Reiterative Mechanisms of Retinoic Acid Signaling during Vertebrate Heart Development


Abstract
1. Introduction
2. Initial Insights into the Roles of RA Signaling during Heart Development
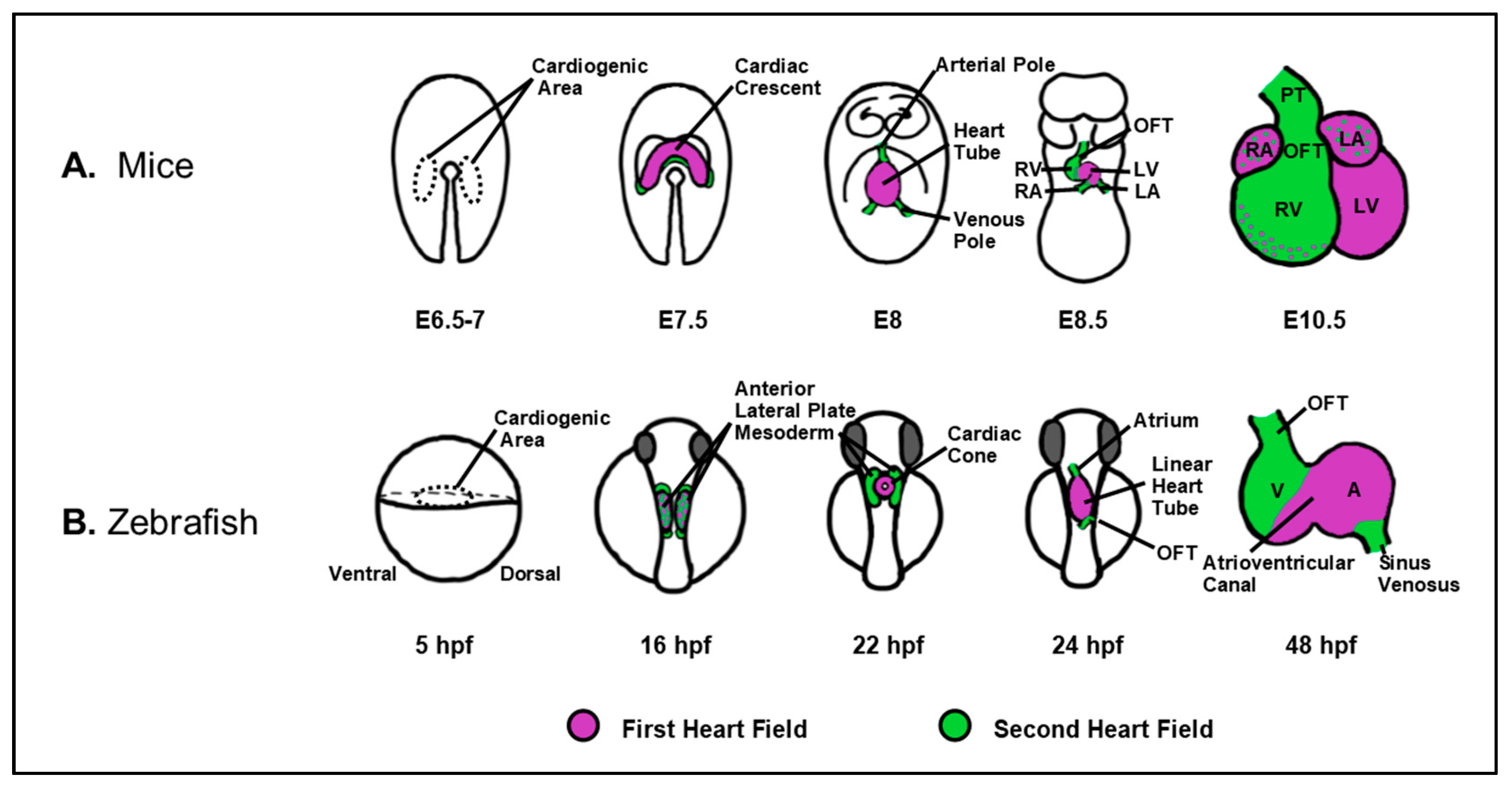
3. RA Signal Restricts Cardiac Progenitor Specification
3.1. Restriction of Cardiac Progenitors Fields within the ALPM
3.2. Coordination of Adjacent Cardiac and Forelimb Progenitor Fields by RA
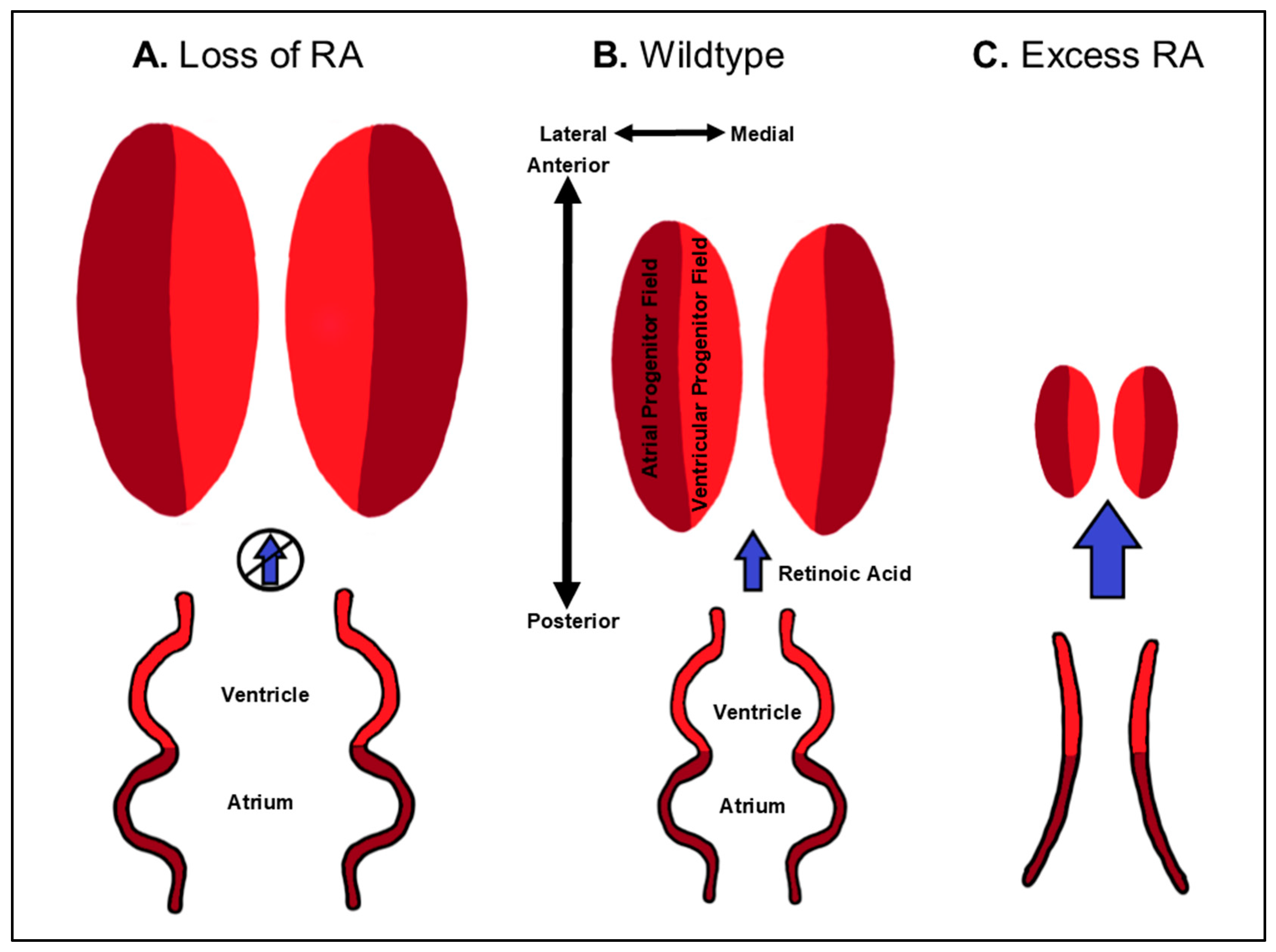
3.3. Dose-Dependent Differential Regulation of Atrial and Ventricular Progenitor Specification by RA
4. Consequences of Perturbing Components of the RA Signaling Pathway during Heart Development
4.1. Congenital Heart Malformations from Loss of RA Synthesis
4.2. Congenital Heart Malformations from the Inability to Limit RA
4.3. Congenital Malformations from Loss of Retinoic Acid Receptors
5. Downstream Effectors of RA Signaling in Vertebrate Heart Development
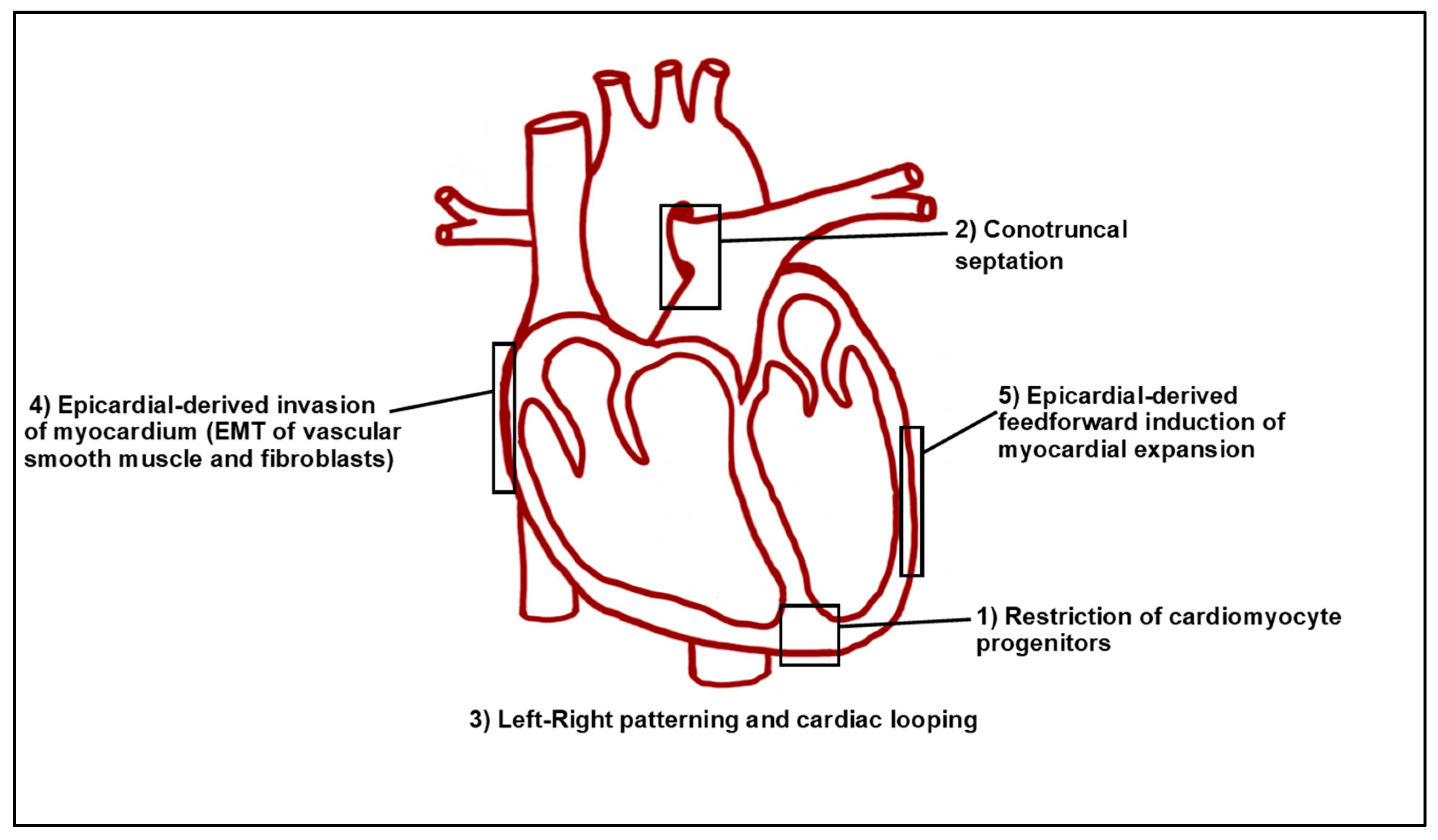
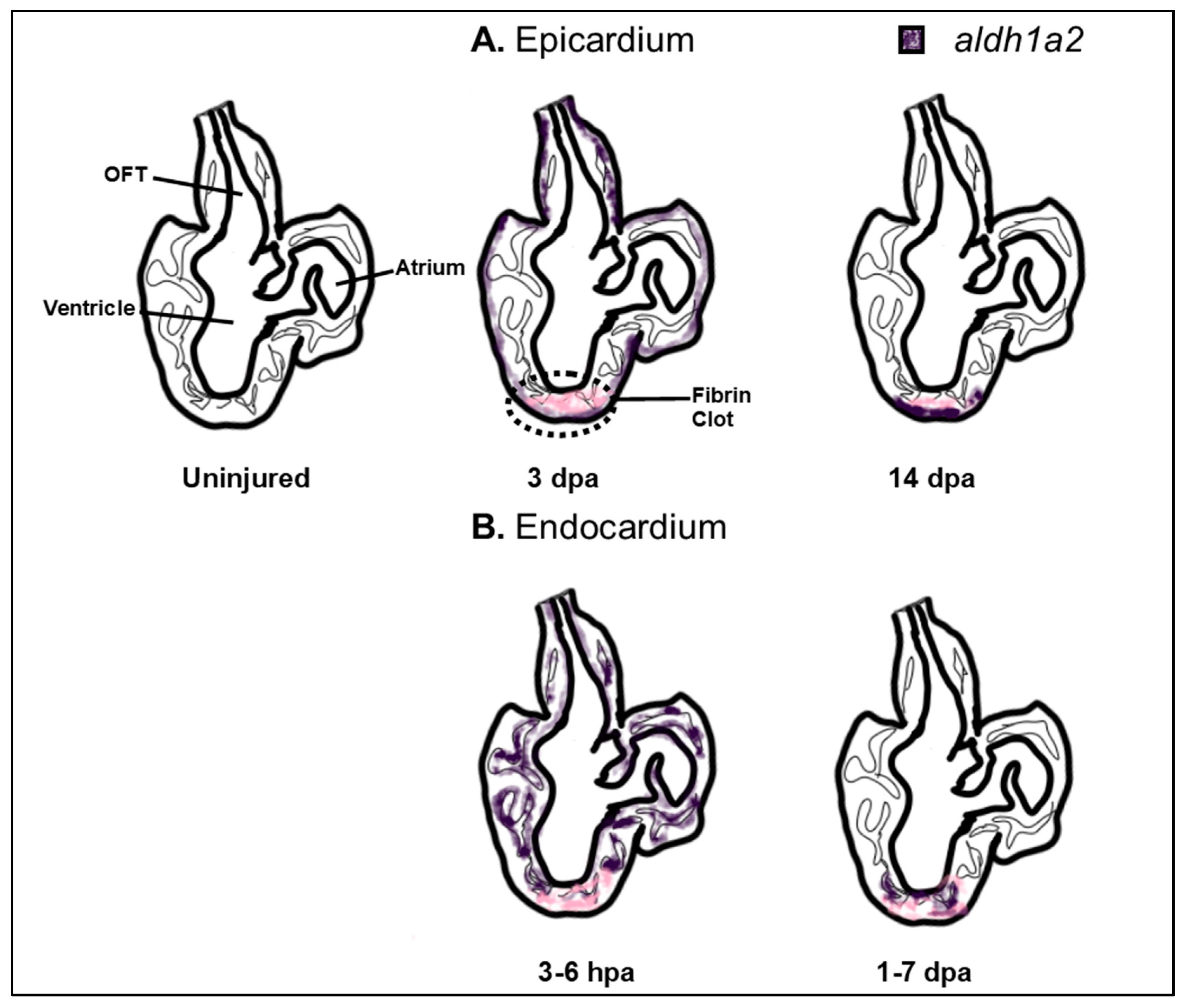
6. Later Requirements of RA Signaling in Heart Development
7. Toward the Future: Understanding RA Signaling in the Vertebrate Heart
Author Contributions
Funding
Conflicts of Interest
References
- Van der Linde, D.; Konings, E.E.M.; Slager, M.A.; Witsenburg, M.; Helbing, W.A.; Takkenberg, J.J.M.; Roos-Hesselink, J.W. Birth Prevalence of Congenital Heart Disease Worldwide: A Systematic Review and Meta-Analysis. J. Am. Coll. Cardiol. 2011, 58, 2241–2247. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, E.J.; Muntner, P.; Alonso, A.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Das, S.R.; et al. Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation 2019, 139, e56–e66. [Google Scholar] [CrossRef] [PubMed]
- D’Aniello, E.; Waxman, J.S. Input overload: Contributions of retinoic acid signaling feedback mechanisms to heart development and teratogenesis. Dev. Dyn. 2015, 244, 513–523. [Google Scholar] [CrossRef]
- Stefanovic, S.; Zaffran, S. Mechanisms of retinoic acid signaling during cardiogenesis. Mech. Dev. 2017, 143, 9–19. [Google Scholar] [CrossRef]
- Wilson, J.G.; Roth, C.B.; Warkany, J. An analysis of the syndrome of malformations induced by maternal vitamin a deficiency. Effects of restoration of vitamin a at various times during gestation. Am. J. Anat. 1953, 92, 189–217. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.G.; Warkany, J. Cardiac and aortic arch anomalies in the offspring of vitamin A deficient rats correlated with similar human anomalies. Pediatrics 1950, 5, 708–725. [Google Scholar]
- Dersch, H.; Zile, M.H. Induction of Normal Cardiovascular Development in the Vitamin A-Deprived Quail Embryo by Natural Retinoids. Dev. Biol. 1993, 160, 424–433. [Google Scholar] [CrossRef]
- Zile, M.H. Vitamin A-not for your eyes only: Requirement for heart formation begins early in embryogenesis. Nutrients 2010, 2, 532–550. [Google Scholar] [CrossRef] [PubMed]
- Finnell, R.H.; Shaw, G.M.; Lammer, E.J.; Brandl, K.L.; Carmichael, S.L.; Rosenquist, T.H. Gene–nutrient interactions: importance of folates and retinoids during early embryogenesis. Toxicol. Appl. Pharmacol. 2004, 198, 75–85. [Google Scholar] [CrossRef]
- World Health Organization. Global Prevalence of Vitamin A Deficiency in Populations at Risk 1995–2005: WHO Global Database on Vitamin A Deficiency; WHO Press: Geneva, Switzerland, 2009. [Google Scholar]
- Lammer, E.J.; Chen, D.T.; Hoar, R.M.; Agnish, N.D.; Benke, P.J.; Braun, J.T.; Curry, C.J.; Fernhoff, P.M.; Grix, A.W.; Lott, I.T.; et al. Retinoic Acid Embryopathy. N. Engl. J. Med. 1985, 313, 837–841. [Google Scholar] [CrossRef] [PubMed]
- Mondal, D.; R Shenoy, S.; Mishra, S. Retinoic acid embryopathy. Int. J. Appl. Basic Med. Res. 2017, 7, 264. [Google Scholar] [CrossRef]
- Doshi, A. The cost of clear skin: balancing the social and safety costs of iPLEDGE with the efficacy of Accutane (isotretinoin). Seton Hall Law Rev. 2007, 37, 625–660. [Google Scholar] [PubMed]
- Duerbeck, N.B.; Dowling, D.D. Vitamin A: Too much of a good thing? Obstet. Gynecol. Surv. 2012, 67, 122–128. [Google Scholar] [CrossRef]
- Wolbach, S.B.; Howe, P.R. Tissue changes following deprivation of fat-soluble A vitamin. J. Exp. Med. 1925, 42, 753–777. [Google Scholar] [CrossRef] [PubMed]
- Hale, F. The Relation of Vitamin a to Anophthalmos in Pigs. Am. J. Ophthalmol. 1935, 18, 1087–1093. [Google Scholar] [CrossRef]
- Wilson, J.G.; Warkany, J. Aortic-arch and cardiac anomalies in the offspring of vitamin A deficient rats. Am. J. Anat. 1949, 85, 113–155. [Google Scholar] [CrossRef]
- Wilson, J.G.; Warkany, J. Congenital anomalies of heart and great vessels in offspring of vitamin A-deficient rats. Am. J. Dis. Child. 1950, 79, 963. [Google Scholar] [CrossRef]
- Cohlan, S.Q. Excessive intake of vitamin A as a cause of congenital anomalies in the rat. Science 1953, 117, 535–536. [Google Scholar] [CrossRef]
- Kalter, H.; Warkany, J. Experimental production of congenital malformations in strains of inbred mice by maternal treatment with hypervitaminosis A. Am. J. Pathol. 1961, 38, 1–21. [Google Scholar]
- Pan, J.; Baker, K.M. Retinoic Acid and the Heart. Vitam. Horm. 2007, 75, 257–283. [Google Scholar]
- Rizzo, R.; Lammer, E.J.; Parano, E.; Pavone, L.; Argyle, J.C. Limb reduction defects in humans associated with prenatal isotretinoin exposure. Teratology 1991, 44, 599–604. [Google Scholar] [CrossRef]
- Rothman, K.J.; Moore, L.L.; Singer, M.R.; Nguyen, U.-S.D.T.; Mannino, S.; Milunsky, A. Teratogenicity of High Vitamin A Intake. N. Engl. J. Med. 1995, 333, 1369–1373. [Google Scholar] [CrossRef]
