Crosstalk of Intercellular Signaling Pathways in the Generation of Midbrain Dopaminergic Neurons In Vivo and from Stem Cells


Abstract
1. Introduction
2. Signaling Pathways in Midbrain Dopaminergic Neuron Generation In Vivo and In Vitro
2.1. FGFs/FGF8 Signaling
2.1.1. FGF Signaling Pathway in the Embryonic Midbrain
2.1.2. FGF-Regulated Developmental Processes in the mDA Neuron Progenitors
2.1.3. FGF Signaling Promotes mDA Neuron Differentiation In Vitro
2.2. SHH Signaling
2.2.1. The SHH Signaling Pathway
2.2.2. Expression of Shh Pathway Components in the Ventral Midbrain
2.2.3. Fate Mapping of SHH-Expressing and SHH-Responding Progenitors in the VM
2.2.4. In Vivo Function of the SHH Signaling Pathway
2.2.5. Function of SHH Signaling in the Generation of mDA Neurons from PSCs
2.3. WNT Signaling
2.3.1. WNT Signaling Pathways and Mechanisms
2.3.2. WNT Signaling in Mammalian mDA Neuron Development In Vivo
2.3.3. WNT Signaling in the Directed Differentiation of PSCs and Direct Conversion of Somatic Cells into mDA Neurons In Vitro
2.4. BMP Signaling in Midbrain Dopaminergic Neuron Generation In Vivo and In Vitro
2.4.1. The BMP Signaling Pathway
2.4.2. The Function of the BMP Signaling Pathway in the Generation of Mammalian mDA Neurons In Vivo
2.4.3. BMPs in Stem Cell-Derived mDA Neuron Maturation
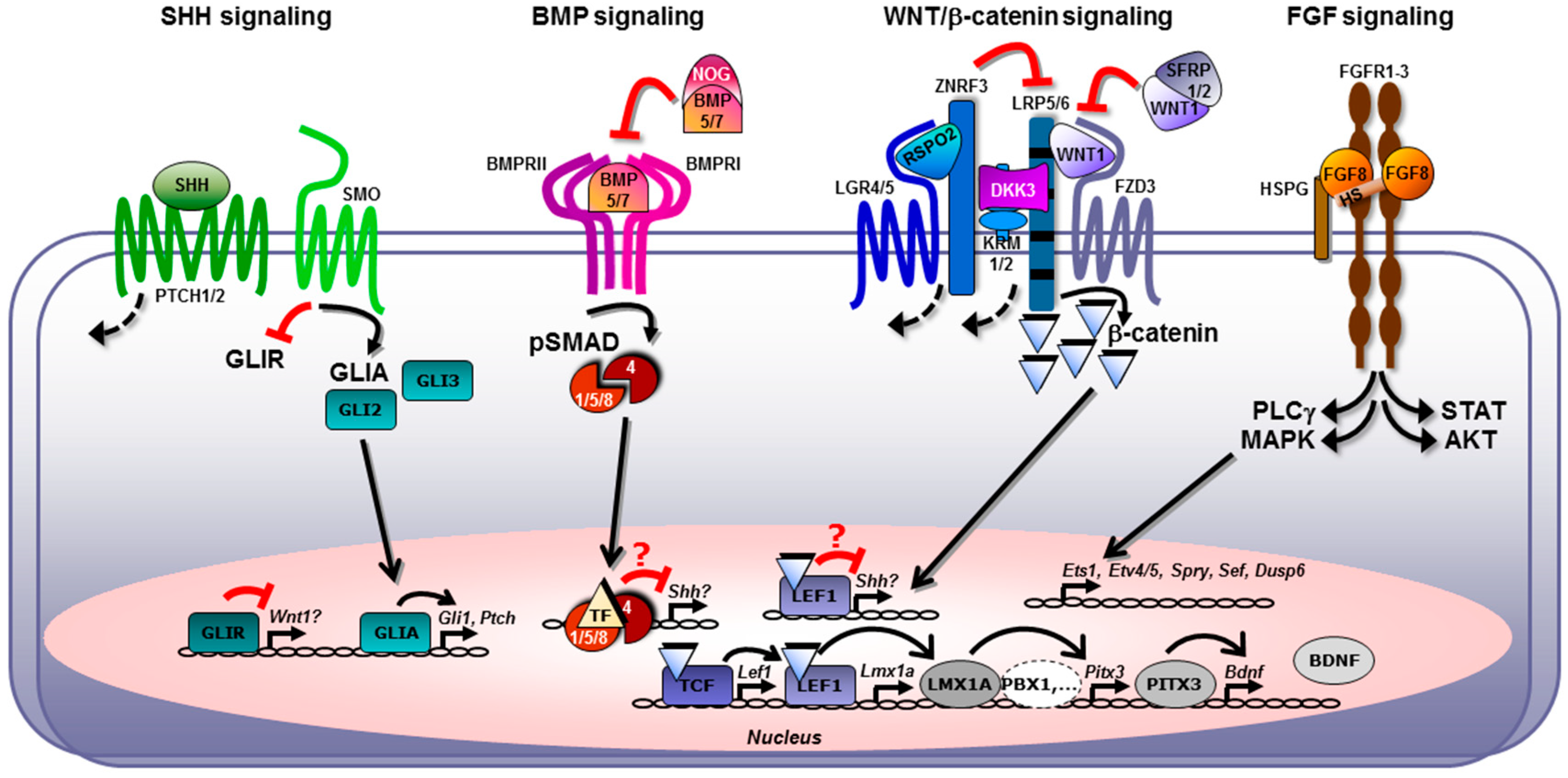
2.5. Cross-Talk between the FGF, SHH, WNT, and BMP/TGFβ Signaling Pathways in Midbrain Dopaminergic Neuron Generation In Vivo and In Vitro
2.5.1. Crosstalk between FGF/FGF8 and WNT Pathways
2.5.2. Crosstalk between SHH and WNT Pathways
2.5.3. Crosstalk between WNT and BMP/TGFβ Pathways
2.5.4. Crosstalk between BMP and SHH Pathways
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bjorklund, A.; Dunnett, S.B. Dopamine neuron systems in the brain: An update. Trends Neurosci. 2007, 30, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Ranganath, A.; Jacob, S.N. Doping the Mind: Dopaminergic Modulation of Prefrontal Cortical Cognition. Neuroscientist 2016, 22, 593–603. [Google Scholar] [CrossRef] [PubMed]
- Grace, A.A. Dysregulation of the dopamine system in the pathophysiology of schizophrenia and depression. Nat. Rev. Neurosci. 2016, 17, 524–532. [Google Scholar] [CrossRef] [PubMed]
- Hadley, J.A.; Nenert, R.; Kraguljac, N.V.; Bolding, M.S.; White, D.M.; Skidmore, F.M.; Visscher, K.M.; Lahti, A.C. Ventral tegmental area/midbrain functional connectivity and response to antipsychotic medication in schizophrenia. Neuropsychopharmacology 2014, 39, 1020–1030. [Google Scholar] [CrossRef]
- Bodea, G.O.; Blaess, S. Establishing diversity in the dopaminergic system. FEBS Lett. 2015, 589, 3773–3785. [Google Scholar] [CrossRef] [PubMed]
- Morello, F.; Partanen, J. Diversity and development of local inhibitory and excitatory neurons associated with dopaminergic nuclei. FEBS Lett. 2015, 589, 3693–3701. [Google Scholar] [CrossRef] [PubMed]
- Anderegg, A.; Poulin, J.-F.; Awatramani, R. Molecular heterogeneity of midbrain dopaminergic neurons--Moving toward single cell resolution. FEBS Lett. 2015, 589, 3714–3726. [Google Scholar] [CrossRef]
- Fu, Y.; Yuan, Y.; Halliday, G.; Rusznak, Z.; Watson, C.; Paxinos, G. A cytoarchitectonic and chemoarchitectonic analysis of the dopamine cell groups in the substantia nigra, ventral tegmental area, and retrorubral field in the mouse. Brain Struct. Funct. 2012, 217, 591–612. [Google Scholar] [CrossRef]
- Blaess, S.; Ang, S.-L. Genetic control of midbrain dopaminergic neuron development. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 113–134. [Google Scholar] [CrossRef]
- Roeper, J. Dissecting the diversity of midbrain dopamine neurons. Trends Neurosci. 2013, 36, 336–342. [Google Scholar] [CrossRef]
- Brignani, S.; Pasterkamp, R.J. Neuronal Subset-Specific Migration and Axonal Wiring Mechanisms in the Developing Midbrain Dopamine System. Front. Neuroanat. 2017, 11, 55. [Google Scholar] [CrossRef] [PubMed]
