wendling, O.; Messaddeq, N.; Dierich, A.; Lampron, C.; Decimo, D.; Viville, S.; Chambon, P.; Mark, M. Contribution of retinoic acid receptor beta isoforms to the formation of the conotruncal septum of the embryonic heart. Dev. Biol. 1998, 198, 303–318. [Google Scholar] [CrossRef]
- Mendelsohn, C.; Lohnes, D.; Decimo, D.; Lufkin, T.; LeMeur, M.; Chambon, P.; Mark, M. Function of the retinoic acid receptors (RARs) during development (II). Multiple abnormalities at various stages of organogenesis in RAR double mutants. Development 1994, 120, 2749–2771. [Google Scholar]
- Lohnes, D.; Mark, M.; Mendelsohn, C.; Dolle, P.; Dierich, A.; Gorry, P.; Gansmuller, A.; Chambon, P. Function of the retinoic acid receptors (RARs) during development (I). Craniofacial and skeletal abnormalities in RAR double mutants. Development 1994, 120, 2723–2748. [Google Scholar]
- Wendling, O.; Dennefeld, C.; Chambon, P.; Mark, M. Retinoid signaling is essential for patterning the endoderm of the third and fourth pharyngeal arches. Development 2000, 127, 1553–1562. [Google Scholar]
- Lohnes, D.; Kastner, P.; Dierich, A.; Mark, M.; LeMeur, M.; Chambon, P. Function of retinoic acid receptor γ in the mouse. Cell 1993, 73, 643–658. [Google Scholar] [CrossRef]
- Iulianella, A.; Lohnes, D. Contribution of retinoic acid receptor gamma to retinoid-induced craniofacial and axial defects. Dev. Dyn. 1997, 209, 92–104. [Google Scholar] [CrossRef]
- Gudas, L.J.; Wagner, J.A. Retinoids regulate stem cell differentiation. J. Cell. Physiol. 2011, 226, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Mark, M.; Ghyselinck, N.B.; Chambon, P. Retinoic acid signalling in the development of branchial arches. Curr. Opin. Genet. Dev. 2004, 14, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Zile, M.H. Vitamin A Requirement for Early Cardiovascular Morphogenesis Specification in the Vertebrate Embryo: Insights from the Avian Embryo. Exp. Biol. Med. 2004, 229, 598–606. [Google Scholar] [CrossRef]
- Maden, M.; Gale, E.; Kostetskii, I.; Zile, M. Vitamin A-deficient quail embryos have half a hindbrain and other neural defects. Curr. Biol. 1996, 6, 417–426. [Google Scholar] [CrossRef]
- Sockanathan, S.; Jessell, T.M. Motor Neuron-Derived Retinoid Signaling Specifies the Subtype Identity of Spinal Motor Neurons. Cell 1998, 94, 503–514. [Google Scholar] [CrossRef]
- Malpel, S.; Mendelsohn, C.; Cardoso, W.V. Regulation of retinoic acid signaling during lung morphogenesis. Development 2000, 127, 3057–3067. [Google Scholar]
- Stratford, T.; Logan, C.; Zile, M.; Maden, M. Abnormal anteroposterior and dorsoventral patterning of the limb bud in the absence of retinoids. Mech. Dev. 1999, 81, 115–125. [Google Scholar] [CrossRef]
- Stratford, T.; Horton, C.; Maden, M. Retinoic acid is required for the initiation of outgrowth in the chick limb bud. Curr. Biol. 1996, 6, 1124–1133. [Google Scholar] [CrossRef]
- Batourina, E.; Gim, S.; Bello, N.; Shy, M.; Clagett-Dame, M.; Srinivas, S.; Costantini, F.; Mendelsohn, C. Vitamin A controls epithelial/mesenchymal interactions through Ret expression. Nat. Genet. 2001, 27, 74–78. [Google Scholar] [CrossRef]
- Mendelsohn, C.; Batourina, E.; Fung, S.; Gilbert, T.; Dodd, J. Stromal cells mediate retinoid-dependent functions essential for renal development. Development 1999, 126, 1139–1148. [Google Scholar] [PubMed]
- Wagner, E.; McCaffery, P.; Drager, U.C. Retinoic Acid in the Formation of the Dorsoventral Retina and Its Central Projections. Dev. Biol. 2000, 222, 460–470. [Google Scholar] [CrossRef] [PubMed]
- Fantel, A.G.; Shepard, T.H.; Newell-Morris, L.L.; Moffett, B.C. Teratogenic effects of retinoic acid in pigtail monkeys (Macaca nemestrina). I. General features. Teratology 1977, 15, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Gale, E.; Zile, M.; Maden, M. Hindbrain respecification in the retinoid-deficient quail. Mech. Dev. 1999, 89, 43–54. [Google Scholar] [CrossRef]
- Happle, R.; Traupe, H.; Bounameaux, Y.; Fisch, T. Teratogenic effects of etretinate in humans. Dtsch. Med. Wochenschr. 1984, 109, 1476–1480. [Google Scholar] [CrossRef] [PubMed]
- Kalter, H.; Warkany, J. Experimental production of congenital malformations in strains of inbred mice by maternal treatment with hypervitaminosis A. Am. J. Pathol. 1961, 38, 1–21. [Google Scholar]
- Kalter, H. The teratogenic effects of hypervitaminosis A upon the face and mouth of inbred mice. Ann. N. Y. Acad. Sci. 1960, 85, 42–55. [Google Scholar] [CrossRef]
- Kistler, A. Teratogenesis of retinoic acid in rats: Susceptible stages and suppression of retinoic acid-induced limb malformations by cycloheximide. Teratology 1981, 23, 25–31. [Google Scholar] [CrossRef]
- Kochhar, D.M.; Johnson, E.M. Morphological and autoradiographic studies of cleft palate induced in rat embryos by maternal hypervitaminosis A. J. Embryol. Exp. Morphol. 1965, 14, 223–238. [Google Scholar]
- Rosa, F.W.; Wilk, A.L.; Kelsey, F.O. Teratogen update: Vitamin A congeners. Teratology 1986, 33, 355–364. [Google Scholar] [CrossRef]
- Shenefelt, R.E. Gross congenital malformations. Animal model: Treatment of various species with a large dose of vitamin A at known stages in pregnancy. Am. J. Pathol. 1972, 66, 589–592. [Google Scholar] [PubMed]
- Iulianella, A.; Beckett, B.; Petkovich, M.; Lohnes, D. A Molecular Basis for Retinoic Acid-Induced Axial Truncation. Dev. Biol. 1999, 205, 33–48. [Google Scholar] [CrossRef]
- Durston, A.J.; Timmermans, J.P.M.; Hage, W.J.; Hendriks, H.F.J.; de Vries, N.J.; Heideveld, M.; Nieuwkoop, P.D. Retinoic acid causes an anteroposterior transformation in the developing central nervous system. Nature 1989, 340, 140–144. [Google Scholar] [CrossRef] [PubMed]
- Dupe, V.; Ghyselinck, N.B.; Wendling, O.; Chambon, P.; Mark, M. Key roles of retinoic acid receptors alpha and beta in the patterning of the caudal hindbrain, pharyngeal arches and otocyst in the mouse. Development 1999, 126, 5051–5059. [Google Scholar] [PubMed]
- Matt, N.; Ghyselinck, N.B.; Wendling, O.; Chambon, P.; Mark, M. Retinoic acid-induced developmental defects are mediated by RARbeta/RXR heterodimers in the pharyngeal endoderm. Development 2003, 130, 2083–2093. [Google Scholar] [CrossRef] [PubMed]
- Mulder, G.B.; Manley, N.; Maggio-Price, L. Retinoic acid-induced thymic abnormalities in the mouse are associated with altered pharyngeal morphology, thymocyte maturation defects, and altered expression of Hoxa3 and Pax1. Teratology 1998, 58, 263–275. [Google Scholar] [CrossRef]
- Mulder, G.B.; Manley, N.; Grant, J.; Schmidt, K.; Zeng, W.; Eckhoff, C.; Maggio-Price, L. Effects of excess vitamin A on development of cranial neural crest-derived structures: A neonatal and embryologic study. Teratology 2000, 62, 214–226. [Google Scholar] [CrossRef]
- Lee, Y.M.; Osumi-Yamashita, N.; Ninomiya, Y.; Moon, C.K.; Eriksson, U.; Eto, K. Retinoic acid stage-dependently alters the migration pattern and identity of hindbrain neural crest cells. Development 1995, 121, 825. [Google Scholar]
- Williams, A.L.; Bohnsack, B.L. What’s retinoic acid got to do with it? Retinoic acid regulation of the neural crest in craniofacial and ocular development. Genesis 2019, 57, e23308. [Google Scholar]
- Arinami, T. Analyses of the associations between the genes of 22q11 deletion syndrome and schizophrenia. J. Hum. Genet. 2006, 51, 1037. [Google Scholar] [CrossRef]
- Gao, S.; Li, X.; Amendt, B.A. Understanding the Role of Tbx1 as a Candidate Gene for 22q11.2 Deletion Syndrome. Curr. Allergy Asthma Rep. 2013, 13, 613–621. [Google Scholar] [CrossRef]
- Papangeli, I.; Scambler, P. The 22q11 deletion: DiGeorge and velocardiofacial syndromes and the role of TBX1. Wiley Interdiscip. Rev. Dev. Biol. 2013, 2, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Scambler, P.J. The 22q11 deletion syndromes. Hum. Mol. Genet. 2000, 9, 2421–2426. [Google Scholar] [CrossRef] [PubMed]
- McDonald-McGinn, D.M.; Sullivan, K.E.; Marino, B.; Philip, N.; Swillen, A.; Vorstman, J.A.S.; Zackai, E.H.; Emanuel, B.S.; Vermeesch, R.J.; Morrow, B.E.; et al. 22q11.2 deletion syndrome. Nat. Rev. Dis. Primers 2015, 1, 15071. [Google Scholar] [CrossRef] [PubMed]
- Conley, M.; Beckwith, J.; Manceer, J.F.K.; Tenckhoff, L. The spectrum of the DiGeorge syndrome. J. Pediatrics 1979, 94, 883–890. [Google Scholar] [CrossRef]
- Wilson, D.I.; Goodship, J.A.; Burn, J.; Cross, I.E.; Scambler, P.J. Deletions within chromosome 22q11 in familial congenital heart disease. Lancet 1992, 340, 573–575. [Google Scholar] [CrossRef]
- Jerome, L.A.; Papaioannou, V.E. DiGeorge syndrome phenotype in mice mutant for the T-box gene, Tbx1. Nat. Genet. 2001, 27, 286–291. [Google Scholar] [CrossRef]
- Lindsay, E.A.; Vitelli, F.; Su, H.; Morishima, M.; Huynh, T.; Pramparo, T.; Jurecic, V.; Ogunrinu, G.; Sutherland, H.F.; Scambler, P.J.; et al. T Tbx1 haploinsufficiency in the DiGeorge syndrome region causes aortic arch defects in mice. Nature 2001, 410, 97–101. [Google Scholar] [CrossRef]
- Merscher, S.; Funke, B.; Epstein, J.A.; Heyer, J.; Puech, A.; Lu, M.M.; Xavier, R.J.; Demay, M.B.; Russell, R.G.; Factor, S.; et al. TBX1 Is Responsible for Cardiovascular Defects in Velo-Cardio-Facial/DiGeorge Syndrome. Cell 2001, 104, 619–629. [Google Scholar] [CrossRef]
- Vitelli, F.; Morishima, M.; Taddei, I.; Lindsay, E.A.; Baldini, A. Tbx1 mutation causes multiple cardiovascular defects and disrupts neural crest and cranial nerve migratory pathways. Hum. Mol. Genet. 2002, 11, 915–922. [Google Scholar] [CrossRef]
- Xu, H.; Morishima, M.; Wylie, J.N.; Schwartz, R.J.; Bruneau, B.G.; Lindsay, E.A.; Baldini, A. Tbx1 has a dual role in the morphogenesis of the cardiac outflow tract. Development 2004, 131, 3217–3227. [Google Scholar] [CrossRef] [PubMed]