- Gliniak, C.M.; Brown, J.M.; Noy, N. The retinol-binding protein receptor STRA6 regulates diurnal insulin responses. J. Biol. Chem. 2017, 292, 15080–15093. [Google Scholar] [CrossRef]
- Heine, U.I.; Roberts, A.B.; Munoz, E.F.; Roche, N.S.; Sporn, M.B. Effects of retinoid deficiency on the development of the heart and vascular system of the quail embryo. Virchows Arch. B. Cell Pathol. Incl. Mol. Pathol. 1985, 50, 135–152. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Huang, W.; Castillo, H.A.; Kane, M.A.; Xavier-Neto, J.; Trainor, P.A.; Moise, A.R. Alterations in retinoic acid signaling affect the development of the mouse coronary vasculature. Dev. Dyn. 2018, 247, 976–991. [Google Scholar] [CrossRef] [PubMed]
- Sugrue, K.F.; Sarkar, A.A.; Leatherbury, L.; Zohn, I.E. The ubiquitin ligase HECTD1 promotes retinoic acid signaling required for development of the aortic arch. Dis. Model. Mech. 2018. [Google Scholar] [CrossRef] [PubMed]
- Niederreither, K.; Subbarayan, V.; Dollé, P.; Chambon, P. Embryonic retinoic acid synthesis is essential for early mouse post-implantation development. Nat. Genet. 1999, 21, 444–448. [Google Scholar] [CrossRef] [PubMed]
- Stainier, D.Y.R.; Fishman, M.C. Patterning the zebrafish heart tube: Acquisition of anteroposterior polarity. Dev. Biol. 1992, 153, 91–101. [Google Scholar] [CrossRef]
- Yutzey, K.E.; Rhee, J.T.; Bader, D. Expression of the atrial-specific myosin heavy chain {AMHC1} and the establishment of anteroposterior polarity in the developing chicken heart. Development 1994, 120, 871–883. [Google Scholar]
- Kostetskii, I.; Yuan, S.-Y.; Kostetskaia, E.; Linask, K.K.; Blanchet, S.; Seleiro, E.; Michaille, J.-J.; Brickell, P.; Zile, M. Initial retinoid requirement for early avian development coincides with retinoid receptor coexpression in the precardiac fields and induction of normal cardiovascular development. Dev. Dyn. 1998, 213, 188–198. [Google Scholar] [CrossRef]
- LaRue, A.C.; Argraves, W.S.; Zile, M.H.; Drake, C.J. Critical role for retinol in the generation/differentiation of angioblasts required for embryonic blood vessel formation. Dev. Dyn. 2004, 230, 666–674. [Google Scholar] [CrossRef]
- Waxman, J.S.; Yelon, D. Increased Hox activity mimics the teratogenic effects of excess retinoic acid signaling. Dev. Dyn. 2009, 238, 1207–1213. [Google Scholar] [CrossRef]
- D’Aniello, E.; Rydeen, A.B.; Anderson, J.L.; Mandal, A.; Waxman, J.S. Depletion of Retinoic Acid Receptors Initiates a Novel Positive Feedback Mechanism that Promotes Teratogenic Increases in Retinoic Acid. PLoS Genet. 2013, 9, e1003689. [Google Scholar] [CrossRef]
- Mendelsohn, C.; Lohnes, D.; Décimo, D.; Lufkin, T.; LeMeur, M.; Chambon, P.; Mark, M. Function of the retinoic acid receptors (RARs) during development (II). Multiple abnormalities at various stages of organogenesis in RAR double mutants. Development 1994, 120, 2749–2771. [Google Scholar] [PubMed]
- Dickman, E.D.; Thaller, C.; Smith, S.M. Temporally-regulated retinoic acid depletion produces specific neural crest, ocular and nervous system defects. Development 1997, 124, 3111–3121. [Google Scholar]
- Duester, G. Retinoic Acid Synthesis and Signaling during Early Organogenesis. Cell 2008, 134, 921–931. [Google Scholar] [CrossRef]
- Niederreither, K.; Dollé, P. Retinoic acid in development: Towards an integrated view. Nat. Rev. Genet. 2008, 9, 541–553. [Google Scholar] [CrossRef]
- Liu, J.; Stainier, D.Y.R. Zebrafish in the Study of Early Cardiac Development. Circ. Res. 2012, 110, 870–874. [Google Scholar] [CrossRef]
- Schilling, T.F.; Nie, Q.; Lander, A.D. Dynamics and precision in retinoic acid morphogen gradients. Curr. Opin. Genet. Dev. 2012, 22, 562–569. [Google Scholar] [CrossRef] [PubMed]
- Waxman, J.S.; Keegan, B.R.; Roberts, R.W.; Poss, K.D.; Yelon, D. Hoxb5b Acts Downstream of Retinoic Acid Signaling in the Forelimb Field to Restrict Heart Field Potential in Zebrafish. Dev. Cell 2008, 15, 923–934. [Google Scholar] [CrossRef]
- Rydeen, A.B.; Waxman, J.S. Cyp26 Enzymes Facilitate Second Heart Field Progenitor Addition and Maintenance of Ventricular Integrity. PLoS Biol. 2016, 14, 1–25. [Google Scholar] [CrossRef] [PubMed]
- De Bono, C.; Thellier, C.; Bertrand, N.; Sturny, R.; Jullian, E.; Cortes, C.; Stefanovic, S.; Zaffran, S.; Théveniau-Ruissy, M.; Kelly, R.G. T-box genes and retinoic acid signaling regulate the segregation of arterial and venous pole progenitor cells in the murine second heart field. Hum. Mol. Genet. 2018, 27, 3747–3760. [Google Scholar] [CrossRef] [PubMed]
- Rydeen, A.B.; Waxman, J.S. Cyp26 enzymes are required to balance the cardiac and vascular lineages within the anterior lateral plate mesoderm. Development 2014, 141, 1638–1648. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Van Bortle, K.; Zhang, Y.; Zhao, M.; Zhang, J.Z.; Geller, B.S.; Gruber, J.J.; Jiang, C.; Wu, J.C.; Snyder, M.P. Disruption of mesoderm formation during cardiac differentiation due to developmental exposure to 13-cis-retinoic acid. Sci. Rep. 2018, 8, 12960. [Google Scholar] [CrossRef] [PubMed]
- Bakkers, J. Zebrafish as a model to study cardiac development and human cardiac disease. Cardiovasc. Res. 2011, 91, 279–288. [Google Scholar] [CrossRef]
- Staudt, D.; Stainier, D. Uncovering the Molecular and Cellular Mechanisms of Heart Development Using the Zebrafish. Annu. Rev. Genet. 2012, 46, 397–418. [Google Scholar] [CrossRef] [PubMed]
- Santini, M.P.; Forte, E.; Harvey, R.P.; Kovacic, J.C. Developmental origin and lineage plasticity of endogenous cardiac stem cells. Development 2016, 143, 1242–1258. [Google Scholar] [CrossRef]