- Arenas, E.; Denham, M.; Villaescusa, J.C. How to make a midbrain dopaminergic neuron. Development 2015, 142, 1918–1936. [Google Scholar] [CrossRef] [PubMed]
- Prakash, N.; Wurst, W. Specification of midbrain territory. Cell Tissue Res. 2004, 318, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Ono, Y.; Nakatani, T.; Sakamoto, Y.; Mizuhara, E.; Minaki, Y.; Kumai, M.; Hamaguchi, A.; Nishimura, M.; Inoue, Y.; Hayashi, H.; et al. Differences in neurogenic potential in floor plate cells along an anteroposterior location: Midbrain dopaminergic neurons originate from mesencephalic floor plate cells. Development 2007, 134, 3213–3225. [Google Scholar] [CrossRef]
- Bonilla, S.; Hall, A.C.; Pinto, L.; Attardo, A.; Gotz, M.; Huttner, W.B.; Arenas, E. Identification of midbrain floor plate radial glia-like cells as dopaminergic progenitors. Glia 2008, 56, 809–820. [Google Scholar] [CrossRef]
- Veenvliet, J.V.; Smidt, M.P. Molecular mechanisms of dopaminergic subset specification: Fundamental aspects and clinical perspectives. Cell. Mol. Life Sci. 2014, 71, 4703–4727. [Google Scholar] [CrossRef] [PubMed]
- Joksimovic, M.; Awatramani, R. Wnt/beta-catenin signaling in midbrain dopaminergic neuron specification and neurogenesis. J. Mol. Cell Biol. 2014, 6, 27–33. [Google Scholar] [CrossRef]
- Wurst, W.; Prakash, N. Wnt1-regulated genetic networks in midbrain dopaminergic neuron development. J. Mol. Cell Biol. 2014, 6, 34–41. [Google Scholar] [CrossRef]
- Jovanovic, V.M.; Salti, A.; Tilleman, H.; Zega, K.; Jukic, M.M.; Zou, H.; Friedel, R.H.; Prakash, N.; Blaess, S.; Edenhofer, F.; et al. BMP/SMAD Pathway Promotes Neurogenesis of Midbrain Dopaminergic Neurons In Vivo and in Human Induced Pluripotent and Neural Stem Cells. J. Neurosci. 2018, 38, 1662–1676. [Google Scholar] [CrossRef]
- Puelles, E.; Acampora, D.; Lacroix, E.; Signore, M.; Annino, A.; Tuorto, F.; Filosa, S.; Corte, G.; Wurst, W.; Ang, S.L.; et al. Otx dose-dependent integrated control of antero-posterior and dorso-ventral patterning of midbrain. Nat. Neurosci. 2003, 6, 453–460. [Google Scholar] [CrossRef]
- Brodski, C.; Weisenhorn, D.M.; Signore, M.; Sillaber, I.; Oesterheld, M.; Broccoli, V.; Acampora, D.; Simeone, A.; Wurst, W. Location and size of dopaminergic and serotonergic cell populations are controlled by the position of the midbrain-hindbrain organizer. J. Neurosci. 2003, 23, 4199–4207. [Google Scholar] [CrossRef]
- Smidt, M.P.; Asbreuk, C.H.; Cox, J.J.; Chen, H.; Johnson, R.L.; Burbach, J.P. A second independent pathway for development of mesencephalic dopaminergic neurons requires Lmx1b. Nat. Neurosci. 2000, 3, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Andersson, E.; Tryggvason, U.; Deng, Q.; Friling, S.; Alekseenko, Z.; Robert, B.; Perlmann, T.; Ericson, J. Identification of intrinsic determinants of midbrain dopamine neurons. Cell 2006, 124, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Metzakopian, E.; Mavromatakis, Y.E.; Gao, N.; Balaskas, N.; Sasaki, H.; Briscoe, J.; Whitsett, J.A.; Goulding, M.; Kaestner, K.H.; et al. Foxa1 and Foxa2 function both upstream of and cooperatively with Lmx1a and Lmx1b in a feedforward loop promoting mesodiencephalic dopaminergic neuron development. Dev. Biol. 2009, 333, 386–396. [Google Scholar] [CrossRef] [PubMed]
- Nakatani, T.; Kumai, M.; Mizuhara, E.; Minaki, Y.; Ono, Y. Lmx1a and Lmx1b cooperate with Foxa2 to coordinate the specification of dopaminergic neurons and control of floor plate cell differentiation in the developing mesencephalon. Dev. Biol. 2010, 339, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.H.; Levesque, M.; Claxton, S.; Johnson, R.L.; Ang, S.L. Lmx1a and lmx1b function cooperatively to regulate proliferation, specification, and differentiation of midbrain dopaminergic progenitors. J. Neurosci. 2011, 31, 12413–12425. [Google Scholar] [CrossRef] [PubMed]
- Deng, Q.; Andersson, E.; Hedlund, E.; Alekseenko, Z.; Coppola, E.; Panman, L.; Millonig, J.H.; Brunet, J.F.; Ericson, J.; Perlmann, T. Specific and integrated roles of Lmx1a, Lmx1b and Phox2a in ventral midbrain development. Development 2011, 138, 3399–3408. [Google Scholar] [CrossRef]