- Arnold, J.S.; Werling, U.; Braunstein, E.M.; Liao, J.; Nowotschin, S.; Edelmann, W.; Hebert, J.M.; Morrow, B.E. Inactivation of Tbx1 in the pharyngeal endoderm results in 22q11DS malformations. Development 2006, 133, 977–987. [Google Scholar] [CrossRef] [PubMed]
- Kochilas, L.K.; Potluri, V.; Gitler, A.; Balasubramanian, K.; Chin, A.J. Cloning and characterization of zebrafish tbx1. Gene Expr. Patterns 2003, 3, 645–651. [Google Scholar] [CrossRef]
- Roberts, C.; Ivins, S.M.; James, C.T.; Scambler, P.J. Retinoic acid down-regulates Tbx1 expression in vivo and in vitro. Dev. Dyn. 2005, 232, 928–938. [Google Scholar] [CrossRef]
- Roberts, C.; Ivins, S.; Cook, A.C.; Baldini, A.; Scambler, P.J. Cyp26 genes a1, b1 and c1 are down-regulated in Tbx1 null mice and inhibition of Cyp26 enzyme function produces a phenocopy of DiGeorge Syndrome in the chick. Hum. Mol. Genet. 2006, 15, 3394–3410. [Google Scholar] [CrossRef]
- Guris, D.L.; Duester, G.; Papaioannou, V.E.; Imamoto, A. Dose-Dependent Interaction of Tbx1 and Crkl and Locally Aberrant RA Signaling in a Model of del22q11 Syndrome. Dev. Cell 2006, 10, 81–92. [Google Scholar] [CrossRef]
- Okubo, T.; Kawamura, A.; Takahashi, J.; Yagi, H.; Morishima, M.; Matsuoka, R.; Takada, S. Ripply3, a Tbx1 repressor, is required for development of the pharyngeal apparatus and its derivatives in mice. Development 2011, 138, 339–348. [Google Scholar] [CrossRef]
- De Bono, C.; Thellier, C.; Bertrand, N.; Sturny, R.; Jullian, E.; Cortes, C.; Stefanovic, S.; Zaffran, S.; Théveniau-Ruissy, M.; Kelly, R.G. T-box genes and retinoic acid signaling regulate the segregation of arterial and venous pole progenitor cells in the murine second heart fieldHuman Molecular Genetics. Hum. Mol. Genet. 2018, 27, 3747–3760. [Google Scholar] [CrossRef]
- Piotrowski, T.; Ahn, D.G.; Schilling, T.F.; Nair, S.; Ruvinsky, I.; Geisler, R.; Rauch, G.J.; Haffter, P.; Zon, L.I.; Zhou, Y.; et al. The zebrafish van gogh mutation disrupts tbx1, which is involved in the DiGeorge deletion syndrome in humans. Development 2003, 130, 5043–5052. [Google Scholar] [CrossRef]
- Williams, P.A.; Cosme, J.; Sridhar, V.; Johnson, E.F.; McRee, D.E. Mammalian Microsomal Cytochrome P450 Monooxygenase: Structural Adaptations for Membrane Binding and Functional Diversity. Mol. Cell 2000, 5, 121–131. [Google Scholar] [CrossRef]
- Cosme, J.; Johnson, E.F. Engineering microsomal cytochrome P450 2C5 to be a soluble, monomeric enzyme: Mutations that alter aggregation, phospholipid dependence of catalysis, and membrane binding. J. Biol. Chem. 2000, 275, 2545–2553. [Google Scholar] [CrossRef] [PubMed]
- Gotoh, O. Substrate recognition sites in cytochrome P450 family 2 (CYP2) proteins inferred from comparative analyses of amino acid and coding nucleotide sequences. J. Biol. Chem. 1992, 267, 83–90. [Google Scholar] [PubMed]
- Graham, S.E.; Peterson, J.A. How Similar Are P450s and What Can. Their Differences Teach. Us? Arch. Biochem. Biophys. 1999, 369, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Gilardi, G.; Di Nardo, G. Heme iron centers in cytochrome P450: Structure and catalytic activity. Rend. Lincei 2017, 28, 159–167. [Google Scholar] [CrossRef]
- Werck-Reichhart, D.; Feyereisen, R. Cytochromes P450: A success story. Genome Biol. 2000, 1, reviews3003-1. [Google Scholar] [CrossRef]
- Thatcher, J.E.; Isoherranen, N. The role of CYP26 enzymes in retinoic acid clearance. Expert Opin. Drug Metab. Toxicol. 2009, 5, 875–886. [Google Scholar] [CrossRef]
- Thatcher, J.E.; Zelter, A.; Isoherranen, N. The relative importance of CYP26A1 in hepatic clearance of all-trans retinoic acid. Biochem. Pharmacol. 2010, 80, 903–912. [Google Scholar] [CrossRef]
- Isoherranen, N.; Zhong, N. Biochemical and physiological importance of the CYP26 retinoic acid hydroxylases. Pharmacol. Ther. 2019, 107400. [Google Scholar] [CrossRef]
- Lutz, J.D.; Dixit, V.; Yeung, C.K.; Dickmann, L.J.; Zelter, A.; Thatcher, J.E.; Nelson, W.L.; Isoherranen, N. Expression and functional characterization of cytochrome P450 26A1, a retinoic acid hydroxylase. Biochem. Pharmacol. 2009, 77, 258–268. [Google Scholar] [CrossRef]
- Taimi, M.; Helvig, C.; Wisniewski, J.; Ramshaw, H.; White, J.; Korczak, B.; Petkovich, M. A Novel Human Cytochrome P450, CYP26C1, Involved in Metabolism of 9-cis and All-trans Isomers of Retinoic Acid. J. Biol. Chem. 2004, 279, 77–85. [Google Scholar] [CrossRef]
- White, J.A.; Guo, Y.D.; Baetz, K.; Beckett-Jones, B.; Bonasoro, J.; Hsu, K.E.; Dilworth, F.J.; Jones, G.; Petkovich, M. Identification of the Retinoic Acid-inducible All-trans-retinoic Acid 4-Hydroxylase. J. Biol. Chem. 1996, 271, 29922–29927. [Google Scholar] [CrossRef] [PubMed]
- Fujii, H.; Sato, T.; Kaneko, S.; Gotoh, O.; Fujii-Kuriyama, Y.; Osawa, K.; Kato, S.; Hamada, H. Metabolic Metabolic inactivation of retinoic acid by a novel P450 differentially expressed in developing mouse embryos. EMBO J. 1997, 16, 4163–4173. [Google Scholar] [CrossRef] [PubMed]
- Ross, A.C.; Zolfaghari, R. Cytochrome P450s in the Regulation of Cellular Retinoic Acid Metabolism. Annu. Rev. Nutr. 2011, 31, 65–87. [Google Scholar] [CrossRef] [PubMed]
- Qian, L.; Zolfaghari, R.; Ross, A.C. Liver-specific cytochrome P450 CYP2C22 is a direct target of retinoic acid and a retinoic acid-metabolizing enzyme in rat liver. J. Lipid Res. 2010, 51, 1781–1792. [Google Scholar] [CrossRef]
- White, J.A.; Ramshaw, H.; Taimi, M.; Stangle, W.; Zhang, A.; Everingham, S.; Creighton, S.; Tam, S.P.; Jones, G.; Petkovich, M. Identification of the human cytochrome P450, P450RAI-2, which is predominantly expressed in the adult cerebellum and is responsible for all-trans-retinoic acid metabolism. Proc. Natl. Acad. Sci. USA 2000, 97, 6403–6408. [Google Scholar] [CrossRef]
- Topletz, A.R.; Tripathy, S.; Foti, R.S.; Shimshoni, J.A.; Nelson, W.L.; Isoherranen, N. Induction of CYP26A1 by metabolites of retinoic acid: Evidence that CYP26A1 is an important enzyme in the elimination of active retinoids. Mol. Pharmacol. 2015, 87, 430–441. [Google Scholar] [CrossRef]
- Topletz, A.R.; Thatcher, J.E.; Zelter, A.; Lutz, J.D.; Tay, S.; Nelson, W.L.; Isoherranen, N. Comparison of the function and expression of CYP26A1 and CYP26B1, the two retinoic acid hydroxylases. Biochem. Pharmacol. 2012, 83, 149–163. [Google Scholar] [CrossRef]
- Stevison, F.; Jing, J.; Tripathy, S.; Isoherranen, N. Chapter Eleven—Role of Retinoic Acid-Metabolizing Cytochrome P450s, CYP26, in Inflammation and Cancer. In Advances in Pharmacology; Hardwick, J.P., Ed.; Academic Press: Cambridge, MA, USA, 2015; pp. 373–412. [Google Scholar]
- Zhong, G.; Ortiz, D.; Zelter, A.; Nath, A.; Isoherranen, N. CYP26C1 Is a Hydroxylase of Multiple Active Retinoids and Interacts with Cellular Retinoic Acid Binding Proteins. Mol. Pharmacol. 2018, 93, 489–503. [Google Scholar] [CrossRef]
- Chithalen, J.V.; Luu, L.; Petkovich, M.; Jones, G. HPLC-MS/MS analysis of the products generated from all-trans-retinoic acid using recombinant human CYP26A. J. Lipid Res. 2002, 43, 1133–1142. [Google Scholar] [CrossRef]
- Samokyszyn, V.M.; Gall, W.E.; Zawada, G.; Freyaldenhoven, M.A.; Chen, G.; Mackenzie, P.I.; Tephly, T.R.; Radominska-Pandya, A. 4-Hydroxyretinoic Acid, a Novel Substrate for Human Liver Microsomal UDP-glucuronosyltransferase(s) and Recombinant UGT2B7. J. Biol. Chem. 2000, 275, 6908–6914. [Google Scholar] [CrossRef]
- Reijntjes, S.; Blentic, A.; Gale, E.; Maden, M. The control of morphogen signalling: Regulation of the synthesis and catabolism of retinoic acid in the developing embryo. Dev. Biol. 2005, 285, 224–237. [Google Scholar] [CrossRef] [PubMed]
- Pijnappel, W.W.M.; Hendriks, H.F.J.; Folkers, G.E.; van den Brink, C.E.; Dekker, E.J.; Edelenbosch, C.; van der Saag, P.T.; Durston, A.J. The retinoid ligand 4-oxo-retinoic acid is a highly active modulator of positional specification. Nature 1993, 366, 340–344. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, K. Teratogenic effects of retinoic acid and related substances on the early development of the zebrafish (Brachydanio rerio) as assessed by a novel scoring system. Toxicol. Vitr. 1995, 9, 267–283. [Google Scholar] [CrossRef]
- Niederreither, K.; Abu-Abed, S.; Schuhbaur, B.; Petkovich, M.; Chambon, P.; Dolle, P. Genetic evidence that oxidative derivatives of retinoic acid are not involved in retinoid signaling during mouse development. Nat. Genet. 2002, 31, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, J.E.; Theodosiou, M.; Chen, J.; Chevret, P.; Alvarez, S.; de Lera, A.R.; Laudet, V.; Croce, J.C.; Schubert, M. Lineage-specific duplication of amphioxus retinoic acid degrading enzymes (CYP26) resulted in sub-functionalization of patterning and homeostatic roles. BMC Evol. Biol. 2017, 17, 24. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, J.E.; Lahaye, F.; Croce, C.J.; Schubert, M. CYP26 function is required for the tissue-specific modulation of retinoic acid signaling during amphioxus development. Int. J. Dev. Biol. 2017, 61, 733–747. [Google Scholar] [CrossRef]
- White, R.J.; Schilling, T.F. How degrading: Cyp26s in hindbrain development. Dev. Dyn. 2008, 237, 2775–2790. [Google Scholar] [CrossRef]
- MacLean, G.; Abu-Abed, S.; Dolle, P.; Tahayatoc, A.; Chambonb, P.; Petkovich, M. Cloning of a novel retinoic-acid metabolizing cytochrome P450, Cyp26B1, and comparative expression analysis with Cyp26A1 during early murine development. Mech. Dev. 2001, 107, 195–201. [Google Scholar] [CrossRef]
- Tahayato, A.; Dolle, P.; Petkovich, M. Cyp26C1 encodes a novel retinoic acid-metabolizing enzyme expressed in the hindbrain, inner ear, first branchial arch and tooth buds during murine development. Gene Expr. Patterns 2003, 3, 449–454. [Google Scholar] [CrossRef]