- Laugwitz, K.-L.; Moretti, A.; Caron, L.; Nakano, A.; Chien, K.R. Islet1 cardiovascular progenitors: A single source for heart lineages? Development 2007, 135, 193–205. [Google Scholar] [CrossRef]
- Buckingham, M. First and Second Heart Field. In Congenital Heart Diseases: The Broken Heart; Springer Vienna: Vienna, Austria, 2016; pp. 25–40. [Google Scholar]
- Durston, A.J.; Timmermans, J.P.M.; Hage, W.J.; Hendriks, H.F.J.; de Vries, N.J.; Heideveld, M.; Nieuwkoop, P.D. Retinoic acid causes an anteroposterior transformation in the developing central nervous system. Nature 1989, 340, 140–144. [Google Scholar] [CrossRef]
- Sive, H.L.; Draper, B.W.; Harland, R.M.; Weintraub, H. Identification of a retinoic acid-sensitive period during primary axis formation in Xenopus laevis. Genes Dev. 1990, 4, 932–942. [Google Scholar] [CrossRef]
- Hochgreb, T. A caudorostral wave of RALDH2 conveys anteroposterior information to the cardiac field. Development 2003, 130, 5363–5374. [Google Scholar] [CrossRef] [PubMed]
- Moss, J.B.; Xavier-Neto, J.; Shapiro, M.D.; Nayeem, S.M.; McCaffery, P.; Dräger, U.C.; Rosenthal, N. Dynamic Patterns of Retinoic Acid Synthesis and Response in the Developing Mammalian Heart. Dev. Biol. 1998, 199, 55–71. [Google Scholar] [CrossRef] [PubMed]
- Niederreither, K.; Fraulob, V.; Garnier, J.-M.; Chambon, P.; Dollé, P. Differential expression of retinoic acid-synthesizing (RALDH) enzymes during fetal development and organ differentiation in the mouse. Mech. Dev. 2002, 110, 165–171. [Google Scholar] [CrossRef]
- Xavier-Neto, J.; Rosenthal, N.; Silva, F.A.; Matos, T.G.F.; Hochgreb, T.; Linhares, V.L.F. Retinoid signaling and cardiac anteroposterior segmentation. Genesis 2001, 31, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Simões-Costa, M.S.; Vasconcelos, M.; Sampaio, A.C.; Cravo, R.M.; Linhares, V.L.; Hochgreb, T.; Yan, C.Y.I.; Davidson, B.; Xavier-Neto, J. The evolutionary origin of cardiac chambers. Dev. Biol. 2005, 277, 1–15. [Google Scholar] [CrossRef]
- Osmond, M.K.; Butler, A.J.; Voon, F.C.T.; Bellairs, R. The effects of retinoic acid on heart formation in the early chick embryo. Development 1991, 113, 1405–1417. [Google Scholar]
- Xavier-Neto, J.; Neville, C.; Shapiro, M.; Houghton, L.; Wang, G.; Nikovits, W.; Stockdale, F.; Rosenthal, N. A retinoic acid-inducible transgenic marker of sino-atrial development in the mouse heart. Development 1999, 126, 2677–2687. [Google Scholar] [PubMed]
- Devalla, H.D.; Schwach, V.; Ford, J.W.; Milnes, J.T.; El-Haou, S.; Jackson, C.; Gkatzis, K.; Elliott, D.A.; Chuva de Sousa Lopes, S.M.; Mummery, C.L.; et al. Atrial-like cardiomyocytes from human pluripotent stem cells are a robust preclinical model for assessing atrial-selective pharmacology. EMBO Mol. Med. 2015, 7, 394–410. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Jiang, J.; Han, P.; Yuan, Q.; Zhang, J.; Zhang, X.; Xu, Y.; Cao, H.; Meng, Q.; Chen, L.; et al. Direct differentiation of atrial and ventricular myocytes from human embryonic stem cells by alternating retinoid signals. Cell Res. 2011, 21, 579–587. [Google Scholar] [CrossRef]
- Gassanov, N.; Er, F.; Zagidullin, N.; Jankowski, M.; Gutkowska, J.; Hoppe, U.C. Retinoid acid-induced effects on atrial and pacemaker cell differentiation and expression of cardiac ion channels. Differentiation 2008, 76, 971–980. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Protze, S.I.; Laksman, Z.; Backx, P.H.; Keller, G.M. Human Pluripotent Stem Cell-Derived Atrial and Ventricular Cardiomyocytes Develop from Distinct Mesoderm Populations. Cell Stem Cell 2017, 21, 179–194.e4. [Google Scholar] [CrossRef] [PubMed]
- Quaranta, R.; Fell, J.; Rühle, F.; Rao, J.; Piccini, I.; Araúzo-Bravo, M.J.; Verkerk, A.O.; Stoll, M.; Greber, B. Revised roles of ISL1 in a hES cell-based model of human heart chamber specification. Elife 2018, 7, e31706. [Google Scholar] [CrossRef]
- Keegan, B.R.; Feldman, J.L.; Begemann, G.; Ingham, P.W.; Yelon, D. Retinoic acid signaling restricts the cardiac progenitor pool. Science 2005, 307, 247–249. [Google Scholar] [CrossRef] [PubMed]
- Collop, A.H.; Broomfield, J.A.S.; Chandraratna, R.A.S.; Yong, Z.; Deimling, S.J.; Kolker, S.J.; Weeks, D.L.; Drysdale, T.A. Retinoic acid signaling is essential for formation of the heart tube in Xenopus. Dev. Biol. 2006, 291, 96–109. [Google Scholar] [CrossRef] [PubMed]
- Ryckebusch, L.; Wang, Z.; Bertrand, N.; Lin, S.-C.; Chi, X.; Schwartz, R.; Zaffran, S.; Niederreither, K. Retinoic acid deficiency alters second heart field formation. Proc. Natl. Acad. Sci. USA 2008, 105, 2913–2918. [Google Scholar] [CrossRef] [PubMed]
- Sirbu, I.O.; Zhao, X.; Duester, G. Retinoic acid controls heart anteroposterior patterning by down-regulatingIsl1 through theFgf8 pathway. Dev. Dyn. 2008, 237, 1627–1635. [Google Scholar] [CrossRef] [PubMed]
- Keegan, B.R.; Meyer, D.; Yelon, D. Organization of cardiac chamber progenitors in the zebrafish blastula. Development 2004, 131, 3081–3091. [Google Scholar] [CrossRef]
- Schoenebeck, J.J.; Keegan, B.R.; Yelon, D. Vessel and Blood Specification Override Cardiac Potential in Anterior Mesoderm. Dev. Cell 2007, 13, 254–267. [Google Scholar] [CrossRef]
- Abu-Issa, R.; Kirby, M.L. Patterning of the heart field in the chick. Dev. Biol. 2008, 319, 223–233. [Google Scholar] [CrossRef]
- Roberts, C.; Ivins, S.; Cook, A.C.; Baldini, A.; Scambler, P.J. Cyp26 genes a1, b1 and c1 are down-regulated in Tbx1 null mice and inhibition of Cyp26 enzyme function produces a phenocopy of DiGeorge Syndrome in the chick. Hum. Mol. Genet. 2006, 15, 3394–3410. [Google Scholar] [CrossRef]