- Sherf, O.; Nashelsky Zolotov, L.; Liser, K.; Tilleman, H.; Jovanovic, V.M.; Zega, K.; Jukic, M.M.; Brodski, C. Otx2 Requires Lmx1b to Control the Development of Mesodiencephalic Dopaminergic Neurons. PLoS ONE 2015, 10, e0139697. [Google Scholar] [CrossRef]
- Andersson, E.; Jensen, J.B.; Parmar, M.; Guillemot, F.; Bjorklund, A. Development of the mesencephalic dopaminergic neuron system is compromised in the absence of neurogenin 2. Development 2006, 133, 507–516. [Google Scholar] [CrossRef]
- Kele, J.; Simplicio, N.; Ferri, A.L.; Mira, H.; Guillemot, F.; Arenas, E.; Ang, S.L. Neurogenin 2 is required for the development of ventral midbrain dopaminergic neurons. Development 2006, 133, 495–505. [Google Scholar] [CrossRef]
- Simon, H.H.; Saueressig, H.; Wurst, W.; Goulding, M.D.; O’Leary, D.D. Fate of midbrain dopaminergic neurons controlled by the engrailed genes. J. Neurosci. 2001, 21, 3126–3134. [Google Scholar] [CrossRef] [PubMed]
- Alberi, L.; Sgado, P.; Simon, H.H. Engrailed genes are cell-autonomously required to prevent apoptosis in mesencephalic dopaminergic neurons. Development 2004, 131, 3229–3236. [Google Scholar] [CrossRef] [PubMed]
- Sgado, P.; Alberi, L.; Gherbassi, D.; Galasso, S.L.; Ramakers, G.M.; Alavian, K.N.; Smidt, M.P.; Dyck, R.H.; Simon, H.H. Slow progressive degeneration of nigral dopaminergic neurons in postnatal Engrailed mutant mice. Proc. Natl. Acad. Sci. USA 2006, 103, 15242–15247. [Google Scholar] [CrossRef] [PubMed]
- Sonnier, L.; Le Pen, G.; Hartmann, A.; Bizot, J.C.; Trovero, F.; Krebs, M.O.; Prochiantz, A. Progressive loss of dopaminergic neurons in the ventral midbrain of adult mice heterozygote for Engrailed1. J. Neurosci. 2007, 27, 1063–1071. [Google Scholar] [CrossRef] [PubMed]
- Castillo, S.O.; Baffi, J.S.; Palkovits, M.; Goldstein, D.S.; Kopin, I.J.; Witta, J.; Magnuson, M.A.; Nikodem, V.M. Dopamine biosynthesis is selectively abolished in substantia nigra/ventral tegmental area but not in hypothalamic neurons in mice with targeted disruption of the Nurr1 gene. Mol. Cell. Neurosci. 1998, 11, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Saucedo-Cardenas, O.; Quintana-Hau, J.D.; Le, W.D.; Smidt, M.P.; Cox, J.J.; de Mayo, F.; Burbach, J.P.; Conneely, O.M. Nurr1 is essential for the induction of the dopaminergic phenotype and the survival of ventral mesencephalic late dopaminergic precursor neurons. Proc. Natl. Acad. Sci. USA 1998, 95, 4013–4018. [Google Scholar] [CrossRef] [PubMed]
- Zetterstrom, R.H.; Solomin, L.; Jansson, L.; Hoffer, B.J.; Olson, L.; Perlmann, T. Dopamine neuron agenesis in Nurr1-deficient mice. Science 1997, 276, 248–250. [Google Scholar] [CrossRef] [PubMed]
- Kadkhodaei, B.; Ito, T.; Joodmardi, E.; Mattsson, B.; Rouillard, C.; Carta, M.; Muramatsu, S.; Sumi-Ichinose, C.; Nomura, T.; Metzger, D.; et al. Nurr1 is required for maintenance of maturing and adult midbrain dopamine neurons. J. Neurosci. 2009, 29, 15923–15932. [Google Scholar] [CrossRef] [PubMed]
- Nunes, I.; Tovmasian, L.T.; Silva, R.M.; Burke, R.E.; Goff, S.P. Pitx3 is required for development of substantia nigra dopaminergic neurons. Proc. Natl. Acad. Sci. USA 2003, 100, 4245–4250. [Google Scholar] [CrossRef] [PubMed]
- Connolly, B.S.; Lang, A.E. Pharmacological treatment of Parkinson disease: A review. JAMA 2014, 311, 1670–1683. [Google Scholar] [CrossRef]
- Barker, R.A.; Parmar, M.; Studer, L.; Takahashi, J. Human Trials of Stem Cell-Derived Dopamine Neurons for Parkinson’s Disease: Dawn of a New Era. Cell Stem Cell 2017, 21, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Chambers, S.M.; Fasano, C.A.; Papapetrou, E.P.; Tomishima, M.; Sadelain, M.; Studer, L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 2009, 27, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Kriks, S.; Shim, J.W.; Piao, J.; Ganat, Y.M.; Wakeman, D.R.; Xie, Z.; Carrillo-Reid, L.; Auyeung, G.; Antonacci, C.; Buch, A.; et al. Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson’s disease. Nature 2011, 480, 547–551. [Google Scholar] [CrossRef]