- White, J.A.; Beckett-Jones, B.; Guo, Y.-D.; Dilworth, F.J.; Bonasoro, J.; Jones, G.; Petkovich, M. cDNA Cloning of Human Retinoic Acid-metabolizing Enzyme (hP450RAI) Identifies a Novel Family of Cytochromes P450 (CYP26). J. Biol. Chem. 1997, 272, 18538–18541. [Google Scholar] [CrossRef]
- Zhao, Q.; Dobbs-McAuliffe, B.; Linney, E. Expression of cyp26b1 during zebrafish early development. Gene Expr. Patterns 2005, 5, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, S.; Kawamura, K. Acquisition of Retinoic Acid Signaling Pathway and Innovation of the Chordate Body Plan. Zool. Sci. 2003, 20, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Blentic, A.; Gale, E.; Maden, M. Retinoic acid signalling centres in the avian embryo identified by sites of expression of synthesising and catabolising enzymes. Dev. Dyn. 2003, 227, 114–127. [Google Scholar] [CrossRef] [PubMed]
- De Roos, K.; Sonneveld, E.; Compaan, B.; Berge, D.T.; Durston, A.J.; van der Saag, P.T. Expression of retinoic acid 4-hydroxylase (CYP26) during mouse and Xenopus laevis embryogenesis. Mech. Dev. 1999, 82, 205–211. [Google Scholar] [CrossRef]
- Reijntjes, S.; Gale, E.; Maden, M. Expression of the retinoic acid catabolising enzyme CYP26B1 in the chick embryo and its regulation by retinoic acid. Gene Expr. Patterns 2003, 3, 621–627. [Google Scholar] [CrossRef]
- Reijntjes, S.; Gale, E.; Maden, M. Generating gradients of retinoic acid in the chick embryo: Cyp26C1 expression and a comparative analysis of the Cyp26 enzymes. Dev. Dyn. 2004, 230, 509–517. [Google Scholar] [CrossRef]
- Swindell, E.C.; Thaller, C.; Sockanathan, S.; Petkovich, M.; Jessell, T.M.; Eichele, G. Complementary Domains of Retinoic Acid Production and Degradation in the Early Chick Embryo. Dev. Biol. 1999, 216, 282–296. [Google Scholar] [CrossRef]
- McCaffery, P.; Wagner, O.N.J.; Petkovich, M.; Drager, U. Dorsal and ventral retinal territories defined by retinoic acid synthesis, break-down and nuclear receptor expression. Mech. Dev. 1999, 82, 119–130. [Google Scholar] [CrossRef]
- Pennimpede, T.; Cameron, D.A.; MacLean, G.A.; Li, H.; Abu-Abed, S.; Petkovich, M. The role of CYP26 enzymes in defining appropriate retinoic acid exposure during embryogenesis. Birth Defects Res. Part Clin. Mol. Teratol. 2010, 88, 883–894. [Google Scholar] [CrossRef]
- Romand, R.; Kondo, T.; Fraulob, V.; Petkovich, M.; Dollé, P.; Hashino, E. Dynamic expression of retinoic acid-synthesizing and -metabolizing enzymes in the developing mouse inner ear. J. Comp. Neurol. 2006, 496, 643–654. [Google Scholar] [CrossRef]
- Stoney, P.N.; Fragoso, Y.D.; Saeed, R.B.; Ashton, A.; Goodman, T.; Simons, C.; Gomaa, M.S. Expression of the retinoic acid catabolic enzyme CYP26B1 in the human brain to maintain signaling homeostasis. Brain Struct. Funct. 2016, 221, 3315–3326. [Google Scholar] [CrossRef] [PubMed]
- Cifelli, C.J.; Ross, A.C. Chronic Vitamin A Status and Acute Repletion with Retinyl Palmitate Are Determinants of the Distribution and Catabolism of all-trans-Retinoic Acid in Rats. J. Nutr. 2007, 137, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Zolfaghari, R.; Ross, A.C. Regulation of CYP26 (cytochrome P450RAI) mRNA expression and retinoic acid metabolism by retinoids and dietary vitamin A in liver of mice and rats. FASEB J. 2000, 14, 2119–2127. [Google Scholar] [CrossRef] [PubMed]
- Chapman, J.S.; Weiss, K.L.; Curley, R.W.; Highland, M.A.; Clagett-Dame, M. Hydrolysis of 4-HPR to atRA occurs in vivo but is not required for retinamide-induced apoptosis. Arch. Biochem. Biophys. 2003, 419, 234–243. [Google Scholar] [CrossRef]
- Wang, Y.; Zolfaghari, R.; Catharine Ross, A. Cloning of rat cytochrome P450RAI (CYP26) cDNA and regulation of its gene expression by all-trans-retinoic acid in vivo. Arch. Biochem. Biophys. 2002, 401, 235–243. [Google Scholar] [CrossRef]
- Zhang, Y.; Zolfaghari, R.; Ross, A.C. Multiple retinoic acid response elements cooperate to enhance the inducibility of CYP26A1 gene expression in liver. Gene 2010, 464, 32–43. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sakai, Y.; Meno, C.; Fujii, H.; Nishino, J.; Shiratori, H.; Saijoh, Y.; Rossant, J.; Hamada, H. The retinoic acid-inactivating enzyme CYP26 is essential for establishing an uneven distribution of retinoic acid along the anterio-posterior axis within the mouse embryo. Genes Dev. 2001, 15, 213–225. [Google Scholar] [CrossRef]
- White, R.J.; Nie, Q.; Lander, A.D.; Schilling, T.F. Complex. Regulation of cyp26a1 Creates a Robust Retinoic Acid Gradient in the Zebrafish Embryo. PLoS Biol. 2007, 5, e304. [Google Scholar] [CrossRef]
- Kinkel, M.D.; Sefton, E.M.; Kikuchi, Y.; Mizoguchi, T.; Ward, A.B.; Prince, V.E. Cyp26 enzymes function in endoderm to regulate pancreatic field size. Proc. Natl. Acad. Sci. USA 2009, 106, 7864–7869. [Google Scholar] [CrossRef] [PubMed]
- Janesick, A.; Tang, W.; Shioda, T.; Blumberg, B. RARγ is required for mesodermal gene expression prior to gastrulation in Xenopus. Development 2018, 145, dev147769. [Google Scholar] [CrossRef]
- Loudig, O.; Maclean, G.A.; Dore, N.L.; Luu, L.; Petkovich, M. Transcriptional co-operativity between distant retinoic acid response elements in regulation of Cyp26A1 inducibility. Biochem. J. 2005, 392, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Loudig, O.; Babichuk, C.; White, J.; Abu-Abed, S.; Mueller, C.; Petkovich, M. Cytochrome P450RAI(CYP26) Promoter: A Distinct Composite Retinoic Acid Response Element Underlies the Complex. Regulation of Retinoic Acid Metabolism. Mol. Endocrinol. 2000, 14, 1483–1497. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Tian, M.; Bao, J.; Xing, G.; Gu, X.; Gao, X.; Linney, E.; Zhao, Q. Retinoid regulation of the zebrafish cyp26a1 promoter. Dev. Dyn. 2008, 237, 3798–3808. [Google Scholar] [CrossRef] [PubMed]
- Lalevee, S.; Anno, Y.N.; Chatagnon, A.; Samarut, E.; Poch, O.; Laudet, V.; Benoit, G.; Lecompte, O.; Rochette-Egly, C. Genome-wide in silico identification of new conserved and functional retinoic acid receptors response elements (directs repeats separated by 5bp). J. Biol. Chem. 2011, 286, 33322–33334. [Google Scholar] [CrossRef] [PubMed]
- Quere, R.; Baudet, A.; Cassinat, B.; Bertrand, G.; Marti, J.; Manchon, L.; Piquemal, D.; Chomienne, C.; Commes, T. Pharmacogenomic analysis of acute promyelocytic leukemia cells highlights CYP26 cytochrome metabolism in differential all-trans retinoic acid sensitivity. Blood 2007, 109, 4450–4460. [Google Scholar] [CrossRef] [PubMed]
- Pozzi, S.; Rossetti, S.; Bistulfi, G.; Sacchi, N. RAR-mediated epigenetic control of the cytochrome P450 Cyp26a1 in embryocarcinoma cells. Oncogene 2006, 25, 1400–1407. [Google Scholar] [CrossRef]
- Kashyap, V.; Gudas, L.J. Epigenetic Regulatory Mechanisms Distinguish Retinoic Acid-mediated Transcriptional Responses in Stem Cells and Fibroblasts. J. Biol. Chem. 2010, 285, 14534–14548. [Google Scholar] [CrossRef]
- Delacroix, L.; Moutier, E.; Altobelli, G.; Legras, S.; Poch, O.; Choukrallah, M.-A.; Bertin, I.; Jost, B.; Davidson, I. Cell-Specific Interaction of Retinoic Acid Receptors with Target. Genes in Mouse Embryonic Fibroblasts and Embryonic Stem Cells. Mol. Cell. Biol. 2010, 30, 231–244. [Google Scholar] [CrossRef]
- Urvalek, A.M.; Gudas, L.J. Retinoic Acid and Histone Deacetylases Regulate Epigenetic Changes in Embryonic Stem Cells. J. Biol. Chem. 2014, 289, 19519–19530. [Google Scholar] [CrossRef]
- Zolfaghari, R.; Cifelli, C.J.; Lieu, S.O.; Chen, Q.; Li, N.-Q.; Ross, A.C. Lipopolysaccharide opposes the induction of CYP26A1 and CYP26B1 gene expression by retinoic acid in the rat liver in vivo. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G1029–G1036. [Google Scholar] [CrossRef][Green Version]
- Wu, L.; Ross, A.C. Acidic retinoids synergize with vitamin A to enhance retinol uptake and STRA6, LRAT, and CYP26B1 expression in neonatal lung. J. Lipid Res. 2010, 51, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Ocaya, P.A.; Elmabsout, A.A.; Olofsson, P.S.; Törmä, H.; Gidlöf, A.C.; Sirsjö, A. CYP26B1 Plays a Major Role in the Regulation of All-trans-Retinoic Acid Metabolism and Signaling in Human Aortic Smooth Muscle Cells. J. Vasc. Res. 2011, 48, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, H.; Yokota, A.; Ohoka, Y.; Iwata, M. Cyp26b1 regulates retinoic acid-dependent signals in T cells and its expression is inhibited by transforming growth factor-β. PLoS ONE 2011, 6, e16089. [Google Scholar] [CrossRef] [PubMed]
- Cambray, S.; Arber, C.; Little, G.; Dougalis, A.G.; de Paola, V.; Ungless, M.A.; Li, M.; Rodríguez, T.A. Activin induces cortical interneuron identity and differentiation in embryonic stem cell-derived telencephalic neural precursors. Nat. Commun. 2012, 3, 841. [Google Scholar] [CrossRef] [PubMed]
- Kipp, J.L.; Golebiowski, A.; Rodriguez, G.; Demczuk, M.; Kilen, S.M.; Mayo, K.E. Gene Expression Profiling Reveals Cyp26b1 to Be an Activin Regulated Gene Involved in Ovarian Granulosa Cell Proliferation. Endocrinology 2011, 152, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Kashimada, K.; Svingen, T.; Feng, C.-W.; Pelosi, E.; Bagheri-Fam, S.; Harley, V.R.; Schlessinger, D.; Bowles, J.; Koopman, P. Antagonistic regulation of Cyp26b1 by transcription factors SOX9/SF1 and FOXL2 during gonadal development in mice. FASEB J. 2011, 25, 3561–3569. [Google Scholar] [CrossRef]
- Tay, S.; Dickmann, L.; Dixit, V.; Isoherranen, N. A Comparison of the Roles of Peroxisome Proliferator-Activated Receptor and Retinoic Acid Receptor on CYP26 Regulation. Mol. Pharmacol. 2010, 77, 218–227. [Google Scholar] [CrossRef]