- Pennimpede, T.; Cameron, D.A.; MacLean, G.A.; Li, H.; Abu-Abed, S.; Petkovich, M. The role of CYP26 enzymes in defining appropriate retinoic acid exposure during embryogenesis. Birth Defects Res. Part A Clin. Mol. Teratol. 2010, 88, 883–894. [Google Scholar] [CrossRef] [PubMed]
- Billings, S.E.; Pierzchalski, K.; Butler Tjaden, N.E.; Pang, X.-Y.; Trainor, P.A.; Kane, M.A.; Moise, A.R. The retinaldehyde reductase DHRS3 is essential for preventing the formation of excess retinoic acid during embryonic development. FASEB J. 2013, 27, 4877–4889. [Google Scholar] [CrossRef]
- Pasutto, F.; Sticht, H.; Hammersen, G.; Gillessen-Kaesbach, G.; FitzPatrick, D.R.; Nürnberg, G.; Brasch, F.; Schirmer-Zimmermann, H.; Tolmie, J.L.; Chitayat, D.; et al. Mutations in STRA6 Cause a Broad Spectrum of Malformations Including Anophthalmia, Congenital Heart Defects, Diaphragmatic Hernia, Alveolar Capillary Dysplasia, Lung Hypoplasia, and Mental Retardation. Am. J. Hum. Genet. 2007, 80, 550–560. [Google Scholar] [CrossRef]
- Golzio, C.; Martinovic-Bouriel, J.; Thomas, S.; Mougou-Zrelli, S.; Grattagliano-Bessières, B.; Bonnière, M.; Delahaye, S.; Munnich, A.; Encha-Razavi, F.; Lyonnet, S.; et al. Matthew-Wood Syndrome Is Caused by Truncating Mutations in the Retinol-Binding Protein Receptor Gene STRA6. Am. J. Hum. Genet. 2007, 80, 1179–1187. [Google Scholar] [CrossRef] [PubMed]
- Noy, N. Vitamin a transport and cell Signaling by the retinol-binding protein receptor STRA6. In Sub-Cellular Biochemistry; Springer: Berlin, Germany, 2016; Volume 81, pp. 77–93. [Google Scholar]
- Ghyselinck, N.; Wendling, O.; Messaddeq, N.; Dierich, A.; Lampron, C.; Décimo, D.; Viville, S.; Chambon, P.; Mark, M. Contribution of retinoic acid receptor β isoforms to the formation of the conotruncal septum of the embryonic heart. Dev. Biol. 1998, 198, 303–318. [Google Scholar] [CrossRef]
- Li, P.; Pashmforoush, M.; Sucov, H.M. Retinoic acid regulates differentiation of the secondary heart field and TGFbeta-mediated outflow tract septation. Dev. Cell 2010, 18, 480–485. [Google Scholar] [CrossRef]
- Abu-Abed, S.; Dollé, P.; Metzger, D.; Beckett, B.; Chambon, P.; Petkovich, M. The retinoic acid-metabolizing enzyme, CYP26A1, is essential for normal hindbrain patterning, vertebral identity, and development of posterior structures. Genes Dev. 2001, 15, 226–240. [Google Scholar] [CrossRef]
- Kudoh, T.; Wilson, S.W.; Dawid, I.B. Distinct roles for Fgf, Wnt and retinoic acid in posteriorizing the neural ectoderm. Development 2002, 129, 4335–4346. [Google Scholar]
- Emoto, Y.; Wada, H.; Okamoto, H.; Kudo, A.; Imai, Y. Retinoic acid-metabolizing enzyme Cyp26a1 is essential for determining territories of hindbrain and spinal cord in zebrafish. Dev. Biol. 2005, 278, 415–427. [Google Scholar] [CrossRef]
- Uehara, M.; Yashiro, K.; Mamiya, S.; Nishino, J.; Chambon, P.; Dolle, P.; Sakai, Y. CYP26A1 and CYP26C1 cooperatively regulate anterior–posterior patterning of the developing brain and the production of migratory cranial neural crest cells in the mouse. Dev. Biol. 2007, 302, 399–411. [Google Scholar] [CrossRef]
- Romeih, M.; Cui, J.; Michaille, J.-J.; Jiang, W.; Zile, M.H. Function of RARgamma and RARalpha2 at the initiation of retinoid signaling is essential for avian embryo survival and for distinct events in cardiac morphogenesis. Dev. Dyn. 2003, 228, 697–708. [Google Scholar] [CrossRef] [PubMed]
- Sandell, L.L.; Sanderson, B.W.; Moiseyev, G.; Johnson, T.; Mushegian, A.; Young, K.; Rey, J.-P.; Ma, J.-X.; Staehling-Hampton, K.; Trainor, P.A. RDH10 is essential for synthesis of embryonic retinoic acid and is required for limb, craniofacial, and organ development. Genes Dev. 2007, 21, 1113–1124. [Google Scholar] [CrossRef]
- Sandell, L.L.; Lynn, M.L.; Inman, K.E.; McDowell, W.; Trainor, P.A. RDH10 Oxidation of Vitamin A Is a Critical Control Step in Synthesis of Retinoic Acid during Mouse Embryogenesis. PLoS ONE 2012, 7, e30698. [Google Scholar] [CrossRef] [PubMed]
- Rhinn, M.; Schuhbaur, B.; Niederreither, K.; Dolle, P. Involvement of retinol dehydrogenase 10 in embryonic patterning and rescue of its loss of function by maternal retinaldehyde treatment. Proc. Natl. Acad. Sci. USA 2011, 108, 16687–16692. [Google Scholar] [CrossRef] [PubMed]
- Cubuk, P.O.; Ho, L.; Reversade, B.; Perçin, E.F. MATTHEW-WOOD SYNDROME: A CASE WITH DEXTROCARDIA AND STREAK GONADS. Genet. Couns. 2016, 27, 405–410. [Google Scholar]
- Dollé, P.; Niederreither, K. The Retinoids; Dollé, P., Neiderreither, K., Eds.; John Wiley & Sons, Inc: Hoboken, NJ, USA, 2015; ISBN 9781118628003. [Google Scholar]
- Zhao, X.; Sirbu, I.O.; Mic, F.A.; Molotkova, N.; Molotkov, A.; Kumar, S.; Duester, G. Retinoic Acid Promotes Limb Induction through Effects on Body Axis Extension but Is Unnecessary for Limb Patterning. Curr. Biol. 2009, 19, 1050–1057. [Google Scholar] [CrossRef]
- Cunningham, T.J.; Zhao, X.; Sandell, L.L.; Evans, S.M.; Trainor, P.A.; Duester, G. Antagonism between Retinoic Acid and Fibroblast Growth Factor Signaling during Limb Development. Cell Rep. 2013, 3, 1503–1511. [Google Scholar] [CrossRef]
- Sorrell, M.R.J.; Waxman, J.S. Restraint of Fgf8 signaling by retinoic acid signaling is required for proper heart and forelimb formation. Dev. Biol. 2011, 358, 44–55. [Google Scholar] [CrossRef]
- Yi Li, Q.; Newbury-Ecob, R.A.; Terrett, J.A.; Wilson, D.I.; Curtis, A.R.J.; Ho Yi, C.; Gebuhr, T.; Bullen, P.J.; Robson, S.C.; Strachan, T.; et al. Holt-Oram syndrome is caused by mutations in TBX5, a member of the Brachyury (T) gene family. Nat. Genet. 1997, 15, 21–29. [Google Scholar] [CrossRef]