- Salti, A.; Nat, R.; Neto, S.; Puschban, Z.; Wenning, G.; Dechant, G. Expression of early developmental markers predicts the efficiency of embryonic stem cell differentiation into midbrain dopaminergic neurons. Stem Cells Dev. 2013, 22, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Deussing, J.M. Targeted mutagenesis tools for modelling psychiatric disorders. Cell Tissue Res. 2013, 354, 9–25. [Google Scholar] [CrossRef]
- La Manno, G.; Gyllborg, D.; Codeluppi, S.; Nishimura, K.; Salto, C.; Zeisel, A.; Borm, L.E.; Stott, S.R.W.; Toledo, E.M.; Villaescusa, J.C.; et al. Molecular Diversity of Midbrain Development in Mouse, Human, and Stem Cells. Cell 2016, 167, 566. [Google Scholar] [CrossRef] [PubMed]
- Breger, L.S.; Fuzzati Armentero, M.T. Genetically engineered animal models of Parkinson’s disease: From worm to rodent. Eur. J. Neurosci. 2018. [Google Scholar] [CrossRef]
- Ornitz, D.M.; Itoh, N. The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 215–266. [Google Scholar] [CrossRef]
- Förthmann, B.; Aletta, J.M.; Lee, Y.-W.; Terranova, C.; Birkaya, B.; Stachowiak, E.K.; Stachowiak, M.K.; Claus, P. Coalition of Nuclear Receptors in the Nervous System. J. Cell. Physiol. 2015, 230, 2875–2880. [Google Scholar] [CrossRef]
- Crossley, P.H.; Martinez, S.; Martin, G.R. Midbrain development induced by FGF8 in the chick embryo. Nature 1996, 380, 66–68. [Google Scholar] [CrossRef]
- Olsen, S.K.; Li, J.Y.H.; Bromleigh, C.; Eliseenkova, A.V.; Ibrahimi, O.A.; Lao, Z.; Zhang, F.; Linhardt, R.J.; Joyner, A.L.; Mohammadi, M. Structural basis by which alternative splicing modulates the organizer activity of FGF8 in the brain. Genes Dev. 2006, 20, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Liu, Z.; Ornitz, D.M. Temporal and spatial gradients of Fgf8 and Fgf17 regulate proliferation and differentiation of midline cerebellar structures. Development 2000, 127, 1833–1843. [Google Scholar]
- Gimeno, L.; Brulet, P.; Martinez, S. Study of Fgf15 gene expression in developing mouse brain. Gene Expr. Patterns 2003, 3, 473–481. [Google Scholar] [CrossRef]
- Fischer, T.; Faus-Kessler, T.; Welzl, G.; Simeone, A.; Wurst, W.; Prakash, N. Fgf15-mediated control of neurogenic and proneural gene expression regulates dorsal midbrain neurogenesis. Dev. Biol. 2011, 350, 496–510. [Google Scholar] [CrossRef]
- Chen, Y.; Mohammadi, M.; Flanagan, J.G. Graded levels of FGF protein span the midbrain and can instruct graded induction and repression of neural mapping labels. Neuron 2009, 62, 773–780. [Google Scholar] [CrossRef] [PubMed]
- Lahti, L.; Saarimaki-Vire, J.; Rita, H.; Partanen, J. FGF signaling gradient maintains symmetrical proliferative divisions of midbrain neuronal progenitors. Dev. Biol. 2011, 349, 270–282. [Google Scholar] [CrossRef] [PubMed]
- Saarimaki-Vire, J.; Peltopuro, P.; Lahti, L.; Naserke, T.; Blak, A.A.; Vogt Weisenhorn, D.M.; Yu, K.; Ornitz, D.M.; Wurst, W.; Partanen, J. Fibroblast growth factor receptors cooperate to regulate neural progenitor properties in the developing midbrain and hindbrain. J. Neurosci. 2007, 27, 8581–8592. [Google Scholar] [CrossRef]
- Lao, Z.; Raju, G.P.; Bai, C.B.; Joyner, A.L. MASTR: A technique for mosaic mutant analysis with spatial and temporal control of recombination using conditional floxed alleles in mice. Cell Rep. 2012, 2, 386–396. [Google Scholar] [CrossRef]
- Jukkola, T.; Lahti, L.; Naserke, T.; Wurst, W.; Partanen, J. FGF regulated gene-expression and neuronal differentiation in the developing midbrain-hindbrain region. Dev. Biol. 2006, 297, 141–157. [Google Scholar] [CrossRef]
- Bottcher, R.T.; Pollet, N.; Delius, H.; Niehrs, C. The transmembrane protein XFLRT3 forms a complex with FGF receptors and promotes FGF signalling. Nat. Cell Biol. 2004, 6, 38–44. [Google Scholar] [CrossRef]