- Stoppie, P.; Borgers, M.; Borghgraef, P.; Dillen, L.; Goossens, J.; Sanz, G.; Szel, H.; van Hove, C.; van Nyen, G.; Nobels, G.; et al. R115866 Inhibits All-trans-Retinoic Acid Metabolism and Exerts Retinoidal Effects in Rodents. J. Pharmacol. Exp. Ther. 2000, 293, 304–312. [Google Scholar]
- Thatcher, J.E.; Buttrick, B.; Shaffer, S.A.; Shimshoni, J.A.; Goodlett, D.R.; Nelson, W.L.; Isoherranen, N. Substrate Specificity and Ligand Interactions of CYP26A1, the Human Liver Retinoic Acid Hydroxylase. Mol. Pharmacol. 2011, 80, 228–239. [Google Scholar] [CrossRef]
- Laue, K.; Pogoda, H.M.; Daniel, P.-á.; van-áHaeringen, A.; Alanay, Y.; von-áAmeln, S.; Rachwalsk, M.; Morgan, T.; Gray, M.-á.; Breuning, M.-á.; et al. Craniosynostosis and Multiple Skeletal Anomalies in Humans and Zebrafish Result from a Defect in the Localized Degradation of Retinoic Acid. Am. J. Hum. Genet. 2011, 89, 595–606. [Google Scholar] [CrossRef]
- Poorendonk, K.M.; Peterson-Maduro, J.; Renn, J.; Trowe, T.; Kranenbarg, S.; Winkler, C.; Schulte-Merker, S. Retinoic acid and Cyp26b1 are critical regulators of osteogenesis in the axial skeleton. Development 2008, 135, 3765–3774. [Google Scholar] [CrossRef] [PubMed]
- Roselló-Díez, A.; Arques, C.G.; Delgado, I.; Giovinazzo, G.; Torres, M. Diffusible signals and epigenetic timing cooperate in late proximo-distal limb patterning. Development 2014, 141, 1534–1543. [Google Scholar] [CrossRef] [PubMed]
- Nelson, C.H.; Buttrick, B.R.; Isoherranen, N. Therapeutic Potential of the Inhibition of the Retinoic Acid Hydroxylases CYP26A1 and CYP26B1 by Xenobiotics. Curr. Top Med. Chem. 2013, 13, 1402–1428. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-J.; Perera, L.; Coulter, S.J.; Mohrenweiser, H.W.; Jetten, A.; Goldstein, J.A. The discovery of new coding alleles of human CYP26A1 that are potentially defective in the metabolism of all-trans retinoic acid and their assessment in a recombinant cDNA expression system. Pharm. Genom. 2007, 17, 169–180. [Google Scholar] [CrossRef]
- Li, H.; Zhang, J.; Chen, S.; Wang, F.; Zhang, T.; Niswander, L. Genetic contribution of retinoid-related genes to neural tube defects. Hum. Mutat. 2018, 39, 550–562. [Google Scholar] [CrossRef]
- Rat, E.; Billaut-Laden, I.; Allorge, D.; Lo-Guidice, J.-M.; Tellier, M.; Cauffiez, C.; Jonckheere, N.; van Seuningen, I.; Lhermitte, M.; Romano, A.; et al. Evidence for a functional genetic polymorphism of the human retinoic acid–metabolizing enzyme CYP26A1, an enzyme that may be involved in spina bifida. Birth Defects Res. Part Clin. Mol. Teratol. 2006, 76, 491–498. [Google Scholar] [CrossRef]
- Wu, S.-J.; Chen, Y.-J.; Shieh, T.-Y.; Chen, C.-M.; Wang, Y.-Y.; Lee, K.-T.; Lin, Y.-M.; Chien, P.-H.; Chen, P.-H. Association Study between Novel CYP26 Polymorphisms and the Risk of Betel Quid-Related Malignant Oral Disorders. Sci. World J. 2015, 2015, 9. [Google Scholar] [CrossRef]
- Chen, P.-H.; Lee, K.-W.; Chen, C.-H.; Shieh, T.-Y.; Ho, P.-S.; Wang, S.-J.; Lee, C.-H.; Yang, S.-F.; Chen, M.-K.; Chiang, S.-L.; et al. CYP26B1 is a novel candidate gene for betel quid-related oral squamous cell carcinoma. Oral Oncol. 2011, 47, 594–600. [Google Scholar] [CrossRef]
- Chen, P.-H.; Lee, K.-W.; Hsu, C.-C.; Chen, J.Y.-F.; Wang, Y.-H.; Chen, K.-K.; Wang, H.-M.D.; Huang, H.-W.; Huang, B. Expression of a splice variant of CYP26B1 in betel quid-related oral cancer. Sci. World J. 2014, 2014, 810561. [Google Scholar] [CrossRef]
- Chang, J.; Zhong, R.; Tian, J.; Li, J.; Zhai, K.; Ke, J.; Lou, J.; Chen, W.; Zhu, B.; Shen, N.; et al. Exome-wide analyses identify low-frequency variant in CYP26B1 and additional coding variants associated with esophageal squamous cell carcinoma. Nat. Genet. 2018, 50, 338–343. [Google Scholar] [CrossRef]
- Meire, F.; Delpierre, I.; Brachet, C.; Roulez, F.; van Nechel, C.; Depasse, F.; Christophe, C.; Menten, B.; de Baere, E. Nonsyndromic bilateral and unilateral optic nerve aplasia: First familial occurrence and potential implication of CYP26A1 and CYP26C1 genes. Mol. Vis. 2011, 17, 2072–2079. [Google Scholar] [PubMed]
- Nilsson, O.; Isoherranen, N.; Guo, M.H.; Lui, J.C.; Jee, Y.H.; Guttmann-Bauman, I.; Acerini, C.; Lee, W.; Allikmets, R.; Yanovski, J.A.; et al. Accelerated Skeletal Maturation in Disorders of Retinoic Acid Metabolism: A Case Report and Focused Review of the Literature. Horm. Metab. Res. 2016, 48, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Elmabsout, A.A.; Kumawat, A.; Saenz-Méndez, P.; Krivospitskaya, O.; Sävenstrand, H.; Olofsson, P.S.; Eriksson, L.A.; Strid, Å.; Valen, G.; Törmä, H.; et al. Cloning and Functional Studies of a Splice Variant of CYP26B1 Expressed in Vascular Cells. PLoS ONE 2012, 7, e36839. [Google Scholar] [CrossRef] [PubMed]
- Krivospitskaya, O.; Elmabsout, A.A.; Sundman, E.; Söderström, Å.L.; Ovchinnikova, O.; Gidlöf, C.A.; Scherbak, N.; Norata, G.D.; Samnegård, A.; Törmä, H.; et al. A CYP26B1 Polymorphism Enhances Retinoic Acid Catabolism and May Aggravate Atherosclerosis. Mol. Med. 2012, 18, 712–718. [Google Scholar] [CrossRef] [PubMed]
- Fransén, K.; Franzén, P.; Magnuson, A.; Elmabsout, A.A.; Nyhlin, N.; Wickbom, A.; Curman, B.; Törkvist, L.; D’Amato, M.; Bohr, J.; et al. Polymorphism in the Retinoic Acid Metabolizing Enzyme CYP26B1 and the Development of Crohn’s Disease. PLoS ONE 2013, 8, e72739. [Google Scholar] [CrossRef] [PubMed]
- Morton, J.E.V.; Frentz, S.; Morgan, T.; Sutherland-Smith, A.J.; Robertson, S.P. Biallelic mutations in CYP26B1: A differential diagnosis for Pfeiffer and Antley–Bixler syndromes. Am. J. Med. Genet. Part 2016, 170, 2706–2710. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Lopes, F.; Soares, G.; Farrell, S.A.; Nelson, C.; Qiao, Y.; Martel, S.; Badukke, C.; Bessa, C.; Ylstra, B.; et al. Phenotypic and functional consequences of haploinsufficiency of genes from exocyst and retinoic acid pathway due to a recurrent microdeletion of 2p13.2. Orphanet J. Rare Dis. 2013, 8, 100. [Google Scholar] [CrossRef]
- Adachi, M.; Tachibana, K.; Asakura, Y.; Yamamoto, T.; Hanaki, K.; Oka, A. Compound heterozygous mutations of cytochrome P450 oxidoreductase gene (POR) in two patients with Antley–Bixler syndrome. Am. J. Med. Genet. Part 2004, 128, 333–339. [Google Scholar] [CrossRef]
- Flück, C.E.; Tajima, T.; Pandey, A.V.; Arlt, W.; Okuhara, K.; Verge, C.F.; Jabs, E.W.; Mendonça, B.B.; Fujieda, K.; Miller, W.L. Mutant P450 oxidoreductase causes disordered steroidogenesis with and without Antley-Bixler syndrome. Nat. Genet. 2004, 36, 228–230. [Google Scholar] [CrossRef]
- Fukami, M.; Ogata, T. Cytochrome P450 oxidoreductase deficiency: Rare congenital disorder leading to skeletal malformations and steroidogenic defects. Pediatrics Int. 2014, 56, 805–808. [Google Scholar] [CrossRef]
- Montalbano, A.; Juergensen, L.; Roeth, R.; Weiss, B.; Fukami, M.; Fricke-Otto, S.; Binder, G.; Ogata, T.; Decker, E.; Nuernberg, G.; et al. Retinoic acid catabolizing enzyme CYP26C1 is a genetic modifier in SHOX deficiency. EMBO Mol. Med. 2016, 8, 1455–1469. [Google Scholar] [CrossRef] [PubMed]
- Montalbano, A.; Juergensen, L.; Fukami, M.; Thiel, C.T.; Hauer, N.H.; Roeth, R.; Weiss, B.; Naiki, Y.; Ogata, T.; Hassel, D.; et al. Functional missense and splicing variants in the retinoic acid catabolizing enzyme CYP26C1 in idiopathic short stature. Eur. J. Hum. Genet. 2018, 26, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- Slavotinek, A.M.; Mehrotra, P.; Nazarenko, I.; Tang, P.L.-F.; Lao, R.; Cameron, D.; Li, B.; Chu, C.; Chou, C.; Marqueling, A.L.; et al. Focal facial dermal dysplasia, type IV, is caused by mutations in CYP26C1. Hum. Mol. Genet. 2012, 22, 696–703. [Google Scholar] [CrossRef] [PubMed]
- Uehara, M.; Yashiro, K.; Takaoka, K.; Yamamoto, M.; Hamada, H. Removal of maternal retinoic acid by embryonic CYP26 is required for correct Nodal expression during early embryonic patterning. Genes Dev. 2009, 23, 1689–1698. [Google Scholar] [CrossRef] [PubMed]
- Ivins, S.; Lammerts van Beuren, K.; Roberts, C.; James, C.; Lindsay, E.; Baldini, A.; Ataliotis, P.; Scambler, P.J. Microarray analysis detects differentially expressed genes in the pharyngeal region of mice lacking Tbx1. Dev. Biol. 2005, 285, 554–569. [Google Scholar] [CrossRef] [PubMed]
- Otto, D.M.E.; Henderson, C.J.; Carrie, D.; Davey, M.; Gundersen, T.E.; Blomhoff, R.; Adams, R.H.; Tickle, C.; Wolf, C.R. Identification of Novel Roles of the Cytochrome P450 System in Early Embryogenesis: Effects on Vasculogenesis and Retinoic Acid Homeostasis. Mol. Cell. Biol. 2003, 23, 6103–6116. [Google Scholar] [CrossRef]
- Ribes, V.; le Roux, I.; Rhinn, M.; Schuhbaur, B.; Dolle, P. Early mouse caudal development relies on crosstalk between retinoic acid, Shh and Fgf signalling pathways. Development 2009, 136, 665–676. [Google Scholar] [CrossRef]
- Shen, A.L.; O’Leary, K.A.; Kasper, C.B. Association of Multiple Developmental Defects and Embryonic Lethality with Loss of Microsomal NADPH-Cytochrome P450 Oxidoreductase. J. Biol. Chem. 2002, 277, 6536–6541. [Google Scholar] [CrossRef]
- Abu-Abed, S.; Dolle, P.; Metzger, D.; Beckett, B.; Chambon, P.; Petkovich, M. The retinoic acid-metabolizing enzyme, CYP26A1, is essential for normal hindbrain patterning, vertebral identity, and development of posterior structures. Genes Dev. 2001, 15, 226–240. [Google Scholar] [CrossRef]
- Yoshikawa, Y.; Fujimori, T.; McMahon, A.P.; Takada, S. Evidence That Absence ofWnt-3aSignaling Promotes Neuralization Instead of Paraxial Mesoderm Development in the Mouse. Dev. Biol. 1997, 183, 234–242. [Google Scholar] [CrossRef]
- Yamaguchi, T.P.; Takada, S.; Yoshikawa, Y.; Wu, N.; McMahon, A.P. T (Brachyury) is a direct target of Wnt3a during paraxial mesoderm specification. Genes Dev. 1999, 13, 3185–3190. [Google Scholar] [CrossRef] [PubMed]