- Basson, C.T.; Bachinsky, D.R.; Lin, R.C.; Levi, T.; Elkins, J.A.; Soults, J.; Grayzel, D.; Kroumpouzou, E.; Traill, T.A.; Leblanc-Straceski, J.; et al. Mutations in human cause limb and cardiac malformation in Holt-Oram syndrome. Nat. Genet. 1997, 15, 30–35. [Google Scholar] [CrossRef]
- Duong, T.B.; Ravisankar, P.; Song, Y.C.; Gafranek, J.T.; Rydeen, A.B.; Dohn, T.E.; Barske, L.A.; Crump, J.G.; Waxman, J.S. Nr2f1a balances atrial chamber and atrioventricular canal size via BMP signaling-independent and -dependent mechanisms. Dev. Biol. 2018, 434, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Barski, A.; Zhao, K. Genomic location analysis by ChIP-Seq. J. Cell. Biochem. 2009, 107, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Duester, G. Retinoic acid controls body axis extension by directly repressing Fgf8 transcription. Development 2014, 141, 2972–2977. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Cunningham, T.J.; Duester, G. Nuclear receptor corepressors Ncor1 and Ncor2 (Smrt) are required for retinoic acid-dependent repression of Fgf8 during somitogenesis. Dev. Biol. 2016, 418, 204–215. [Google Scholar] [CrossRef] [PubMed]
- Rudnicki, M.A.; McBurney, M.W. Teratocarcinomas and Embryonic Stem Cells: A practical Approach; Robertson, E., Ed.; Oxford: Washington, D.C., USA, 1987; ISBN 1852210044. [Google Scholar]
- Edwards, M.K.; McBurney, M.W. The concentration of retinoic acid determines the differentiated cell types formed by a teratocarcinoma cell line. Dev. Biol. 1983, 98, 187–191. [Google Scholar] [CrossRef]
- Wobus, A.M.; Rohwedel, J.; Maltsev, V.; Hescheler, J. In vitro differentiation of embryonic stem cells into cardiomyocytes or skeletal muscle cells is specifically modulated by retinoic acid. Roux’s Arch. Dev. Biol. 1994, 204, 36–45. [Google Scholar] [CrossRef]
- Hernandez, R.E.; Putzke, A.P.; Myers, J.P.; Margaretha, L.; Moens, C.B. Cyp26 enzymes generate the retinoic acid response pattern necessary for hindbrain development. Development 2007, 134, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Samarut, E.; Fraher, D.; Gibert, Y. ZebRA: An overview of retinoic acid signaling during zebrafish development. Biochim. Biophys. Acta Gene Regul. Mech. 2015, 1849, 73–83. [Google Scholar] [CrossRef]
- White, R.J.; Nie, Q.; Lander, A.D.; Schilling, T.F. Complex Regulation of cyp26a1 Creates a Robust Retinoic Acid Gradient in the Zebrafish Embryo. PLoS Biol. 2007, 5, e304. [Google Scholar] [CrossRef]
- Sosnik, J.; Zheng, L.; Rackauckas, C.V.; Digman, M.; Gratton, E.; Nie, Q.; Schilling, T.F. Noise modulation in retinoic acid signaling sharpens segmental boundaries of gene expression in the embryonic zebrafish hindbrain. Elife 2016, 5, e14034. [Google Scholar] [CrossRef]
- Shimozono, S.; Iimura, T.; Kitaguchi, T.; Higashijima, S.-I.; Miyawaki, A. Visualization of an endogenous retinoic acid gradient across embryonic development. Nature 2013, 496, 363–366. [Google Scholar] [CrossRef]
- Ruiz, A.; Mark, M.; Jacobs, H.; Klopfenstein, M.; Hu, J.; Lloyd, M.; Habib, S.; Tosha, C.; Radu, R.A.; Ghyselinck, N.B.; et al. Retinoid content, visual responses, and ocular morphology are compromised in the retinas of mice lacking the retinol-binding protein receptor, STRA6. Investig. Ophthalmol. Vis. Sci. 2012, 53, 3027–3039. [Google Scholar] [CrossRef]
- Berry, D.C.; Jacobs, H.; Marwarha, G.; Gely-Pernot, A.; O’Byrne, S.M.; DeSantis, D.; Klopfenstein, M.; Feret, B.; Dennefeld, C.; Blaner, W.S.; et al. The STRA6 receptor is essential for retinol-binding protein-induced insulin resistance but not for maintaining vitamin A homeostasis in tissues other than the eye. J. Biol. Chem. 2013, 288, 24528–24539. [Google Scholar] [CrossRef]
- Amengual, J.; Zhang, N.; Kemerer, M.; Maeda, T.; Palczewski, K.; Von Lintig, J. STRA6 is critical for cellular vitamin A uptake and homeostasis. Hum. Mol. Genet. 2014, 23, 5402–5417. [Google Scholar] [CrossRef]
- Quadro, L.; Blaner, W.S.; Salchow, D.J.; Vogel, S.; Piantedosi, R.; Gouras, P.; Freeman, S.; Cosma, M.P.; Colantuoni, V.; Gottesman, M.E. Impaired retinal function and vitamin A availability in mice lacking retinol-binding protein. EMBO J. 1999, 18, 4633–4644. [Google Scholar] [CrossRef]
- Batten, M.L.; Imanishi, Y.; Maeda, T.; Tu, D.C.; Moise, A.R.; Bronson, D.; Possin, D.; Van Gelder, R.N.; Baehr, W.; Palczewski, K. Lecithin-retinol Acyltransferase Is Essential for Accumulation of All- trans -Retinyl Esters in the Eye and in the Liver. J. Biol. Chem. 2004, 279, 10422–10432. [Google Scholar] [CrossRef]
- Kedishvili, N.Y. Enzymology of retinoic acid biosynthesis and degradation. J. Lipid Res. 2013, 54, 1744–1760. [Google Scholar] [CrossRef]
- Chatzi, C.; Cunningham, T.J.; Duester, G. Investigation of retinoic acid function during embryonic brain development using retinaldehyde-rescued Rdh10 knockout mice. Dev. Dyn. 2013, 242, 1056–1065. [Google Scholar] [CrossRef]
- D’Aniello, E.; Ravisankar, P.; Waxman, J.S. Rdh10a Provides a Conserved Critical Step in the Synthesis of Retinoic Acid during Zebrafish Embryogenesis. PLoS ONE 2015, 10, e0138588. [Google Scholar] [CrossRef]
- Ryckebüsch, L.; Bertrand, N.; Mesbah, K.; Bajolle, F.; Niederreither, K.; Kelly, R.G.; Zaffran, S. Decreased levels of embryonic retinoic acid synthesis accelerate recovery from arterial growth delay in a mouse model of DiGeorge syndrome. Circ. Res. 2010, 106, 686–694. [Google Scholar] [CrossRef]
- Théveniau-Ruissy, M.; Dandonneau, M.; Mesbah, K.; Ghez, O.; Mattei, M.-G.; Miquerol, L.; Kelly, R.G. The del22q11.2 Candidate Gene Tbx1 Controls Regional Outflow Tract Identity and Coronary Artery Patterning. Circ. Res. 2008, 103, 142–148. [Google Scholar] [CrossRef]