- Haines, B.P.; Wheldon, L.M.; Summerbell, D.; Heath, J.K.; Rigby, P.W.J. Regulated expression of FLRT genes implies a functional role in the regulation of FGF signalling during mouse development. Dev. Biol. 2006, 297, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Hirate, Y.; Okamoto, H. Canopy1, a novel regulator of FGF signaling around the midbrain-hindbrain boundary in zebrafish. Curr. Biol. 2006, 16, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Matsui, T.; Thitamadee, S.; Murata, T.; Kakinuma, H.; Nabetani, T.; Hirabayashi, Y.; Hirate, Y.; Okamoto, H.; Bessho, Y. Canopy1, a positive feedback regulator of FGF signaling, controls progenitor cell clustering during Kupffer’s vesicle organogenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 9881–9886. [Google Scholar] [CrossRef] [PubMed]
- Neben, C.L.; Lo, M.; Jura, N.; Klein, O.D. Feedback regulation of RTK signaling in development. Dev. Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Harada, H.; Sato, T.; Nakamura, H. Fgf8 signaling for development of the midbrain and hindbrain. Dev. Growth Differ. 2016, 58, 437–445. [Google Scholar] [CrossRef]
- Ye, W.; Shimamura, K.; Rubenstein, J.L.; Hynes, M.A.; Rosenthal, A. FGF and Shh signals control dopaminergic and serotonergic cell fate in the anterior neural plate. Cell 1998, 93, 755–766. [Google Scholar] [CrossRef]
- Chi, C.L.; Martinez, S.; Wurst, W.; Martin, G.R. The isthmic organizer signal FGF8 is required for cell survival in the prospective midbrain and cerebellum. Development 2003, 130, 2633–2644. [Google Scholar] [CrossRef] [PubMed]
- Trokovic, R.; Jukkola, T.; Saarimaki, J.; Peltopuro, P.; Naserke, T.; Weisenhorn, D.M.; Trokovic, N.; Wurst, W.; Partanen, J. Fgfr1-dependent boundary cells between developing mid- and hindbrain. Dev. Biol. 2005, 278, 428–439. [Google Scholar] [CrossRef]
- Sunmonu, N.A.; Li, K.; Guo, Q.; Li, J.Y. Gbx2 and Fgf8 are sequentially required for formation of the midbrain-hindbrain compartment boundary. Development 2011, 138, 725–734. [Google Scholar] [CrossRef]
- Liu, A.; Losos, K.; Joyner, A.L. FGF8 can activate Gbx2 and transform regions of the rostral mouse brain into a hindbrain fate. Development 1999, 126, 4827–4838. [Google Scholar]
- Martinez, S.; Crossley, P.H.; Cobos, I.; Rubenstein, J.L.; Martin, G.R. FGF8 induces formation of an ectopic isthmic organizer and isthmocerebellar development via a repressive effect on Otx2 expression. Development 1999, 126, 1189–1200. [Google Scholar] [PubMed]
- Sato, T.; Araki, I.; Nakamura, H. Inductive signal and tissue responsiveness defining the tectum and the cerebellum. Development 2001, 128, 2461–2469. [Google Scholar] [PubMed]
- Araki, I.; Nakamura, H. Engrailed defines the position of dorsal di-mesencephalic boundary by repressing diencephalic fate. Development 1999, 126, 5127–5135. [Google Scholar] [PubMed]
- Liu, A.; Joyner, A.L. EN and GBX2 play essential roles downstream of FGF8 in patterning the mouse mid/hindbrain region. Development 2001, 128, 181–191. [Google Scholar] [PubMed]
- Lahti, L.; Peltopuro, P.; Piepponen, T.P.; Partanen, J. Cell-autonomous FGF signaling regulates anteroposterior patterning and neuronal differentiation in the mesodiencephalic dopaminergic progenitor domain. Development 2012, 139, 894–905. [Google Scholar] [CrossRef] [PubMed]
- Kee, N.; Volakakis, N.; Kirkeby, A.; Dahl, L.; Storvall, H.; Nolbrant, S.; Lahti, L.; Bjorklund, A.K.; Gillberg, L.; Joodmardi, E.; et al. Single-Cell Analysis Reveals a Close Relationship between Differentiating Dopamine and Subthalamic Nucleus Neuronal Lineages. Cell Stem Cell 2017, 20, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Nouri, N.; Awatramani, R. A novel floor plate boundary defined by adjacent En1 and Dbx1 microdomains distinguishes midbrain dopamine and hypothalamic neurons. Development 2017, 144, 916–927. [Google Scholar] [CrossRef]
- Asbreuk, C.H.; Vogelaar, C.F.; Hellemons, A.; Smidt, M.P.; Burbach, J.P. CNS expression pattern of Lmx1b and coexpression with ptx genes suggest functional cooperativity in the development of forebrain motor control systems. Mol. Cell. Neurosci. 2002, 21, 410–420. [Google Scholar] [CrossRef]