- Takada, S.; Stark, K.L.; Shea, M.J.; Vassileva, G.; McMahon, J.A.; McMahon, A.P. Wnt-3a regulates somite and tailbud formation in the mouse embryo. Genes Dev. 1994, 8, 174–189. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, D.G.; Bhatt, S.; Herrmann, B.G. Expression pattern of the mouse T gene and its role in mesoderm formation. Nature 1990, 343, 657–659. [Google Scholar] [CrossRef] [PubMed]
- Abu-Abed, S.; Dollé, P.; Metzger, D.; Wood, C.; MacLean, G.; Chambon, P.; Petkovich, M. Developing with lethal RA levels: Genetic ablation of Rarg can restore the viability of mice lacking Cyp26a1. Development 2003, 130, 1449–1459. [Google Scholar] [CrossRef]
- Abu-Abed, S.S.; Beckett, B.R.; Chiba, H.; Chithalen, J.V.; Jones, G.; Metzger, D.; Chambon, P.; Petkovich, M. Mouse P450RAI (CYP26) Expression and Retinoic Acid-inducible Retinoic Acid Metabolism in F9 Cells Are Regulated by Retinoic Acid Receptor γ and Retinoid X Receptor α. J. Biol. Chem. 1998, 273, 2409–2415. [Google Scholar] [CrossRef]
- Ribes, V.; Fraulob, V.; Petkovich, M.; Dollé, P. The oxidizing enzyme CYP26a1 tightly regulates the availability of retinoic acid in the gastrulating mouse embryo to ensure proper head development and vasculogenesis. Dev. Dyn. 2007, 236, 644–653. [Google Scholar] [CrossRef] [PubMed]
- Emoto, Y.; Wada, H.; Okamoto, H.; Kudo, A.; Imai, Y. Retinoic acid-metabolizing enzyme Cyp26a1 is essential for determining territories of hindbrain and spinal cord in zebrafish. Dev. Biol. 2005, 278, 415–427. [Google Scholar] [CrossRef] [PubMed]
- Uehara, M.; Yashiro, K.; Mamiya, S.; Nishino, J.; Chambon, P.; Dolle, P.; Sakai, Y. CYP26A1 and CYP26C1 cooperatively regulate anterior-posterior patterning of the developing brain and the production of migratory cranial neural crest cells in the mouse. Dev. Biol. 2007, 302, 399–411. [Google Scholar] [CrossRef]
- Ribes, V.; Otto, D.M.E.; Dickmann, L.; Schmidt, K.; Schuhbaur, B.; Henderson, C.; Blomhoff, R.; Wolf, C.R.; Tickle, C.; Dollé, P. Rescue of cytochrome P450 oxidoreductase (Por) mouse mutants reveals functions in vasculogenesis, brain and limb patterning linked to retinoic acid homeostasis. Dev. Biol. 2007, 303, 66–81. [Google Scholar] [CrossRef]
- Dranse, H.J.; Sampaio, A.V.; Petkovich, M.; Underhill, T.M. Genetic deletion of Cyp26b1 negatively impacts limb skeletogenesis by inhibiting chondrogenesis. J. Cell Sci. 2011, 124, 2723–2734. [Google Scholar] [CrossRef] [PubMed]
- Pennimpede, T.; Cameron, D.A.; MacLean, G.A.; Petkovich, M. Analysis of Cyp26b1/Rarg compound-null mice reveals two genetically separable effects of retinoic acid on limb outgrowth. Dev. Biol. 2010, 339, 179–186. [Google Scholar] [CrossRef]
- Probst, S.; Kraemer, C.; Demougin, P.; Sheth, R.; Martin, G.R.; Shiratori, H.; Hamada, H.; Iber, D.; Zeller, R.; Zuniga, A.E. SHH propagates distal limb bud development by enhancing CYP26B1-mediated retinoic acid clearance via AER-FGF signalling. Development 2011, 138, 1913–1923. [Google Scholar] [CrossRef] [PubMed]
- Yashiro, K.; Zhao, X.; Uehara, M.; Yamashita, K.; Nishijima, M.; Nishino, J.; Saijoh, Y.; Sakai, Y.; Hamada, H. Regulation of Retinoic Acid Distribution Is Required for Proximodistal Patterning and Outgrowth of the Developing Mouse Limb. Dev. Cell 2004, 6, 411–422. [Google Scholar] [CrossRef]
- MacLean, G.; Doll, P.; Petkovich, M. Genetic disruption of CYP26B1 severely affects development of neural crest derived head structures, but does not compromise hindbrain patterning. Dev. Dyn. 2009, 238, 732–745. [Google Scholar] [CrossRef] [PubMed]
- Okano, J.; Kimura, W.; Papaionnou, V.E.; Miura, N.; Yamada, G.; Shiota, K.; Sakai, Y. The regulation of endogenous retinoic acid level through CYP26B1 is required for elevation of palatal shelves. Dev. Dyn. 2012, 241, 1744–1756. [Google Scholar] [CrossRef] [PubMed]
- Reijntjes, S.; Rodaway, A.; Maden, M. The retinoic acid metabolising gene, CYP26B1, patterns the cartilaginous cranial neural crest in zebrafish. Int. J. Dev. Biol. 2007, 51, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Kudoh, T.; Wilson, S.W.; Dawid, I.B. Distinct roles for Fgf, Wnt and retinoic acid in posteriorizing the neural ectoderm. Development 2002, 129, 4335–4346. [Google Scholar]
- Hernandez, R.E.; Putzke, A.P.; Myers, J.P.; Margaretha, L.; Moens, C.B. Cyp26 enzymes generate the retinoic acid response pattern necessary for hindbrain development. Development 2007, 134, 177–187. [Google Scholar] [CrossRef]
- Rydeen, A.B.; Waxman, J.S. Cyp26 enzymes are required to balance the cardiac and vascular lineages within the anterior lateral plate mesoderm. Development 2014, 141, 1638–1648. [Google Scholar] [CrossRef]
- Okuda, Y.; Ogura, E.; Kondoh, H.; Kamachi, Y. B1 SOX Coordinate Cell Specification with Patterning and Morphogenesis in the Early Zebrafish Embryo. PLoS Genet. 2010, 6, e1000936. [Google Scholar] [CrossRef]
- Chen, C.; Stedman, A.; Havis, E.; Anselme, I.; Onichtchouk, D.; Giudicelli, F.; Schneider-Maunoury, S. Initiation of cyp26a1 Expression in the Zebrafish Anterior Neural Plate by a Novel Cis-Acting Element. PLoS ONE 2016, 11, e0150639. [Google Scholar] [CrossRef] [PubMed]
- Maurus, D.; Harris, W.A. Zic-associated holoprosencephaly: Zebrafish Zic1 controls midline formation and forebrain patterning by regulating Nodal, Hedgehog, and retinoic acid signaling. Genes Dev. 2009, 23, 1461–1473. [Google Scholar] [CrossRef] [PubMed]
- El-Jaick, K.B.; Powers, S.E.; Bartholin, L.; Myers, K.R.; Hahn, J.; Orioli, I.M.; Ouspenskaia, M.; Lacbawan, F.; Roessler, E.; Wotton, D.; et al. Functional analysis of mutations in TGIF associated with holoprosencephaly. Mol. Genet. Metab. 2007, 90, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Gripp, K.W.; Wotton, D.; Edwards, M.C.; Roessler, E.; Ades, L.; Meinecke, P.; Richieri-Costa, A.; Zackai, E.H.; Massagué, J.; Muenke, M.; et al. Mutations in TGIF cause holoprosencephaly and link NODAL signalling to human neural axis determination. Nat. Genet. 2000, 25, 205–208. [Google Scholar] [CrossRef]
- Aguilella, C.; Dubourg, C.; Attia-Sobol, J.; Vigneron, J.; Blayau, M.; Pasquier, L.; Lazaro, L.; Odent, S.; David, V. Molecular screening of the TGIF gene in holoprosencephaly: Identification of two novel mutations. Hum. Genet. 2003, 112, 131–134. [Google Scholar] [CrossRef]
- Kantaputra, P.N.; Limwongse, C.; Tochareontanaphol, C.; Mutirangura, A.; Mevatee, U.; Praphanphoj, V. Contiguous gene syndrome of holoprosencephaly and hypotrichosis simplex: Association with an 18p11.3 deletion. Am. J. Med. Genet. Part 2006, 140, 2598–2602. [Google Scholar] [CrossRef]
- Chen, M.; Kuo, S.-J.; Liu, C.-S.; Chen, W.-L.; Ko, T.-M.; Chen, T.-H.; Chang, S.-P.; Huang, C.-H.; Chang, Y.-Y.; Wang, B.-T. A novel heterozygous missense mutation 377T > C (V126A) of TGIF gene in a family segregated with holoprosencephaly and moyamoya disease. Prenat. Diagn. 2006, 26, 226–230. [Google Scholar] [CrossRef]
- Wallis, D.; Muenke, M. Mutations in holoprosencephaly. Hum. Mutat. 2000, 16, 99–108. [Google Scholar] [CrossRef]
- Ferrand, N.; Demange, C.; Prunier, C.; Seo, S.R.; Atfi, A. A mechanism for mutational inactivation of the homeodomain protein TGIF in holoprosencephaly. FASEB J. 2007, 21, 488–496. [Google Scholar] [CrossRef]
- Bartholin, L.; Powers, S.E.; Melhuish, T.A.; Lasse, S.; Weinstein, M.; Wotton, D. TGIF Inhibits Retinoid Signaling. Mol. Cell. Biol. 2006, 26, 990–1001. [Google Scholar] [CrossRef]
- Jin, J.-Z.; Gu, S.; McKinney, P.; Ding, J. Expression and functional analysis of Tgif during mouse midline development. Dev. Dyn. 2006, 235, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Walsh, C.A. Targeted Disruption of Tgif, the Mouse Ortholog of a Human Holoprosencephaly Gene, Does Not. Result in Holoprosencephaly in Mice. Mol. Cell. Biol. 2005, 25, 3639–3647. [Google Scholar] [CrossRef] [PubMed]
- Kuang, C.; Xiao, Y.; Yang, L.; Chen, Q.; Wang, Z.; Conway, S.J.; Chen, Y. Intragenic deletion of Tgif causes defectsin brain development. Hum. Mol. Genet. 2006, 15, 3508–3519. [Google Scholar] [CrossRef] [PubMed]
- Gongal, P.A.; Waskiewicz, A.J. Zebrafish model of holoprosencephaly demonstrates a key role for TGIF in regulating retinoic acid metabolism. Hum. Mol. Genet. 2007, 17, 525–538. [Google Scholar] [CrossRef]
- Bertolino, E.; Reimund, B.; Wildt-Perinic, D.; Clerc, R.G. A Novel Homeobox Protein Which Recognizes a TGT Core and Functionally Interferes with a Retinoid-responsive Motif. J. Biol. Chem. 1995, 270, 31178–31188. [Google Scholar] [CrossRef]
- Ribes, V.; Wang, Z.; Dollé, P.; Niederreither, K. Retinaldehyde dehydrogenase 2 (RALDH2)-mediated retinoic acid synthesis regulates early mouse embryonic forebrain development by controlling FGF and sonic hedgehog signaling. Development 2006, 133, 351–361. [Google Scholar] [CrossRef]
- Halilagic, A.; Ribes, V.; Ghyselinck, N.B.; Zile, M.H.; Dollé, P.; Studer, M. Retinoids control anterior and dorsal properties in the developing forebrain. Dev. Biol. 2007, 303, 362–375. [Google Scholar] [CrossRef]
- Molotkova, N.; Molotkov, A.; Duester, G. Role of retinoic acid during forebrain development begins late when Raldh3 generates retinoic acid in the ventral subventricular zone. Dev. Biol. 2007, 303, 601–610. [Google Scholar] [CrossRef]
- Gupta, S.; Sen, J. Retinoic acid signaling regulates development of the dorsal forebrain midline and the choroid plexus in the chick. Development 2015, 142, 1293–1298. [Google Scholar] [CrossRef]
- Mallo, M. Reassessing the Role of Hox Genes during Vertebrate Development and Evolution. Trends Genet. 2018, 34, 209–217. [Google Scholar] [CrossRef]
- Lescroat, F.; Zaffran, S. Hox and Tale transcription factors in heart development and disease. Int. J. Dev. Biol. 2018, 62, 837–846. [Google Scholar] [CrossRef] [PubMed]