- Rana, M.S.; Théveniau-Ruissy, M.; De Bono, C.; Mesbah, K.; Francou, A.; Rammah, M.; Domínguez, J.N.; Roux, M.; Laforest, B.; Anderson, R.H.; et al. Tbx1 Coordinates Addition of Posterior Second Heart Field Progenitor Cells to the Arterial and Venous Poles of the Heart. Circ. Res. 2014, 115, 790–799. [Google Scholar] [CrossRef]
- El Robrini, N.; Etchevers, H.C.; Ryckebüsch, L.; Faure, E.; Eudes, N.; Niederreither, K.; Zaffran, S.; Bertrand, N. Cardiac outflow morphogenesis depends on effects of retinoic acid signaling on multiple cell lineages. Dev. Dyn. 2016, 245, 388–401. [Google Scholar] [CrossRef]
- Dobbs-McAuliffe, B.; Zhao, Q.; Linney, E. Feedback mechanisms regulate retinoic acid production and degradation in the zebrafish embryo. Mech. Dev. 2004, 121, 339–350. [Google Scholar] [CrossRef]
- Yagi, H.; Furutani, Y.; Hamada, H.; Sasaki, T.; Asakawa, S.; Minoshima, S.; Ichida, F.; Joo, K.; Kimura, M.; Imamura, S.; et al. Role of TBX1 in human del22q11.2 syndrome. Lancet 2003, 362, 1366–1373. [Google Scholar] [CrossRef]
- Guris, D.L.; Duester, G.; Papaioannou, V.E.; Imamoto, A. Dose-Dependent Interaction of Tbx1 and Crkl and Locally Aberrant RA Signaling in a Model of del22q11 Syndrome. Dev. Cell 2006, 10, 81–92. [Google Scholar] [CrossRef]
- Roberts, C.; Ivins, S.M.; James, C.T.; Scambler, P.J. Retinoic acid down-regulatesTbx1 expression in vivo and in vitro. Dev. Dyn. 2005, 232, 928–938. [Google Scholar] [CrossRef]
- Maynard, T.M.; Gopalakrishna, D.; Meechan, D.W.; Paronett, E.M.; Newbern, J.M.; LaMantia, A.-S. 22q11 Gene dosage establishes an adaptive range for sonic hedgehog and retinoic acid signaling during early development. Hum. Mol. Genet. 2013, 22, 300–312. [Google Scholar] [CrossRef]
- Niederreither, K.; Abu-Abed, S.; Schuhbaur, B.; Petkovich, M.; Chambon, P.; Dollé, P. Genetic evidence that oxidative derivatives of retinoic acid are not involved in retinoid signaling during mouse development. Nat. Genet. 2002, 31, 84–88. [Google Scholar] [CrossRef]
- Sakai, Y.; Meno, C.; Fujii, H.; Nishino, J.; Shiratori, H.; Saijoh, Y.; Rossant, J.; Hamada, H. The retinoic acid-inactivating enzyme CYP26 is essential for establishing an uneven distribution of retinoic acid along the anterio-posterior axis within the mouse embryo. Genes Dev. 2001, 15, 213–225. [Google Scholar] [CrossRef]
- Adams, M.K.; Belyaeva, O.V.; Wu, L.; Kedishvili, N.Y. The retinaldehyde reductase activity of DHRS3 is reciprocally activated by retinol dehydrogenase 10 to control retinoid homeostasis. J. Biol. Chem. 2014, 289, 14868–14880. [Google Scholar] [CrossRef]
- Feng, L.; Hernandez, R.E.; Waxman, J.S.; Yelon, D.; Moens, C.B. Dhrs3a regulates retinoic acid biosynthesis through a feedback inhibition mechanism. Dev. Biol. 2010, 338, 1–14. [Google Scholar] [CrossRef]
- Bastien, J.; Rochette-Egly, C. Nuclear retinoid receptors and the transcription of retinoid-target genes. Gene 2004, 328, 1–16. [Google Scholar] [CrossRef]
- Dupé, V.; Ghyselinck, N.B.; Wendling, O.; Chambon, P.; Mark, M. Key roles of retinoic acid receptors alpha and beta in the patterning of the caudal hindbrain, pharyngeal arches and otocyst in the mouse. Development 1999, 126, 5051–5059. [Google Scholar]
- Sucov, H.M.; Dyson, E.; Gumeringer, C.L.; Price, J.; Chien, K.R.; Evans, R.M. RXR alpha mutant mice establish a genetic basis for vitamin A signaling in heart morphogenesis. Genes Dev. 1994, 8, 1007–1018. [Google Scholar] [CrossRef]
- Gruber, P.J.; Kubalak, S.W.; Pexieder, T.; Sucov, H.M.; Evans, R.M.; Chien, K.R. RXR alpha deficiency confers genetic susceptibility for aortic sac, conotruncal, atrioventricular cushion, and ventricular muscle defects in mice. J. Clin. Investig. 1996, 98, 1332–1343. [Google Scholar] [CrossRef]
- Kastner, P.; Grondona, J.M.; Mark, M.; Gansmuller, A.; LeMeur, M.; Decimo, D.; Vonesch, J.-L.; Dollé, P.; Chambon, P. Genetic analysis of RXRα developmental function: Convergence of RXR and RAR signaling pathways in heart and eye morphogenesis. Cell 1994, 78, 987–1003. [Google Scholar] [CrossRef]
- Li, E.; Sucov, H.M.; Lee, K.F.; Evans, R.M.; Jaenisch, R. Normal development and growth of mice carrying a targeted disruption of the alpha 1 retinoic acid receptor gene. Proc. Natl. Acad. Sci. USA 1993, 90, 1590–1594. [Google Scholar] [CrossRef]
- Kastner, P.; Messaddeq, N.; Mark, M.; Wendling, O.; Grondona, J.M.; Ward, S.; Ghyselinck, N.; Chambon, P. Vitamin A deficiency and mutations of RXRalpha, RXRbeta and RARalpha lead to early differentiation of embryonic ventricular cardiomyocytes. Development 1997, 124. [Google Scholar]
- Chen, J.; Kubalak, S.W.; Chien, K.R. Ventricular muscle-restricted targeting of the RXRalpha gene reveals a non-cell-autonomous requirement in cardiac chamber morphogenesis. Development 1998, 125, 1943–1949. [Google Scholar]
- Xin, M.; Davis, C.A.; Molkentin, J.D.; Lien, C.-L.; Duncan, S.A.; Richardson, J.A.; Olson, E.N. A threshold of GATA4 and GATA6 expression is required for cardiovascular development. Proc. Natl. Acad. Sci. USA 2006, 103, 11189–11194. [Google Scholar] [CrossRef]
- Sakabe, M.; Kokubo, H.; Nakajima, Y.; Saga, Y. Ectopic retinoic acid signaling affects outflow tract cushion development through suppression of the myocardial Tbx2-Tgf 2 pathway. Development 2012, 139, 385–395. [Google Scholar] [CrossRef]
- Bertrand, S.; Thisse, B.; Tavares, R.; Sachs, L.; Chaumot, A.; Bardet, P.-L.; Escrivà, H.; Duffraisse, M.; Marchand, O.; Safi, R.; et al. Unexpected Novel Relational Links Uncovered by Extensive Developmental Profiling of Nuclear Receptor Expression. PLoS Genet. 2007, 3, e188. [Google Scholar] [CrossRef]