- Ellisor, D.; Rieser, C.; Voelcker, B.; Machan, J.T.; Zervas, M. Genetic dissection of midbrain dopamine neuron development in vivo. Dev. Biol. 2012, 372, 249–262. [Google Scholar] [CrossRef]
- Poulin, J.-F.; Zou, J.; Drouin-Ouellet, J.; Kim, K.-Y.A.; Cicchetti, F.; Awatramani, R.B. Defining midbrain dopaminergic neuron diversity by single-cell gene expression profiling. Cell Rep. 2014, 9, 930–943. [Google Scholar] [CrossRef]
- McGowan, L.D.; Alaama, R.A.; Striedter, G.F. FGF2 delays tectal neurogenesis, increases tectal cell numbers, and alters tectal lamination in embryonic chicks. PLoS ONE 2013, 8, e79949. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Shimazaki, T.; Naka, H.; Fukami, S.-I.; Satoh, Y.; Okano, H.; Lax, I.; Schlessinger, J.; Gotoh, N. FRS2α regulates Erk levels to control a self-renewal target Hes1 and proliferation of FGF-responsive neural stem/progenitor cells. Stem Cells 2010, 28, 1661–1673. [Google Scholar] [CrossRef] [PubMed]
- Kameda, Y.; Saitoh, T.; Fujimura, T. Hes1 regulates the number and anterior-posterior patterning of mesencephalic dopaminergic neurons at the mid/hindbrain boundary (isthmus). Dev. Biol. 2011, 358, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.; Li, J.Y.; Bromleigh, C.; Lao, Z.; Niswander, L.A.; Joyner, A.L. FGF17b and FGF18 have different midbrain regulatory properties from FGF8b or activated FGF receptors. Development 2003, 130, 6175–6185. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, K.; Mizushima, S.; Tamada, A.; Yamamoto, N.; Takashima, S.; Murakami, F. FGF8 signaling regulates growth of midbrain dopaminergic axons by inducing semaphorin 3F. J. Neurosci. 2009, 29, 4044–4055. [Google Scholar] [CrossRef] [PubMed]
- Ohmachi, S.; Watanabe, Y.; Mikami, T.; Kusu, N.; Ibi, T.; Akaike, A.; Itoh, N. FGF-20, a novel neurotrophic factor, preferentially expressed in the substantia nigra pars compacta of rat brain. Biochem. Biophys. Res. Commun. 2000, 277, 355–360. [Google Scholar] [CrossRef]
- Ohmachi, S.; Mikami, T.; Konishi, M.; Miyake, A.; Itoh, N. Preferential neurotrophic activity of fibroblast growth factor-20 for dopaminergic neurons through fibroblast growth factor receptor-1c. J. Neurosci. Res. 2003, 72, 436–443. [Google Scholar] [CrossRef]
- Murase, S.; McKay, R.D. A specific survival response in dopamine neurons at most risk in Parkinson’s disease. J. Neurosci. 2006, 26, 9750–9760. [Google Scholar] [CrossRef]
- Sleeman, I.J.; Boshoff, E.L.; Duty, S. Fibroblast growth factor-20 protects against dopamine neuron loss in vitro and provides functional protection in the 6-hydroxydopamine-lesioned rat model of Parkinson’s disease. Neuropharmacology 2012, 63, 1268–1277. [Google Scholar] [CrossRef]
- IPDGC; WTCCC2. A two-stage meta-analysis identifies several new loci for Parkinson’s disease. PLoS Genet. 2011, 7, e1002142. [Google Scholar] [CrossRef]
- Murphy, M.; Drago, J.; Bartlett, P.F. Fibroblast growth factor stimulates the proliferation and differentiation of neural precursor cells in vitro. J. Neurosci. Res. 1990, 25, 463–475. [Google Scholar] [CrossRef] [PubMed]
- Tropepe, V.; Sibilia, M.; Ciruna, B.G.; Rossant, J.; Wagner, E.F.; van der Kooy, D. Distinct neural stem cells proliferate in response to EGF and FGF in the developing mouse telencephalon. Dev. Biol. 1999, 208, 166–188. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Lumelsky, N.; Studer, L.; Auerbach, J.M.; McKay, R.D. Efficient generation of midbrain and hindbrain neurons from mouse embryonic stem cells. Nat. Biotechnol. 2000, 18, 675–679. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Pernaute, R.; Studer, L.; Bankiewicz, K.S.; Major, E.O.; McKay, R.D. In vitro generation and transplantation of precursor-derived human dopamine neurons. J. Neurosci. Res. 2001, 65, 284–288. [Google Scholar] [CrossRef] [PubMed]