- Roux, M.; Zaffran, S. Hox Genes in Cardiovascular Development and Diseases. J. Dev. Biol. 2016, 4, 14. [Google Scholar] [CrossRef] [PubMed]
- Parker, H.J.; Bronner, M.E.; Krumlauf, R. The vertebrate Hox gene regulatory network for hindbrain segmentation: Evolution and diversification. Bioessays 2016, 38, 526–538. [Google Scholar] [CrossRef] [PubMed]
- Krumlauf, R. Chapter Thirty-Four—Hox Genes and the Hindbrain: A Study in Segments. In Current Topics in Developmental Biology; Wassarman, P.M., Ed.; Academic Press: Cambridge, MA, USA, 2016; pp. 581–596. [Google Scholar]
- Parker, H.J.; Pushel, I.; Krumlauf, R. Coupling the roles of Hox genes to regulatory networks patterning cranial neural crest. Dev. Biol. 2018, 444, S67–S78. [Google Scholar] [CrossRef]
- Parker, H.J.; Krumlauf, R. Segmental arithmetic: Summing up the Hox gene regulatory network for hindbrain development in chordates. Wiley Interdiscip. Rev. Dev. Biol. 2017, 6, e286. [Google Scholar] [CrossRef]
- Alexander, T.; Nolte, C.; Krumlauf, R. Hox Genes and Segmentation of the Hindbrain and Axial Skeleton. Annu. Rev. Cell Dev. Biol. 2009, 25, 431–456. [Google Scholar] [CrossRef]
- Tümpel, S.; Wiedemann, L.M.; Krumlauf, R. Chapter 8 Hox Genes and Segmentation of the Vertebrate Hindbrain. In Current Topics in Developmental Biology; Academic Press: Cambridge, MA, USA, 2019; pp. 103–137. [Google Scholar]
- Prin, F.; Serpente, P.; Itasaki, N.; Gould, A.P. Hox proteins drive cell segregation and non-autonomous apical remodelling during hindbrain segmentation. Development 2014, 141, 1492–1502. [Google Scholar] [CrossRef]
- Shimozono, S.; Iimura, T.; Kitaguchi, T.; Higashijima, S.-I.; Miyawaki, A. Visualization of an endogenous retinoic acid gradient across embryonic development. Nature 2013, 496, 363. [Google Scholar] [CrossRef]
- Maves, L.; Kimmel, C.B. Dynamic and sequential patterning of the zebrafish posterior hindbrain by retinoic acid. Dev. Biol. 2005, 285, 593–605. [Google Scholar] [CrossRef]
- Sirbu, I.O.; Gresh, L.; Barra, J.; Duester, G. Shifting boundaries of retinoic acid activity control hindbrain segmental gene expression. Development 2005, 132, 2611–2622. [Google Scholar] [CrossRef]
- Shiotsugu, J.; Katsuyama, Y.; Arima, K.; Baxter, A.; Koide, T.; Song, J.; Chandraratna, R.A.S.; Blumberg, B. Multiple points of interaction between retinoic acid and FGF signaling during embryonic axis formation. Development 2004, 131, 2653–2667. [Google Scholar] [CrossRef] [PubMed]
- Schilling, T.F.; Sosnik, J.; Nie, Q. Visualizing retinoic acid morphogen gradients. Methods Cell Biol. 2016, 133, 139–163. [Google Scholar] [PubMed]
- Schilling, T.F.; Nie, Q.; Lander, A.D. Dynamics and precision in retinoic acid morphogen gradients. Curr. Opin. Genet. Dev. 2012, 22, 562–569. [Google Scholar] [CrossRef]
- Labalette, C.; Wassef, M.A.; Desmarquet-Trin, C.; Bouchoucha, Y.X.; Men, J.L.; Charnay, P.; Gilardi-Hebenstreit, P. Molecular dissection of segment formation in the developing hindbrain. Development 2015, 142, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Radtke, K.; Zheng, L.; Cai, A.Q.; Schilling, T.F.; Nie, Q. Noise drives sharpening of gene expression boundaries in the zebrafish hindbrain. Mol. Syst. Biol. 2012, 8, 613. [Google Scholar] [CrossRef] [PubMed]
- Niethamer, T.K.; Bush, J.Q. Getting direction(s): The Eph/ephrin signaling system in cell positioning. Dev. Biol. 2019, 447, 42–57. [Google Scholar] [CrossRef]
- Cayuso, J.; Xu, Q.; Wilkinson, D.G. Mechanisms of boundary formation by Eph receptor and ephrin signaling. Dev. Biol. 2015, 401, 122–131. [Google Scholar] [CrossRef]
- Kitazawa, T.; Rijli, F.M. Integrating into the Rhombomere Community across the Border. Dev. Cell 2018, 45, 546–548. [Google Scholar] [CrossRef]
- Addison, M.; Xu, Q.; Cayuso, J.; Wilkinson, D.G. Cell Identity Switching Regulated by Retinoic Acid Signaling Maintains Homogeneous Segments in the Hindbrain. Dev. Cell 2018, 45, 606–620. [Google Scholar] [CrossRef]
- Wilkinson, D.G. Establishing sharp and homogeneous segments in the hindbrain. F1000Research 2018, 7. [Google Scholar] [CrossRef]
- Pourquié, O. Vertebrate Segmentation: From Cyclic Gene Networks to Scoliosis. Cell 2011, 145, 650–663. [Google Scholar] [CrossRef] [PubMed]
- Aulehla, A.; Pourquié, O. Signaling Gradients during Paraxial Mesoderm Development. Cold Spring Harb. Perspect. Biol. 2010, 2, a000869. [Google Scholar] [CrossRef] [PubMed]
- Sirbu, I.O.; Duester, G. Retinoic-acid signalling in node ectoderm and posterior neural plate directs left–right patterning of somitic mesoderm. Nat. Cell Biol. 2006, 8, 271–277. [Google Scholar] [CrossRef] [PubMed]
- El Shahawy, M.; Reibring, C.-G.; Hallberg, K.; Neben, C.L.; Marangoni, P.; Harfe, B.D.; Klein, O.D.; Linde, A.; Gritli-Linde, A. Sonic Hedgehog Signaling Is Required for Cyp26 Expression during Embryonic Development. Int. J. Mol. Sci. 2019, 20, 2275. [Google Scholar] [CrossRef]
- Wilson, V.; Olivera-Martinez, I.; Storey, K.G. Stem cells, signals and vertebrate body axis extension. Development 2009, 136, 1591–1604. [Google Scholar] [CrossRef]
- Tzouanacou, E.; Wegener, A.; Wymeersch, F.J.; Wilson, V.; Nicolas, J.-F. Redefining the Progression of Lineage Segregations during Mammalian Embryogenesis by Clonal Analysis. Dev. Cell 2009, 17, 365–376. [Google Scholar] [CrossRef]
- Henrique, D.; Abranches, E.; Verrier, L.; Storey, K.G. Neuromesodermal progenitors and the making of the spinal cord. Development 2015, 142, 2864–2875. [Google Scholar] [CrossRef]
- Kimelman, D. Chapter Twenty-Nine—Tales of Tails (and Trunks): Forming the Posterior Body in Vertebrate Embryos. In Current Topics in Developmental Biology; Wassarman, P.M., Ed.; Academic Press: Cambridge, MA, USA, 2016; pp. 517–536. [Google Scholar]
- Attardi, A.; Fulton, T.; Florescu, M.; Shah, G.; Muresan, L.; Lenz, M.O.; Lancaster, C.; Huisken, J.; van Oudenaarden, A.; Steventon, B. Neuromesodermal progenitors are a conserved source of spinal cord with divergent growth dynamics. Development 2018, 145, dev166728. [Google Scholar] [CrossRef]
- Cambray, N.; Wilson, V. Axial progenitors with extensive potency are localised to the mouse chordoneural hinge. Development 2002, 129, 4855–4866. [Google Scholar]
- Cambray, N.; Wilson, V. Two distinct sources for a population of maturing axial progenitors. Development 2007, 134, 2829–2840. [Google Scholar] [CrossRef]
- Delfino-Machín, M.; Lunn, J.S.; Breitkreuz, D.N.; Akai, J.; Storey, K.G. Specification and maintenance of the spinal cord stem zone. Development 2005, 132, 4273–4283. [Google Scholar] [CrossRef] [PubMed]
- Olivera-Martinez, I.; Harada, H.; Halley, P.A.; Storey, K.G. Loss of FGF-Dependent Mesoderm Identity and Rise of Endogenous Retinoid Signalling Determine Cessation of Body Axis Elongation. PLoS Biol. 2012, 10, e1001415. [Google Scholar] [CrossRef] [PubMed]
- Kondoh, H.; Takemoto, T. Axial stem cells deriving both posterior neural and mesodermal tissues during gastrulation. Curr. Opin. Genet. Dev. 2012, 22, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Kondoh, H.; Takada, S.; Takemoto, T. Axial level-dependent molecular and cellular mechanisms underlying the genesis of the embryonic neural plate. Dev. Growth Differ. 2016, 58, 427–436. [Google Scholar] [CrossRef]
- Del Corral, R.D.; Olivera-Martinez, I.; Goriely, A.; Gale, E.; Maden, M.; Storey, K. Opposing FGF and Retinoid Pathways Control. Ventral Neural Pattern, Neuronal Differentiation, and Segmentation during Body Axis Extension. Neuron 2003, 40, 65–79. [Google Scholar] [CrossRef]
- Martin, B.L.; Kimelman, D. Brachyury establishes the embryonic mesodermal progenitor niche. Genes Dev. 2010, 24, 2778–2783. [Google Scholar] [CrossRef]
- Martin, B.L.; Kimelman, D. Canonical Wnt Signaling Dynamically Controls Multiple Stem Cell Fate Decisions during Vertebrate Body Formation. Dev. Cell 2012, 22, 223–232. [Google Scholar] [CrossRef]
- Garriock, R.J.; Chalamalasetty, R.B.; Kennedy, M.W.; Canizales, L.C.; Lewandoski, M.; Yamaguchi, T.P. Lineage tracing of neuromesodermal progenitors reveals novel Wnt-dependent roles in trunk progenitor cell maintenance and differentiation. Development 2015, 142, 1628–1638. [Google Scholar] [CrossRef]
- Savory, J.G.A.; Bouchard, N.; Pierre, V.; Rijli, F.M.; de Repentigny, Y.; Kothary, R.; Lohnes, D. Cdx2 regulation of posterior development through non-Hox targets. Development 2009, 136, 4099–4110. [Google Scholar] [CrossRef]
- Young, T.; Rowland, J.E.; van de Ven, C.; Bialecka, M.; Novoa, A.; Carapuco, M.; van Nes, J.; de Graaff, W.; Duluc, I.; Freund, J.-N.; et al. Cdx and Hox Genes Differentially Regulate Posterior Axial Growth in Mammalian Embryos. Dev. Cell 2009, 17, 516–526. [Google Scholar] [CrossRef]
- Young, T.; Deschamps, J. Chapter 8 Hox, Cdx, and Anteroposterior Patterning in the Mouse Embryo. In Current Topics in Developmental Biology; Academic Press: Cambridge, MA, USA, 2009; pp. 235–255. [Google Scholar]
- Koch, F.; Scholze, M.; Wittler, L.; Schifferl, D.; Sudheer, S.; Grote, P.; Timmermann, B.; Macura, K.; Herrmann, B.G. Antagonistic Activities of Sox2 and Brachyury Control. the Fate Choice of Neuro-Mesodermal Progenitors. Dev. Cell 2017, 42, 514–526. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, T.J.; Duester, G. Mechanisms of retinoic acid signalling and its roles in organ and limb development. Nat. Rev. Mol. Cell Biol. 2015, 16, 110–123. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, T.J.; Colas, A.; Duester, G. Early molecular events during retinoic acid induced differentiation of neuromesodermal progenitors. Biol. Open 2016, 5, 1821–1833. [Google Scholar] [CrossRef] [PubMed]