- Waxman, J.S.; Yelon, D. Comparison of the expression patterns of newly identified zebrafish retinoic acid and retinoid X receptors. Dev. Dyn. 2007, 236, 587–595. [Google Scholar] [CrossRef]
- Linville, A.; Radtke, K.; Waxman, J.S.; Yelon, D.; Schilling, T.F. Combinatorial roles for zebrafish retinoic acid receptors in the hindbrain, limbs and pharyngeal arches. Dev. Biol. 2009, 325, 60–70. [Google Scholar] [CrossRef]
- Huang, D.; Chen, S.; Langston, A.; Gudas, L. A conserved retinoic acid responsive element in the murine Hoxb-1 gene is required for expression in the developing gut. Development 1998, 125, 3235–3246. [Google Scholar]
- Marshall, H.; Studer, M.; Pöpperl, H.; Aparicio, S.; Kuroiwa, A.; Brenner, S.; Krumlauf, R. A conserved retinoic acid response element required for early expression of the homeobox gene Hoxb-1. Nature 1994, 370, 567–571. [Google Scholar] [CrossRef]
- Oosterveen, T.; van Vliet, P.; Deschamps, J.; Meijlink, F. The Direct Context of a Hox Retinoic Acid Response Element Is Crucial for its Activity. J. Biol. Chem. 2003, 278, 24103–24107. [Google Scholar] [CrossRef]
- Langston, A.W.; Thompson, J.R.; Gudas, L.J. Retinoic Acid-responsive Enhancers Located 3′ of the Hox A and Hox B Homeobox Gene Clusters. J. Biol. Chem. 1997, 272, 2167–2175. [Google Scholar] [CrossRef]
- LaRosa, G.J.; Gudas, L.J. Early retinoic acid-induced F9 teratocarcinoma stem cell gene ERA-1: alternate splicing creates transcripts for a homeobox-containing protein and one lacking the homeobox. Mol. Cell. Biol. 1988, 8, 3906–3917. [Google Scholar] [CrossRef]
- Dupé, V.; Davenne, M.; Brocard, J.; Dollé, P.; Mark, M.; Dierich, A.; Chambon, P.; Rijli, F.M. In vivo functional analysis of the Hoxa-1 3’ retinoic acid response element (3’RARE). Development 1997, 124, 399–410. [Google Scholar]
- Roux, M.; Laforest, B.; Capecchi, M.; Bertrand, N.; Zaffran, S. Hoxb1 regulates proliferation and differentiation of second heart field progenitors in pharyngeal mesoderm and genetically interacts with Hoxa1 during cardiac outflow tract development. Dev. Biol. 2015, 406, 247–258. [Google Scholar] [CrossRef]
- Bertrand, N.; Roux, M.; Ryckebüsch, L.; Niederreither, K.; Dollé, P.; Moon, A.; Capecchi, M.; Zaffran, S. Hox genes define distinct progenitor sub-domains within the second heart field. Dev. Biol. 2011, 353, 266–274. [Google Scholar] [CrossRef]
- Nolte, C.; Jinks, T.; Wang, X.; Martinez Pastor, M.T.; Krumlauf, R. Shadow enhancers flanking the HoxB cluster direct dynamic Hox expression in early heart and endoderm development. Dev. Biol. 2013, 383, 158–173. [Google Scholar] [CrossRef]
- Jonk, L.J.C.; de Jonge, M.E.J.; Vervaart, J.M.A.; Wissink, S.; Kruijer, W. Isolation and Developmental Expression of Retinoic-Acid-Induced Genes. Dev. Biol. 1994, 161, 604–614. [Google Scholar] [CrossRef]
- van der Wees, J.; Matharu, P.J.; de Roos, K.; Destre´e, O.H.J.; Godsave, S.F.; Durston, A.J.; Sweeney, G.E. Developmental expression and differential regulation by retinoic acid ofXenopus COUP-TF-A andCOUP-TF-B. Mech. Dev. 1996, 54, 173–184. [Google Scholar] [CrossRef]
- Pereira, F.A.; Tsai, M.J.; Tsai, S.Y. COUP-TF orphan nuclear receptors in development and differentiation. Cell. Mol. Life Sci. 2000, 57, 1388–1398. [Google Scholar] [CrossRef]
- Love, C.E.; Prince, V.E. Expression and retinoic acid regulation of the zebrafish nr2f orphan nuclear receptor genes. Dev. Dyn. 2012, 241, 1603–1615. [Google Scholar] [CrossRef]
- Laursen, K.B.; Mongan, N.P.; Zhuang, Y.; Ng, M.M.; Benoit, Y.D.; Gudas, L.J. Polycomb recruitment attenuates retinoic acid–induced transcription of the bivalent NR2F1 gene. Nucleic Acids Res. 2013, 41, 6430–6443. [Google Scholar] [CrossRef]
- Dohn, T.E.; Ravisankar, P.; Tirera, F.T.; Martin, K.E.; Gafranek, J.T.; Duong, T.B.; VanDyke, T.L.; Touvron, M.; Barske, L.A.; Crump, J.G.; et al. Nr2f-dependent allocation of ventricular cardiomyocyte and pharyngeal muscle progenitors. PLoS Genet. 2019, 15, e1007962. [Google Scholar] [CrossRef]
- Lin, F.-J.; Qin, J.; Tang, K.; Tsai, S.Y.; Tsai, M.-J. Coup d’Etat: An Orphan Takes Control. Endocr. Rev. 2011, 32, 404–421. [Google Scholar] [CrossRef]
- Pereira, F.A.; Qiu, Y.; Zhou, G.; Tsai, M.-J.; Tsai, S.Y. The orphan nuclear receptor COUP-TFII is required for angiogenesis and heart development. Genes Dev. 1999, 13, 1037–1049. [Google Scholar] [CrossRef]
- Wu, S.P.; Cheng, C.M.; Lanz, R.B.; Wang, T.; Respress, J.L.; Ather, S.; Chen, W.; Tsai, S.J.; Wehrens, X.H.T.; Tsai, M.J.; et al. Atrial Identity Is Determined by a COUP-TFII Regulatory Network. Dev. Cell 2013, 25, 417–426. [Google Scholar] [CrossRef]
- Begemann, G.; Schilling, T.F.; Rauch, G.J.; Geisler, R.; Ingham, P.W. The zebrafish neckless mutation reveals a requirement for raldh2 in mesodermal signals that pattern the hindbrain. Development 2001, 128, 3081–3094. [Google Scholar]
- Niederreither, K. The regional pattern of retinoic acid synthesis by RALDH2 is essential for the development of posterior pharyngeal arches and the enteric nervous system. Development 2003, 130, 2525–2534. [Google Scholar] [CrossRef]
- Meilhac, S.M.; Lescroart, F.; Blanpain, C.; Buckingham, M.E. Cardiac cell lineages that form the heart. Cold Spring Harb. Perspect. Med. 2014, 4, a013888. [Google Scholar] [CrossRef]
- Lescroart, F.; Meilhac, S.M. Cell lineages, growth and repair of the mouse heart. In Mouse Development. Results and Problems in Cell Differentiation; Kubiak, J., Ed.; Springer: Berlin, Germany, 2012; pp. 263–289. ISBN 978-3-642-30406-4. [Google Scholar]