- Friling, S.; Andersson, E.; Thompson, L.H.; Jonsson, M.E.; Hebsgaard, J.B.; Nanou, E.; Alekseenko, Z.; Marklund, U.; Kjellander, S.; Volakakis, N.; et al. Efficient production of mesencephalic dopamine neurons by Lmx1a expression in embryonic stem cells. Proc. Natl. Acad. Sci. USA 2009, 106, 7613–7618. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Su, S.C.; Wang, H.; Cheng, A.W.; Cassady, J.P.; Lodato, M.A.; Lengner, C.J.; Chung, C.Y.; Dawlaty, M.M.; Tsai, L.H.; et al. Functional integration of dopaminergic neurons directly converted from mouse fibroblasts. Cell Stem Cell 2011, 9, 413–419. [Google Scholar] [CrossRef]
- Kirkeby, A.; Nolbrant, S.; Tiklova, K.; Heuer, A.; Kee, N.; Cardoso, T.; Ottosson, D.R.; Lelos, M.J.; Rifes, P.; Dunnett, S.B.; et al. Predictive Markers Guide Differentiation to Improve Graft Outcome in Clinical Translation of hESC-Based Therapy for Parkinson’s Disease. Cell Stem Cell 2017, 20, 135–148. [Google Scholar] [CrossRef]
- Jaeger, I.; Arber, C.; Risner-Janiczek, J.R.; Kuechler, J.; Pritzsche, D.; Chen, I.C.; Naveenan, T.; Ungless, M.A.; Li, M. Temporally controlled modulation of FGF/ERK signaling directs midbrain dopaminergic neural progenitor fate in mouse and human pluripotent stem cells. Development 2011, 138, 4363–4374. [Google Scholar] [CrossRef]
- Nolbrant, S.; Heuer, A.; Parmar, M.; Kirkeby, A. Generation of high-purity human ventral midbrain dopaminergic progenitors for in vitro maturation and intracerebral transplantation. Nat. Protoc. 2017, 12, 1962–1979. [Google Scholar] [CrossRef]
- Briscoe, J.; Thérond, P.P. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat. Rev. Mol. Cell Biol. 2013, 14, 416–429. [Google Scholar] [CrossRef]
- Pereira, J.; Johnson, W.E.; O’Brien, S.J.; Jarvis, E.D.; Zhang, G.; Gilbert, M.T.P.; Vasconcelos, V.; Antunes, A. Evolutionary genomics and adaptive evolution of the Hedgehog gene family (Shh, Ihh and Dhh) in vertebrates. PLoS ONE 2014, 9, e74132. [Google Scholar] [CrossRef]
- Belgacem, Y.H.; Hamilton, A.M.; Shim, S.; Spencer, K.A.; Borodinsky, L.N. The Many Hats of Sonic Hedgehog Signaling in Nervous System Development and Disease. J. Dev. Biol. 2016, 4, 35. [Google Scholar] [CrossRef] [PubMed]
- Petrov, K.; Wierbowski, B.M.; Salic, A. Sending and Receiving Hedgehog Signals. Annu. Rev. Cell Dev. Biol. 2017, 33, 145–168. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Fallon, J.F.; Beachy, P.A. Hedgehog-regulated processing of Gli3 produces an anterior/posterior repressor gradient in the developing vertebrate limb. Cell 2000, 100, 423–434. [Google Scholar] [CrossRef]
- Pan, Y.; Bai, C.B.; Joyner, A.L.; Wang, B. Sonic hedgehog signaling regulates Gli2 transcriptional activity by suppressing its processing and degradation. Mol. Cell. Biol. 2006, 26, 3365–3377. [Google Scholar] [CrossRef] [PubMed]
- Persson, M.; Stamataki, D.; te Welscher, P.; Andersson, E.; Böse, J.; Rüther, U.; Ericson, J.; Briscoe, J. Dorsal-ventral patterning of the spinal cord requires Gli3 transcriptional repressor activity. Genes Dev. 2002, 16, 2865–2878. [Google Scholar] [CrossRef]
- Wijgerde, M.; McMahon, J.A.; Rule, M.; McMahon, A.P. A direct requirement for Hedgehog signaling for normal specification of all ventral progenitor domains in the presumptive mammalian spinal cord. Genes Dev. 2002, 16, 2849–2864. [Google Scholar] [CrossRef]
- Bai, C.B.; Stephen, D.; Joyner, A.L. All mouse ventral spinal cord patterning by hedgehog is Gli dependent and involves an activator function of Gli3. Dev. Cell 2004, 6, 103–115. [Google Scholar] [CrossRef]
- Bai, C.B.; Auerbach, W.; Lee, J.S.; Stephen, D.; Joyner, A.L. Gli2, but not Gli1, is required for initial Shh signaling and ectopic activation of the Shh pathway. Development 2002, 129, 4753–4761. [Google Scholar]
- Matise, M.P.; Epstein, D.J.; Park, H.L.; Platt, K.A.; Joyner, A.L. Gli2 is required for induction of floor plate and adjacent cells, but not most ventral neurons in the mouse central nervous system. Development 1998, 125, 2759–2770. [Google Scholar]