- Goto, H.; Kimmey, S.C.; Row, R.H.; Matus, D.Q.; Martin, B.L. FGF and canonical Wnt signaling cooperate to induce paraxial mesoderm from tailbud neuromesodermal progenitors through regulation of a two-step epithelial to mesenchymal transition. Development 2017, 144, 1412–1424. [Google Scholar] [CrossRef] [PubMed]
- Neijts, R.; Amin, S.; van Rooijen, C.; Deschamps, J. Cdx is crucial for the timing mechanism driving colinear Hox activation and defines a trunk segment in the Hox cluster topology. Dev. Biol. 2017, 422, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Martin, B.L. Factors that coordinate mesoderm specification from neuromesodermal progenitors with segmentation during vertebrate axial extension. Semin. Cell Dev. Biol. 2016, 49, 59–67. [Google Scholar] [CrossRef]
- Steventon, B.; Martinez Arias, A. Evo-engineering and the cellular and molecular origins of the vertebrate spinal cord. Dev. Biol. 2017, 432, 3–13. [Google Scholar] [CrossRef]
- Diez del Corral, R.; Morales, A.V. The Multiple Roles of FGF Signaling in the Developing Spinal Cord. Front. Cell Dev. Biol. 2017, 5, 58. [Google Scholar] [CrossRef]
- Wymeersch, F.J.; Skylaki, S.; Huang, Y.; Watson, J.A.; Economou, C.; Marek-Johnston, C.; Tomlinson, S.R.; Wilson, V. Transcriptionally dynamic progenitor populations organised around a stable niche drive axial patterning. Development 2019, 146, dev168161. [Google Scholar] [CrossRef]
- Gouti, M.; Tsakiridis, A.; Wymeersch, F.J.; Huang, Y.; Kleinjung, J.; Wilson, V.; Briscoe, J. In Vitro Generation of Neuromesodermal Progenitors Reveals Distinct Roles for Wnt Signalling in the Specification of Spinal Cord and Paraxial Mesoderm Identity. PLoS Biol. 2014, 12, e1001937. [Google Scholar] [CrossRef]
- Turner, D.A.; Hayward, P.C.; Baillie-Johnson, P.; Rué, P.; Broome, R.; Faunes, F.; Arias, A.M. Wnt/β-catenin and FGF signalling direct the specification and maintenance of a neuromesodermal axial progenitor in ensembles of mouse embryonic stem cells. Development 2014, 141, 4243–4253. [Google Scholar] [CrossRef] [PubMed]
- Tsakiridis, A.; Huang, Y.; Blin, G.; Skylaki, S.; Wymeersch, F.; Osorno, R.; Economou, C.; Karagianni, E.; Zhao, S.; Lowell, S.; et al. Distinct Wnt-driven primitive streak-like populations reflect in vivo lineage precursors. Development 2014, 141, 1209–1221. [Google Scholar] [CrossRef] [PubMed]
- Lippmann, E.S.; Williams, C.E.; Ruhl, D.A.; Estevez-Silva, M.C.; Chapman, E.R.; Coon, J.J.; Ashton, S.R. Deterministic HOX patterning in human pluripotent stem cell-derived neuroectoderm. Stem Cell Rep. 2015, 4, 632–644. [Google Scholar] [CrossRef] [PubMed]
- Gouti, M.; Delile, J.; Stamataki, D.; Wymeersch, F.J.; Huang, Y.; Kleinjung, J.; Wilson, V.; Briscoe, J. A Gene Regulatory Network Balances Neural and Mesoderm Specification during Vertebrate Trunk Development. Dev. Cell 2017, 41, 243–261. [Google Scholar] [CrossRef] [PubMed]
- Edri, S.; Hayward, P.; Jawaid, W.; Arias, A.M. Neuro-mesodermal progenitors (NMPs): A comparative study between pluripotent stem cells and embryo-derived populations. Development 2019, 146, dev180190. [Google Scholar] [CrossRef] [PubMed]
- Edri, S.; Hayward, P.; Baillie-Johnson, P.; Steventon, B.J.; Arias, A.M. An epiblast stem cell-derived multipotent progenitor population for axial extension. Development 2019, 146, dev168187. [Google Scholar] [CrossRef]
- Janesick, A.; Nguyen, T.T.L.; Aisaki, K.-I.; Igarashi, K.; Kitajima, S.; Chandraratna, R.A.S.; Kanno, J.; Blumberg, B. Active repression by RARγ signaling is required for vertebrate axial elongation. Development 2014, 141, 2260–2270. [Google Scholar] [CrossRef]
- Cooper, K.L.; Hu, J.K.-H.; Berge, D.T.; Fernandez-Teran, M.; Ros, M.A.; Tabin, C.J. Initiation of Proximal-Distal Patterning in the Vertebrate Limb by Signals and Growth. Science 2011, 332, 1083–1086. [Google Scholar] [CrossRef]
- Zuniga, A. Next generation limb development and evolution: Old questions, new perspectives. Development 2015, 142, 3810–3820. [Google Scholar] [CrossRef]
- Saiz-Lopez, P.; Chinnaiya, K.; Campa, V.M.; Delgado, I.; Ros, M.A.; Towers, M. An intrinsic timer specifies distal structures of the vertebrate limb. Nat. Commun. 2015, 6, 8108. [Google Scholar] [CrossRef]
- Laue, K.; Janicke, M.; Plaster, N.; Sonntag, C.; Hammerschmidt, M. Restriction of retinoic acid activity by Cyp26b1 is required for proper timing and patterning of osteogenesis during zebrafish development. Development 2008, 135, 3775–3787. [Google Scholar] [CrossRef] [PubMed]
- Jeradi, S.; Hammerschmidt, M. Retinoic acid-induced premature osteoblast-to-preosteocyte transitioning has multiple effects on calvarial development. Development 2016, 143, 1205–1216. [Google Scholar] [CrossRef] [PubMed]
- Pogoda, H.-M.; Riedl-Quinkertz, I.; Löhr, H.; Waxman, J.S.; Dale, R.M.; Topczewski, J.; Schulte-Merker, S.; Hammerschmidt, M. Direct activation of chordoblasts by retinoic acid is required for segmented centra mineralization during zebrafish spine development. Development 2018, 145, dev159418. [Google Scholar] [CrossRef] [PubMed]
- Hutson, M.R.; Kirby, M.L. Model systems for the study of heart development and disease: Cardiac neural crest and conotruncal malformations. Semin. Cell Dev. Biol. 2007, 18, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Plein, A.; Fantin, A.; Ruhrberg, C. Chapter Six—Neural Crest Cells in Cardiovascular Development. In Current Topics in Developmental Biology; Trainor, P.A., Ed.; Academic Press: Cambridge, MA, USA, 2015; pp. 183–200. [Google Scholar]
- Diogo, R.; Kelly, R.G.; Christiaen, L.; Levine, M.; Ziermann, J.M.; Molnar, J.L.; Noden, D.M.; Tzahor, E. A new heart for a new head in vertebrate cardiopharyngeal evolution. Nature 2015, 520, 466. [Google Scholar] [CrossRef]
- Kelly, R.G.; Buckingham, M.E.; Moorman, A.F. Heart Fields and Cardiac Morphogenesis. Cold Spring Harb. Perspect. Med. 2014, 4, a015750. [Google Scholar] [CrossRef]
- McGurk, P.D.; Swartz, M.E.; Chen, J.W.; Galloway, J.L.; Eberhart, J.K. In vivo zebrafish morphogenesis shows Cyp26b1 promotes tendon condensation and musculoskeletal patterning in the embryonic jaw. PLoS Genet. 2017, 13, e1007112. [Google Scholar] [CrossRef]
- El Shahawy, M.; Reibring, C.-G.; Neben, C.L.; Hallberg, K.; Marangoni, P.; Harfe, B.D.; Klein, O.D.; Linde, A.; Gritli-Linde, A. Cell fate specification in the lingual epithelium is controlled by antagonistic activities of Sonic hedgehog and retinoic acid. PLoS Genet. 2017, 13, e1006914. [Google Scholar] [CrossRef]
- Hasten, E.; Morrow, B.E. Tbx1 and Foxi3 genetically interact in the pharyngeal pouch endoderm in a mouse model for 22q11.2 deletion syndrome. PLoS Genet. 2019, 15, e1008301. [Google Scholar] [CrossRef]
- Rydeen, A.B.; Waxman, J.S. Cyp26 Enzymes Facilitate Second Heart Field Progenitor Addition and Maintenance of Ventricular Integrity. PLoS Biol. 2016, 14, e2000504. [Google Scholar] [CrossRef]
- Waxman, J.S.; Keegan, B.R.; Roberts, R.W.; Poss, K.D.; Yelon, D. Hoxb5b acts downstream of retinoic acid signaling in the forelimb field to restrict heart field potential in zebrafish. Dev. Biol. 2008, 319, 482. [Google Scholar] [CrossRef]
- Waxman, J.S.; Yelon, D. Increased Hox activity mimics the teratogenic effects of excess retinoic acid signaling. Dev. Dyn. 2009, 238, 1207–1213. [Google Scholar] [CrossRef] [PubMed]
- D’Aniello, E.; Rydeen, A.B.; Anderson, J.L.; Mandal, A.; Waxman, J.S. Depletion of Retinoic Acid Receptors Initiates a Novel Positive Feedback Mechanism that Promotes Teratogenic Increases in Retinoic Acid. PLoS Genet. 2013, 9, e1003689. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Jiang, J.; Han, P.; Yuan, Q.; Zhang, J.; Zhang, X.; Xu, Y.; Cao, H.; Meng, Q.; Chen, L.; et al. Direct differentiation of atrial and ventricular myocytes from human embryonic stem cells by alternating retinoid signals. Cell Res. 2010, 21, 579. [Google Scholar] [CrossRef]
- Devalla, H.D.; Schwach, V.; Ford, J.W.; Milnes, J.T.; El-Haou, S.; Jackson, C.; Gkatzis, K.; Elliott, D.A.; Lopes, S.M.C.D.; Mummery, C.L.; et al. Atrial-like cardiomyocytes from human pluripotent stem cells are a robust preclinical model for assessing atrial-selective pharmacology. EMBO Mol. Med. 2015, 7, 394–410. [Google Scholar] [CrossRef]
- Lee, J.H.; Protze, S.I.; Laksman, Z.; Backx, P.H.; Keller, G.M. Human Pluripotent Stem Cell-Derived Atrial and Ventricular Cardiomyocytes Develop from Distinct Mesoderm Populations. Cell Stem Cell 2017, 21, 179–194. [Google Scholar] [CrossRef]
- Perl, E.; Waxman, J.S. Reiterative Mechanisms of Retinoic Acid Signaling during Vertebrate Heart Development. J. Dev. Biol. 2019, 7, 11. [Google Scholar] [CrossRef]
- Kinkel, M.D.; Eames, S.C.; Alonzo, M.R.; Prince, V.E. Cdx4 is required in the endoderm to localize the pancreas and limitβ -cell number. Development 2008, 135, 919–929. [Google Scholar] [CrossRef]
- Naylor, R.W.; Skvarca, L.B.; Thisse, C.; Thisse, B.; Hukriede, N.A.; Davidson, A.J. BMP and retinoic acid regulate anterior–posterior patterning of the non-axial mesoderm across the dorsal–ventral axis. Nat. Commun. 2016, 7, 12197. [Google Scholar] [CrossRef]
- Wingert, R.A.; Selleck, R.; Yu, J.; Song, H.-D.; Chen, Z.; Song, A.; Zhou, Y.; Thisse, B.; Thisse, C.; McMahon, A.P.; et al. The cdx Genes and Retinoic Acid Control. the Positioning and Segmentation of the Zebrafish Pronephros. PLoS Genet. 2007, 3, e189. [Google Scholar] [CrossRef]
- Brendolan, A.; Ferretti, E.; Salsi, V.; Moses, K.; Quaggin, S.; Blasi, F.; Cleary, M.L.; Selleri, L. A Pbx1-dependent genetic and transcriptional network regulates spleen ontogeny. Development 2005, 132, 3113–3126. [Google Scholar] [CrossRef] [PubMed]
- Lenti, E.; Farinello, D.; Yokoyama, K.K.; Penkov, D.; Castagnaro, L.; Lavorgna, G.; Wuputra, K.; Sandell, L.L.; Tjaden, N.E.B.; Bernassola, F.; et al. Transcription factor TLX1 controls retinoic acid signaling to ensure spleen development. J. Clin. Investig. 2016, 126, 2452–2464. [Google Scholar] [CrossRef] [PubMed]