- Niederreither, K.; Vermot, J.; Messaddeq, N.; Schuhbaur, B.; Chambon, P.; Dollé, P. Embryonic retinoic acid synthesis is essential for heart morphogenesis in the mouse. Development 2001, 128, 1019–1031. [Google Scholar]
- Von Gise, A.; Zhou, B.; Honor, L.B.; Ma, Q.; Petryk, A.; Pu, W.T. WT1 regulates epicardial epithelial to mesenchymal transition through β-catenin and retinoic acid signaling pathways. Dev. Biol. 2011, 356, 421–431. [Google Scholar] [CrossRef]
- Braitsch, C.M.; Combs, M.D.; Quaggin, S.E.; Yutzey, K.E. Pod1/Tcf21 is regulated by retinoic acid signaling and inhibits differentiation of epicardium-derived cells into smooth muscle in the developing heart. Dev. Biol. 2012, 368, 345–357. [Google Scholar] [CrossRef]
- Xavier-Neto, J.; Shapiro, M.D.; Houghton, L.; Rosenthal, N. Sequential programs of retinoic acid synthesis in the myocardial and epicardial layers of the developing avian heart. Dev. Biol. 2000, 219, 129–141. [Google Scholar] [CrossRef]
- Pérez-Pomares, J.M.; Phelps, A.; Sedmerova, M.; Carmona, R.; González-Iriarte, M.; Muoz-Chápuli, R.; Wessels, A. Experimental studies on the spatiotemporal expression of WT1 and RALDH2 in the embryonic avian heart: A model for the regulation of myocardial and valvuloseptal development by epicardially derived cells (EPDCs). Dev. Biol. 2002, 247, 307–326. [Google Scholar] [CrossRef]
- Guadix, J.A.; Ruiz-Villalba, A.; Lettice, L.; Velecela, V.; Munoz-Chapuli, R.; Hastie, N.D.; Perez-Pomares, J.M.; Martinez-Estrada, O.M. Wt1 controls retinoic acid signalling in embryonic epicardium through transcriptional activation of Raldh2. Development 2011, 138, 1093–1097. [Google Scholar] [CrossRef]
- Brade, T.; Kumar, S.; Cunningham, T.J.; Chatzi, C.; Zhao, X.; Cavallero, S.; Li, P.; Sucov, H.M.; Ruiz-Lozano, P.; Duester, G. Retinoic acid stimulates myocardial expansion by induction of hepatic erythropoietin which activates epicardial Igf2. Development 2011, 138, 139–148. [Google Scholar] [CrossRef]
- Stuckmann, I.; Evans, S.; Lassar, A.B. Erythropoietin and retinoic acid, secreted from the epicardium, are required for cardiac myocyte proliferation. Dev. Biol. 2003, 255, 334–349. [Google Scholar] [CrossRef]
- Kang, J.O.; Sucov, H.M. Convergent proliferative response and divergent morphogenic pathways induced by epicardial and endocardial signaling in fetal heart development. Mech. Dev. 2005, 122, 57–65. [Google Scholar] [CrossRef]
- Lin, S.-C.; Dolle, P.; Ryckebusch, L.; Noseda, M.; Zaffran, S.; Schneider, M.D.; Niederreither, K. Endogenous retinoic acid regulates cardiac progenitor differentiation. Proc. Natl. Acad. Sci. USA 2010, 107, 9234–9239. [Google Scholar] [CrossRef]
- Merki, E.; Zamora, M.; Raya, A.; Kawakami, Y.; Wang, J.; Zhang, X.; Burch, J.; Kubalak, S.W.; Kaliman, P.; Belmonte, J.C.I.; et al. Epicardial retinoid X receptor is required for myocardial growth and coronary artery formation. Proc. Natl. Acad. Sci. USA 2005, 102, 18455–18460. [Google Scholar] [CrossRef]
- Lavine, K.J.; Yu, K.; White, A.C.; Zhang, X.; Smith, C.; Partanen, J.; Ornitz, D.M. Endocardial and epicardial derived FGF signals regulate myocardial proliferation and differentiation in vivo. Dev. Cell 2005, 8, 85–95. [Google Scholar] [CrossRef]
- Itou, J.; Oishi, I.; Kawakami, H.; Glass, T.J.; Richter, J.; Johnson, A.; Lund, T.C.; Kawakami, Y. Migration of cardiomyocytes is essential for heart regeneration in zebrafish. Development 2012, 139, 4133–4142. [Google Scholar] [CrossRef]
- Lepilina, A.; Coon, A.N.; Kikuchi, K.; Holdway, J.E.; Roberts, R.W.; Burns, C.G.; Poss, K.D. A Dynamic Epicardial Injury Response Supports Progenitor Cell Activity during Zebrafish Heart Regeneration. Cell 2006, 127, 607–619. [Google Scholar] [CrossRef]
- Kikuchi, K.; Holdway, J.E.; Major, R.J.; Blum, N.; Dahn, R.D.; Begemann, G.; Poss, K.D. Retinoic acid production by endocardium and epicardium is an injury response essential for zebrafish heart regeneration. Dev. Cell 2011, 20, 397–404. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cardiac Defect | Genes | Species | References |
|---|---|---|---|
| Increased CM specification/Cardiomegaly | ALDH1A2 | Mouse | [28] |
| Zebrafish | [65] | ||
| CYP26A1;CYP26C1 | Zebrafish | [42,44] | |
| RARαb1 | Zebrafish | [34] | |
| Outflow-tract defects | CYP26A1 | Human | [72,73] |
| DHRS3 | Mouse | [26,74] | |
| STRA6 | Human | [75,76,77] | |
| RARα1;RARβ | |||
| RARα1;RARγ | |||
| RARα1;RXRα | Mouse | [35,78,79] | |
| RARβ1;RXRα | |||
| RARβ2;RXRα | |||
| Atrioventricular septal defects | DHRS3 | Mouse | [74] |
| STRA6 | Human | [76] | |
| Asymmetry/ Looping defects | ALDH1A2 | Mouse | [28,53,54,55,67,68] |
| CYP26A1 | Mouse | [80,81,82] | |
| CYP26A1;CYP26C1 | Mouse | [83] | |
| RARγ | Chicken | [84] | |
| RDH10 | Mouse | [85,86,87] | |
| STRA6 | Human | [88] | |
| Inflow-tract defects | RARα2 | Chicken | [84] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perl, E.; Waxman, J.S. Reiterative Mechanisms of Retinoic Acid Signaling during Vertebrate Heart Development. J. Dev. Biol. 2019, 7, 11. https://doi.org/10.3390/jdb7020011
Perl E, Waxman JS. Reiterative Mechanisms of Retinoic Acid Signaling during Vertebrate Heart Development. Journal of Developmental Biology. 2019; 7(2):11. https://doi.org/10.3390/jdb7020011
Chicago/Turabian StylePerl, Eliyahu, and Joshua S. Waxman. 2019. "Reiterative Mechanisms of Retinoic Acid Signaling during Vertebrate Heart Development" Journal of Developmental Biology 7, no. 2: 11. https://doi.org/10.3390/jdb7020011
APA StylePerl, E., & Waxman, J. S. (2019). Reiterative Mechanisms of Retinoic Acid Signaling during Vertebrate Heart Development. Journal of Developmental Biology, 7(2), 11. https://doi.org/10.3390/jdb7020011