- Bowles, J.; Secker, G.; Nguyen, C.; Kazenwadel, J.; Truong, V.; Frampton, E.; Curtis, C.; Skoczylas, R.; Davidson, T.L.; Miura, N.; et al. Control. of retinoid levels by CYP26B1 is important for lymphatic vascular development in the mouse embryo. Dev. Biol. 2014, 386, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Okano, J.; Lichti, U.; Mamiya, S.; Aronova, M.; Zhang, G.; Yuspa, S.H.; Hamada, H.; Sakai, Y.; Morasso, M.I. Increased retinoic acid levels through ablation of Cyp26b1 determine the processes of embryonic skin barrier formation and peridermal development. J. Cell Sci. 2012, 125, 1827–1836. [Google Scholar] [CrossRef] [PubMed]
- Chanda, B.; Ditadi, A.; Iscove, N.N.; Keller, G. Retinoic Acid Signaling Is Essential for Embryonic Hematopoietic Stem Cell Development. Cell 2013, 155, 215–227. [Google Scholar] [CrossRef]
- Ghiaur, G.; Yegnasubramanian, S.; Perkins, B.; Gucwa, J.L.; Gerber, J.M.; Jones, R.J. Regulation of human hematopoietic stem cell self-renewal by the microenvironment’s control of retinoic acid signaling. Proc. Natl. Acad. Sci. USA 2013, 110, 16121–16126. [Google Scholar] [CrossRef] [PubMed]
- Bowles, J.; Knight, D.; Smith, C.; Wilhelm, D.; Richman, J.; Mamiya, S.; Yashiro, K.; Chawengsaksophak, K.; Wilson, M.J.; Rossant, J.; et al. Retinoid Signaling Determines Germ Cell Fate in Mice. Science 2006, 312, 596–600. [Google Scholar] [CrossRef]
- Koubova, J.; Menke, D.B.; Zhou, Q.; Capel, B.; Griswold, M.D.; Page, D.C. Retinoic acid regulates sex-specific timing of meiotic initiation in mice. Proc. Natl. Acad. Sci. USA 2006, 103, 2474–2479. [Google Scholar] [CrossRef]
- Yadu, N.; Kumar, P.G. Retinoic acid signaling in regulation of meiosis during embryonic development in mice. Genesis 2019, 57, e23327. [Google Scholar] [CrossRef]
- Saba, R.; Wu, Q.; Saga, Y. CYP26B1 promotes male germ cell differentiation by suppressing STRA8-dependent meiotic and STRA8-independent mitotic pathways. Dev. Biol. 2014, 389, 173–181. [Google Scholar] [CrossRef]
- Bowles, J.; Feng, C.-W.; Ineson, J.; Miles, K.; Spiller, C.M.; Harley, V.R.; Sinclair, A.H.; Koopman, P. Retinoic Acid Antagonizes Testis Development in Mice. Cell Rep. 2018, 24, 1330–1341. [Google Scholar] [CrossRef] [PubMed]
- Blum, N.; Begemann, G. Retinoic acid signaling controls the formation, proliferation and survival of the blastema during adult zebrafish fin regeneration. Development 2012, 139, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Blum, N.; Begemann, G. The roles of endogenous retinoid signaling in organ and appendage regeneration. Cell. Mol. Life Sci. 2013, 70, 3907–3927. [Google Scholar] [CrossRef] [PubMed]
- Blum, N.; Begemann, G. Osteoblast de- and redifferentiation are controlled by a dynamic response to retinoic acid during zebrafish fin regeneration. Development 2015, 142, 2894–2903. [Google Scholar] [CrossRef]
- Lee, Y.; Hami, D.; de Val, S.; Kagermeier-Schenk, B.; Wills, A.A.; Black, B.L.; Weidinger, G.; Poss, K.D. Maintenance of blastemal proliferation by functionally diverse epidermis in regenerating zebrafish fins. Dev. Biol. 2009, 331, 270–280. [Google Scholar] [CrossRef]
- Blum, N.; Begemann, G. Retinoic acid signaling spatially restricts osteoblasts and controls ray-interray organization during zebrafish fin regeneration. Development 2015, 142, 2888–2893. [Google Scholar] [CrossRef]
- Thomas, A.G.; Henry, J.J. Retinoic acid regulation by CYP26 in vertebrate lens regeneration. Dev. Biol. 2014, 386, 291–301. [Google Scholar] [CrossRef]
- Thomas, A.G.; Adil, M.T.; Henry, J.J. Understanding the basis of CYP26 mediated regulation of lens regeneration using ex vivo eye cultures and 4-oxo-RA. bioRxiv 2019, 631994. [Google Scholar] [CrossRef]
- Rubbini, D.; Robert-Moreno, À.; Hoijman, E.; Alsina, B. Retinoic Acid Signaling Mediates Hair Cell Regeneration by Repressing p27kip and sox2 in Supporting Cells. J. Neurosci. 2015, 35, 15752–15766. [Google Scholar] [CrossRef]
- Lee, L.M.Y.; Leung, C.-Y.; Tang, W.W.C.; Choi, H.-L.; Leung, Y.-C.; McCaffery, P.J.; Wang, C.-C.; Woolf, A.S.; Shum, A.S.W. A paradoxical teratogenic mechanism for retinoic acid. Proc. Natl. Acad. Sci. USA 2012, 109, 13668–13673. [Google Scholar] [CrossRef]
- D’Aniello, E.; Waxman, J.S. Input overload: Contributions of retinoic acid signaling feedback mechanisms to heart development and teratogenesis. Dev. Dyn. 2015, 244, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Rydeen, A.; Voisin, N.; D’Aniello, E.; Ravisankar, P.; Devignes, C.-S.; Waxman, J.S. Excessive feedback of Cyp26a1 promotes cell non-autonomous loss of retinoic acid signaling. Dev. Biol. 2015, 405, 47–55. [Google Scholar] [CrossRef] [PubMed]









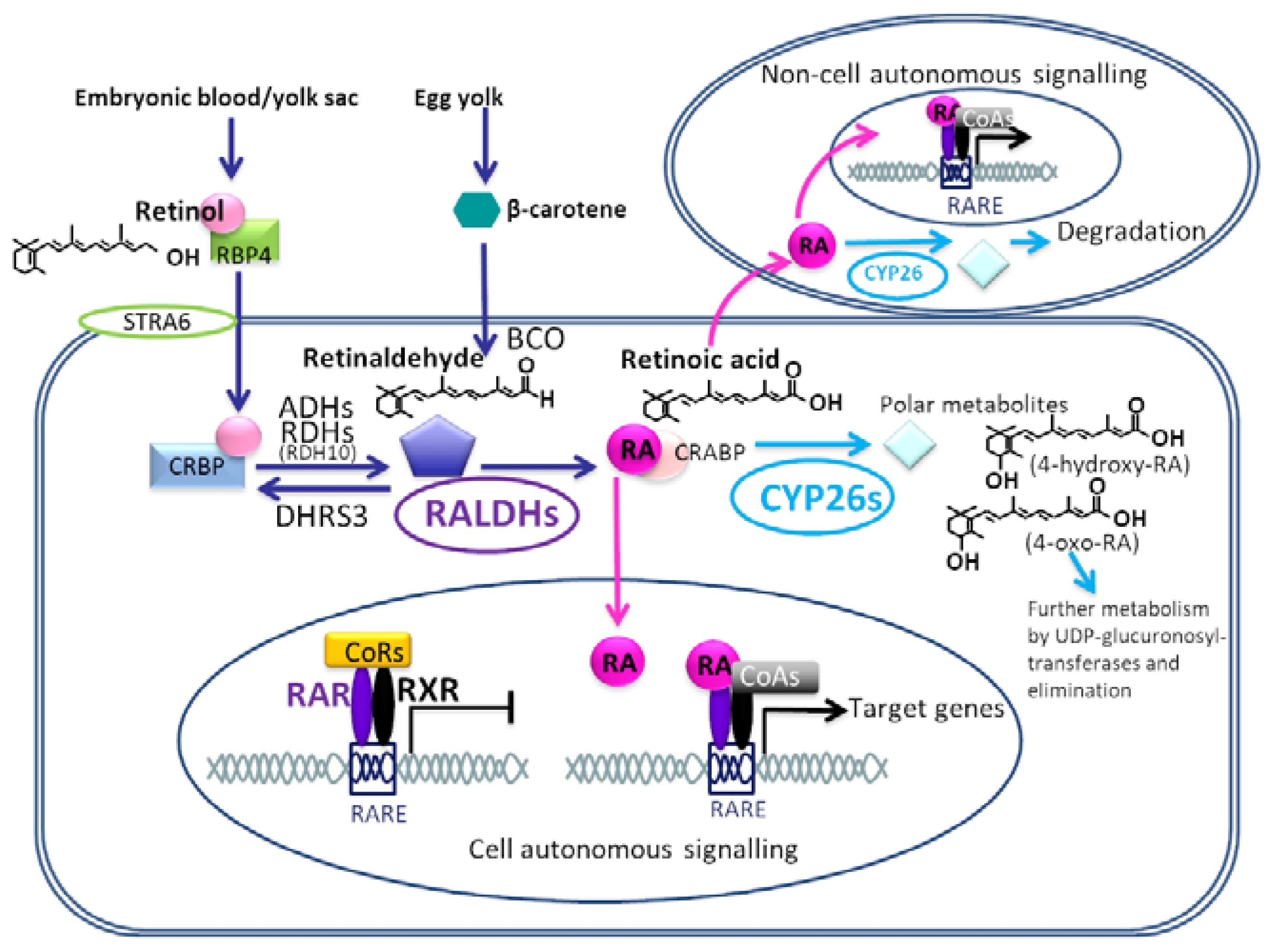{kind=link}
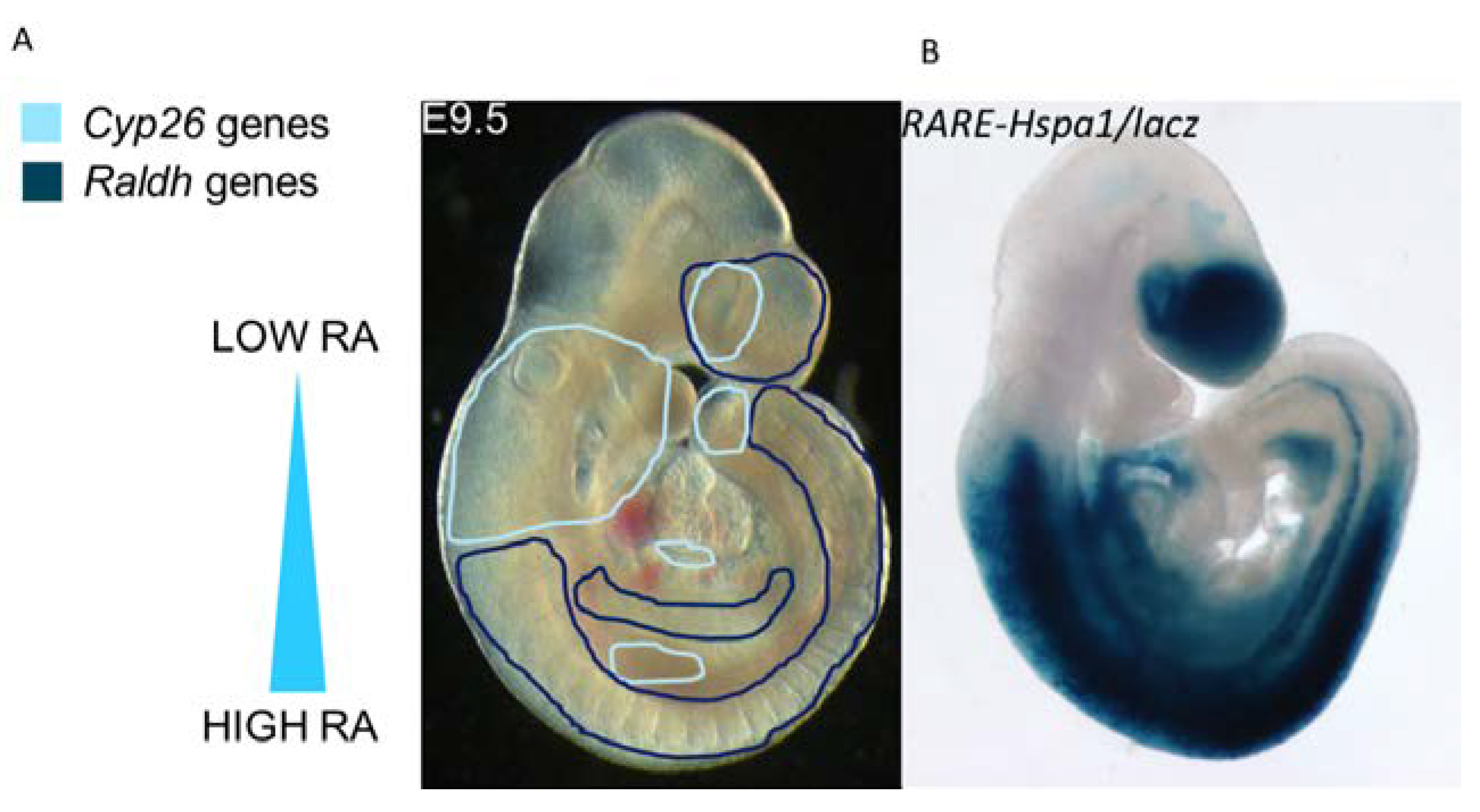{kind=link}
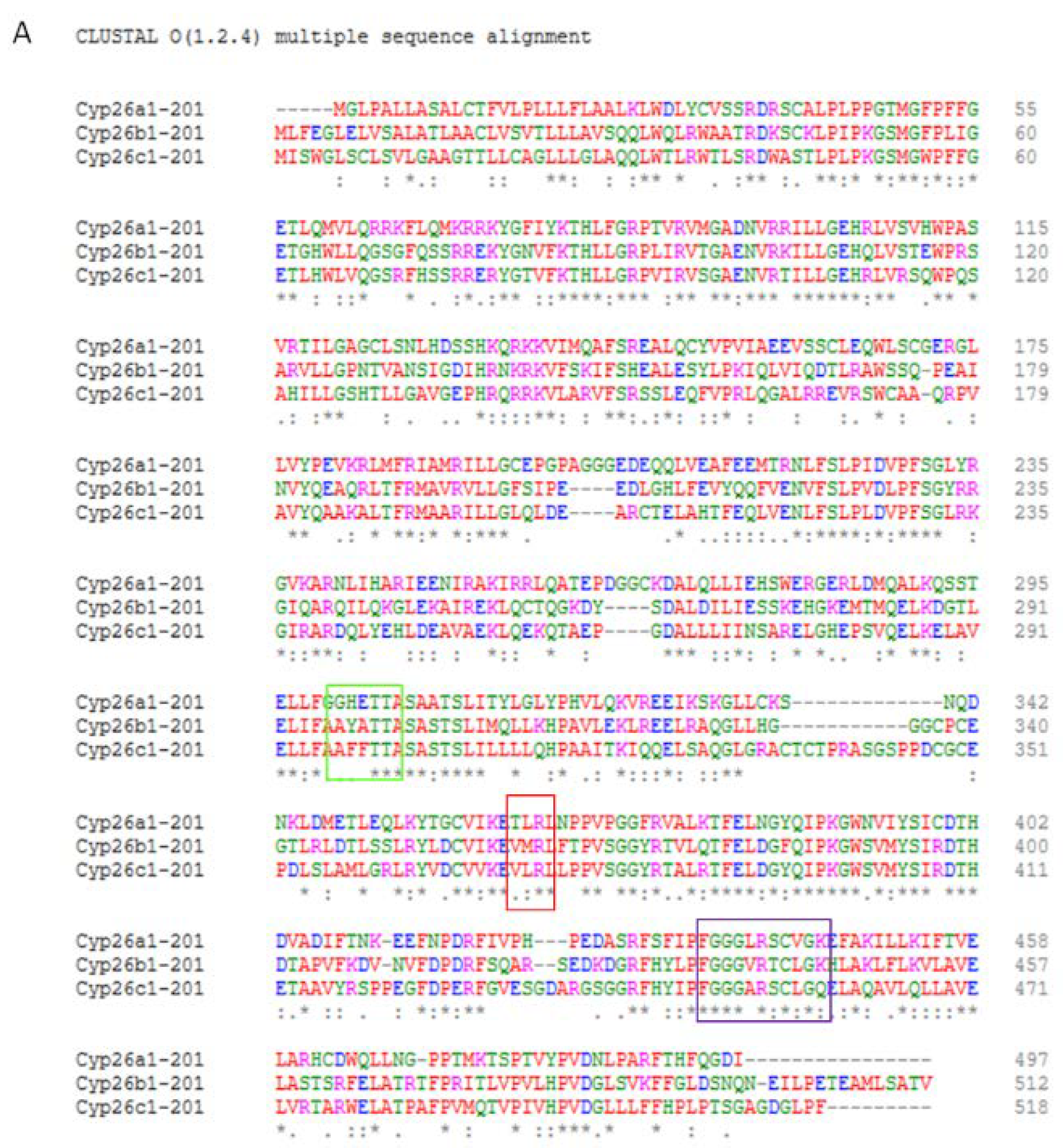{kind=link}
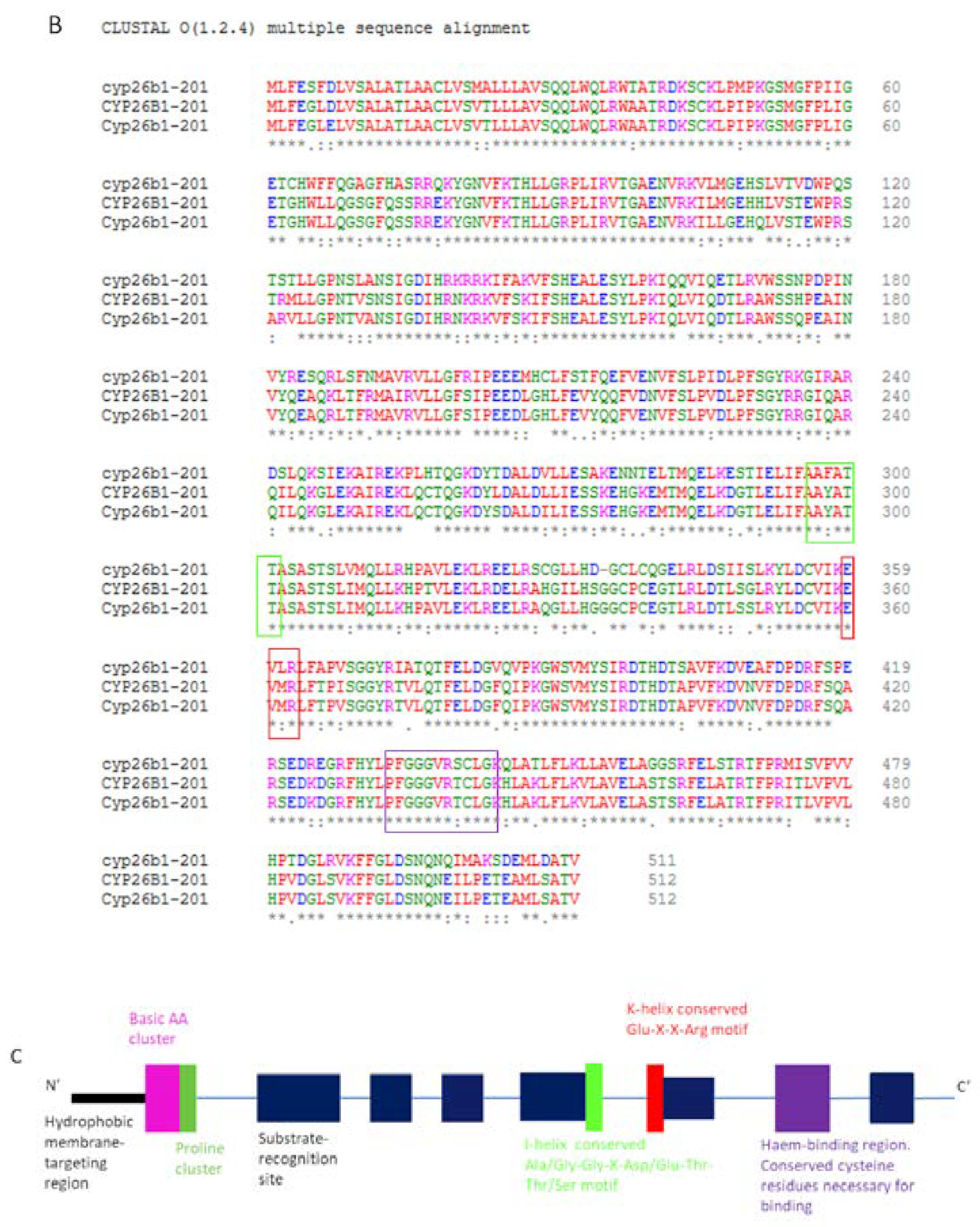{kind=link}
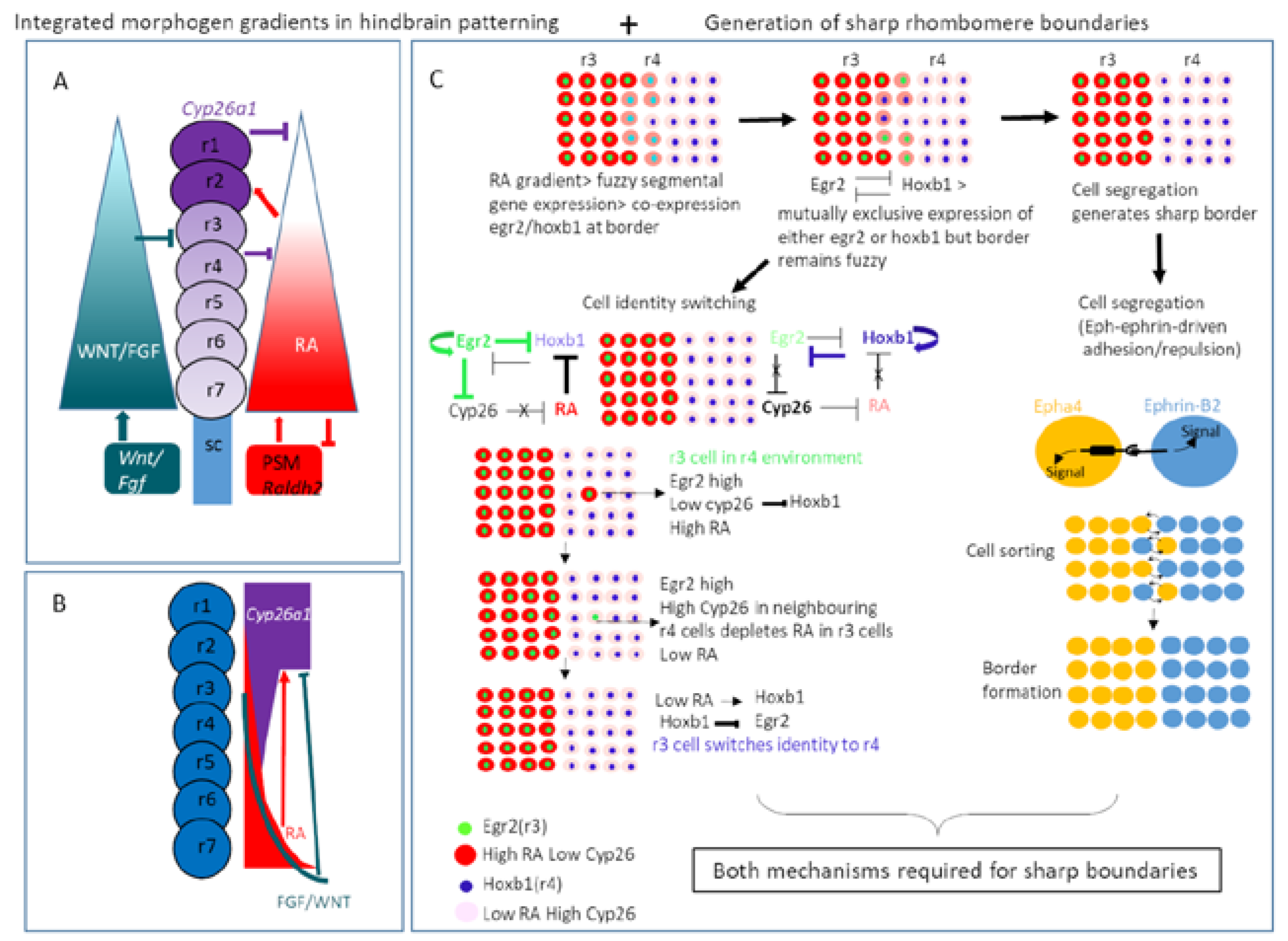{kind=link}
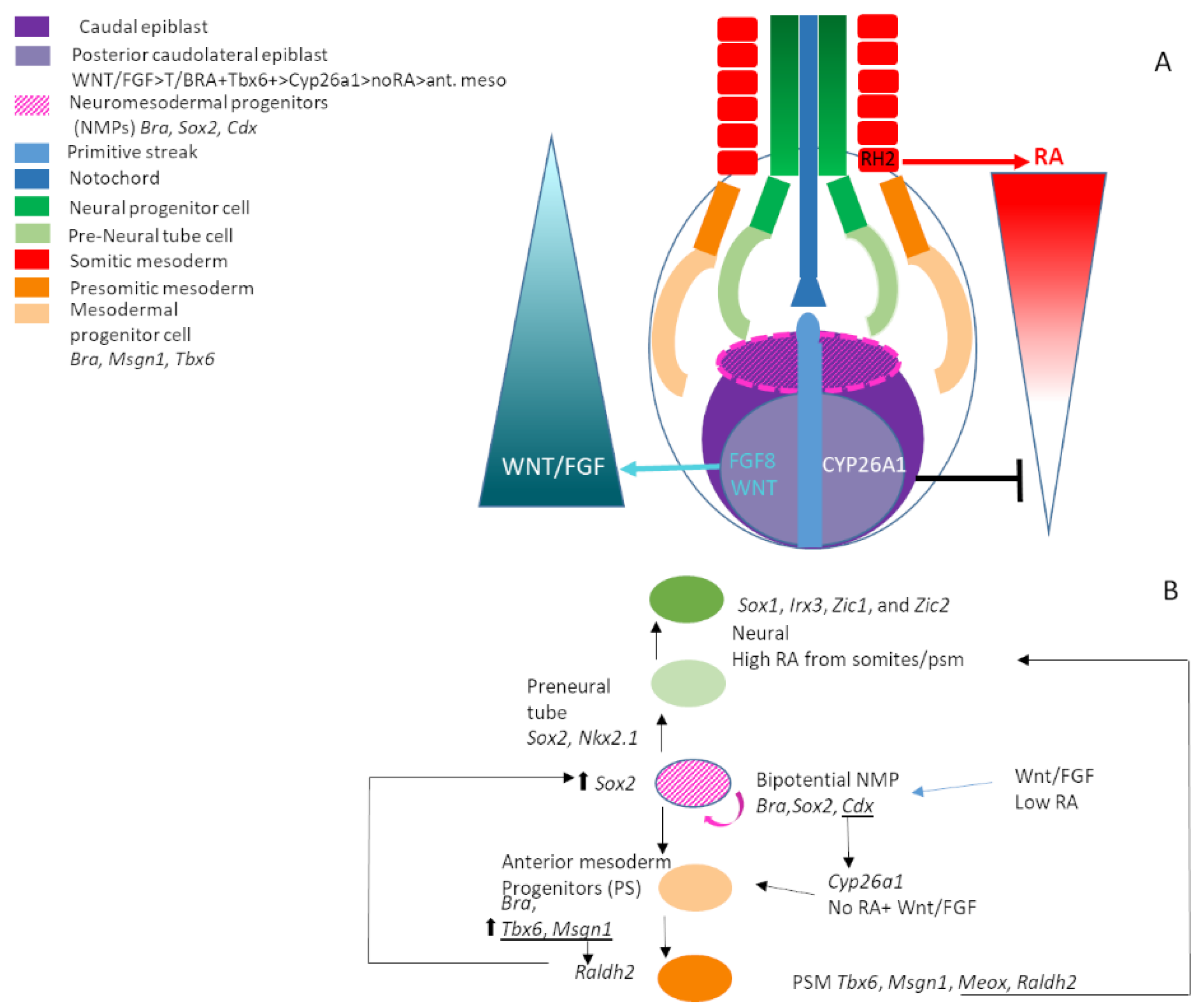{kind=link}
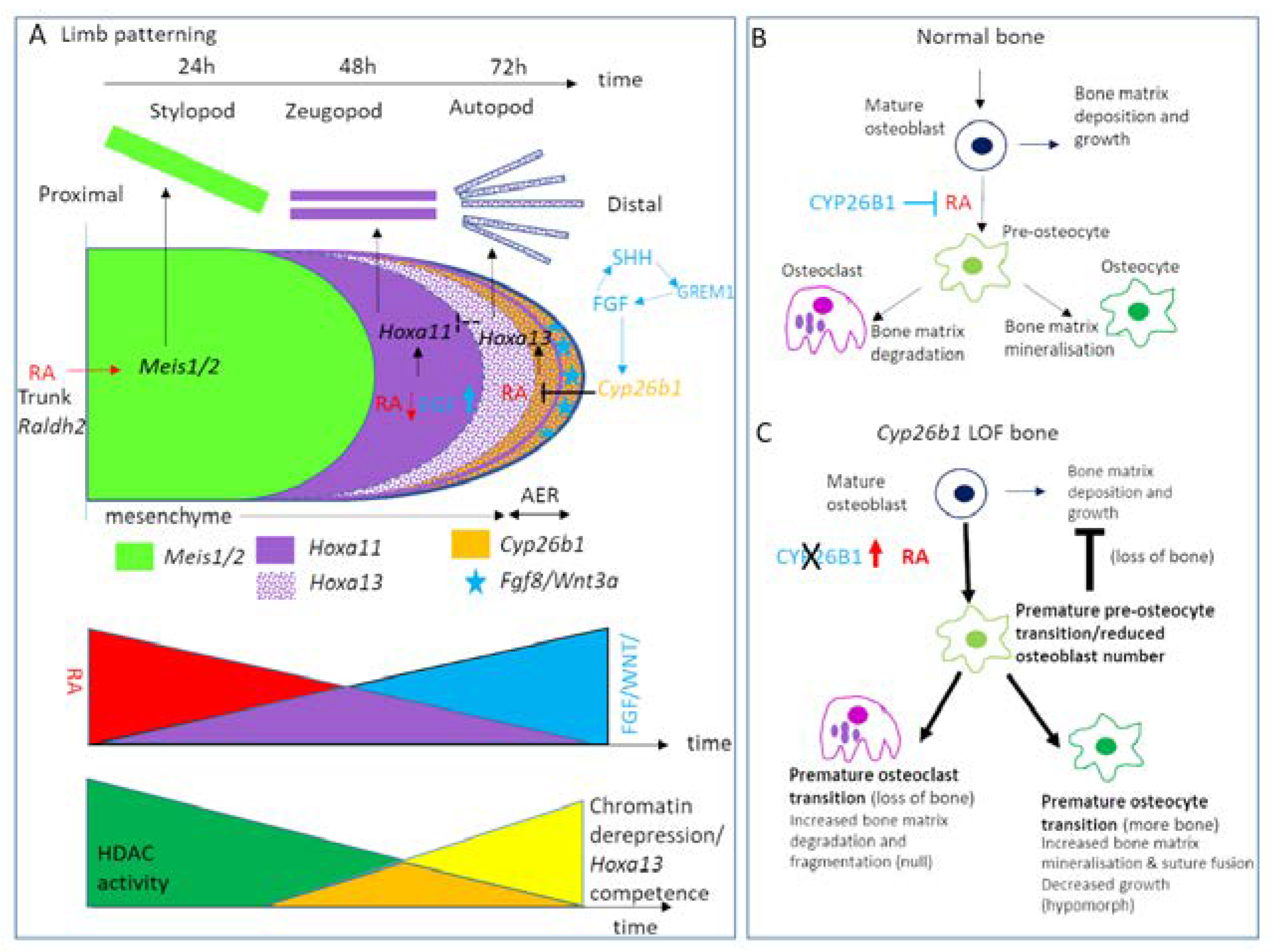{kind=link}
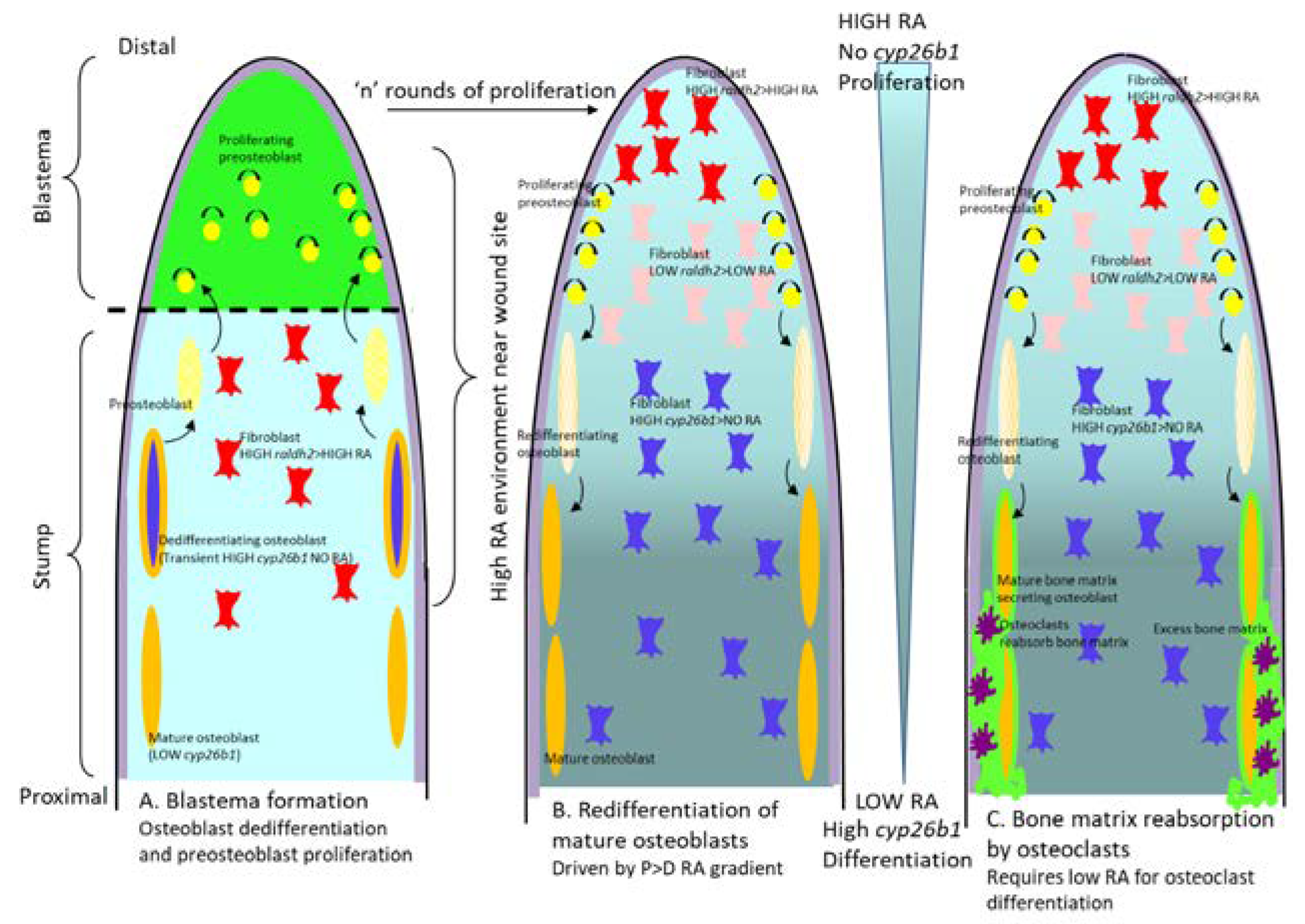{kind=link}
| Gene | Nucleotide/AA Change | Change in Enzyme Function | Phenotype | Reference |
|---|---|---|---|---|
| CYP26A1 | R173S | ? | None reported | [206] |
| F186L | 40–80% reduced atRA metabolising activity in COS cells | |||
| C358R | ||||
| CYP26A1 | g.3116delT | reduced atRA metabolising activity | Associated with spina bifida | [208] |
| premature stop | ||||
| CYP26A1 | rs4411227 C/G or C/C | ? | Increased risk oral and pharyngeal cancer | [209,210,211] |
| CYP26A1 and CYP26C1 | Microdeletion of up to 249–363 kb of chrs. 10q23.33 | Haploinsufficiency | Optic nerve aplasia | [213] |
| CYP26A1, CYP26C1, EXOC6 | ||||
| CYP26A1 and CYP26C1 | 8.3 Mb microdel. Chrs 10q23.2–23.33. The 79 deleted genes included CYP26A1 and C1, | Haploinsufficiency CYP26A1, CYP26C1 + 79 other genes | Premature ageing skeletal and dental development, retinal scarring, and autism-spectrum | [214] |
| Raised plasma RA levels | ||||
| CYP26B1 | Nine missense or splicing changes | 100% loss of function | Neural tube, limb, craniofacial, skeletal, heart, kidney and lung defects | [207] |
| (samples collected during gestation) | ||||
| 583C > T Arg195Met (1) | ||||
| 589c > A Leu197Met (3) | ||||
| 704G > A Arg235Gln (1) | ||||
| 712C > G Gln238Glu (1) | ||||
| 715G > A Ala237Thr (3) | ||||
| CYP26B1 | Splicing variant with loss of exon 2 | 30% loss of function | Expressed in atherosclerotic lesion vascular cells | [215] |
| CYP26B1 | rs3768647/9309462 | ? | Increased risk oral and pharyngeal cancer | [209,210,211,212] |
| C/C or C/T | ||||
| rs138478634 G/A change in exon 5 | ||||
| CYP26B1 | rs2241057T/T (major allele | Lower CYP26B1 activity | Increased risk of Crohn’s disease | [217] |
| rs2241057C/C (minor allele) | Higher CYP26B1 activity | Larger macrophage-rich atherosclerotic | [216] | |
| L264S | lesions | |||
| CYP26B1 | 3 c.1088G > T | 100% loss of function by affecting the K-helix | Craniofacial, skull, pelvic, limb long bone skeletal defects | [202] |
| homozygous | ||||
| p.Arg363Leu | ||||
| 1 died in utero/2 terminations | ||||
| CYP26B1 | c.436T > C | 31% loss of function | Defects similar to Antley–Bixler and Pfeiffer Syndromes | [202] |
| homozygous | Skull, digit and joint skeletal defects | |||
| p.Ser146Pro | ||||
| Died at 5 months | ||||
| CYP26B1 | c.1303G > A | Predicted loss of function | Skull and long bone skeletal defects, intellectual disability | [218] |
| p.Gly435Ser | ||||
| homozygous | ||||
| (survived to adulthood) | ||||
| CYP26B1 | 0.78–4 Mb microdeletion 0.78–4 Mb chrs.2p13.2–13.3 | 8% induction of CYP26B1 mRNA by RA compared to controls | Intellectual disability, language delay, hyperactivity, dysmorphic facies and vertebral and/or craniofacial abnormalities | [219] |
| CYP26B1 and EXOC6B haploinsufficient | ||||
| CYP26C1 | Missense p.Phe508Cys | Loss of function | Confers increased severity of SHOX p.Val161Ala mutation skeletal and short stature phenotype | [223] |
| CYP26C1 | Missense c.148C > T, Pro50Ser; | Loss of function | Short stature phenotype | [224] |
| c.356A > C, p.Gln119Pro; c.910G > A, Ala304 The | ||||
| Splice variant truncation | ||||
| c.706-A > C | ||||
| CYP26C1 | Duplication > frameshift | 100% loss of function | Focal facial dermal dysplasia | [225] |
| > premature stop | ||||
| c.844–851dupCCATGCA | ||||
| p.Glu284fsX128 (maternal) | ||||
| homozygous and compound heterozygous | ||||
| Missense c.1433G > A p.Arg478His (paternal) | ||||
| compound heterozygous | ||||
| POR | Missense, nonsense, frameshift, splicing and exon deletions | Loss of function | Antley–Bixler like skeletal defects and deficient steroidal profiles | [220,221,222] |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roberts, C. Regulating Retinoic Acid Availability during Development and Regeneration: The Role of the CYP26 Enzymes. J. Dev. Biol. 2020, 8, 6. https://doi.org/10.3390/jdb8010006
Roberts C. Regulating Retinoic Acid Availability during Development and Regeneration: The Role of the CYP26 Enzymes. Journal of Developmental Biology. 2020; 8(1):6. https://doi.org/10.3390/jdb8010006
Chicago/Turabian StyleRoberts, Catherine. 2020. "Regulating Retinoic Acid Availability during Development and Regeneration: The Role of the CYP26 Enzymes" Journal of Developmental Biology 8, no. 1: 6. https://doi.org/10.3390/jdb8010006
APA StyleRoberts, C. (2020). Regulating Retinoic Acid Availability during Development and Regeneration: The Role of the CYP26 Enzymes. Journal of Developmental Biology, 8(1), 6. https://doi.org/10.3390/jdb8010006