- Park, H.L.; Bai, C.; Platt, K.A.; Matise, M.P.; Beeghly, A.; Hui, C.C.; Nakashima, M.; Joyner, A.L. Mouse Gli1 mutants are viable but have defects in SHH signaling in combination with a Gli2 mutation. Development 2000, 127, 1593–1605. [Google Scholar] [PubMed]
- Dessaud, E.; Yang, L.L.; Hill, K.; Cox, B.; Ulloa, F.; Ribeiro, A.; Mynett, A.; Novitch, B.G.; Briscoe, J. Interpretation of the sonic hedgehog morphogen gradient by a temporal adaptation mechanism. Nature 2007, 450, 717–720. [Google Scholar] [CrossRef]
- Harfe, B.D.; Scherz, P.J.; Nissim, S.; Tian, H.; McMahon, A.P.; Tabin, C.J. Evidence for an expansion-based temporal Shh gradient in specifying vertebrate digit identities. Cell 2004, 118, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.; Joyner, A.L. Dynamic changes in the response of cells to positive hedgehog signaling during mouse limb patterning. Cell 2004, 118, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Bangs, F.; Anderson, K.V. Primary Cilia and Mammalian Hedgehog Signaling. Cold Spring Harb. Perspect. Biol. 2017, 9, a028175. [Google Scholar] [CrossRef] [PubMed]
- Allen, B.L.; Song, J.Y.; Izzi, L.; Althaus, I.W.; Kang, J.-S.; Charron, F.; Krauss, R.S.; McMahon, A.P. Overlapping roles and collective requirement for the coreceptors GAS1, CDO, and BOC in SHH pathway function. Dev. Cell 2011, 20, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Izzi, L.; Lévesque, M.; Morin, S.; Laniel, D.; Wilkes, B.C.; Mille, F.; Krauss, R.S.; McMahon, A.P.; Allen, B.L.; Charron, F. Boc and Gas1 each form distinct Shh receptor complexes with Ptch1 and are required for Shh-mediated cell proliferation. Dev. Cell 2011, 20, 788–801. [Google Scholar] [CrossRef] [PubMed]
- Ang, S.L.; Wierda, A.; Wong, D.; Stevens, K.A.; Cascio, S.; Rossant, J.; Zaret, K.S. The formation and maintenance of the definitive endoderm lineage in the mouse: Involvement of HNF3/forkhead proteins. Development 1993, 119, 1301–1315. [Google Scholar]
- Metzakopian, E.; Lin, W.; Salmon-Divon, M.; Dvinge, H.; Andersson, E.; Ericson, J.; Perlmann, T.; Whitsett, J.A.; Bertone, P.; Ang, S.L. Genome-wide characterization of Foxa2 targets reveals upregulation of floor plate genes and repression of ventrolateral genes in midbrain dopaminergic progenitors. Development 2012, 139, 2625–2634. [Google Scholar] [CrossRef] [PubMed]
- Blaess, S.; Corrales, J.D.; Joyner, A.L. Sonic hedgehog regulates Gli activator and repressor functions with spatial and temporal precision in the mid/hindbrain region. Development 2006, 133, 1799–1809. [Google Scholar] [CrossRef] [PubMed]
- Fogel, J.L.; Chiang, C.; Huang, X.; Agarwala, S. Ventral specification and perturbed boundary formation in the mouse midbrain in the absence of Hedgehog signaling. Dev. Dyn. 2008, 237, 1359–1372. [Google Scholar] [CrossRef] [PubMed]
- Hayes, L.; Ralls, S.; Wang, H.; Ahn, S. Duration of Shh signaling contributes to mDA neuron diversity. Dev. Biol. 2013, 374, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Blaess, S.; Bodea, G.O.; Kabanova, A.; Chanet, S.; Mugniery, E.; Derouiche, A.; Stephen, D.; Joyner, A.L. Temporal-spatial changes in Sonic Hedgehog expression and signaling reveal different potentials of ventral mesencephalic progenitors to populate distinct ventral midbrain nuclei. Neural Dev. 2011, 6, 29. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Luo, S.X.; Tang, V.; Huang, E.J. Temporal and spatial requirements of Smoothened in ventral midbrain neuronal development. Neural Dev. 2013, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Perez-Balaguer, A.; Puelles, E.; Wurst, W.; Martinez, S. Shh dependent and independent maintenance of basal midbrain. Mech. Dev. 2009, 126, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Mavromatakis, Y.E.; Lin, W.; Metzakopian, E.; Ferri, A.L.; Yan, C.H.; Sasaki, H.; Whisett, J.; Ang, S.L. Foxa1 and Foxa2 positively and negatively regulate Shh signalling to specify ventral midbrain progenitor identity. Mech. Dev. 2011, 128, 90–103. [Google Scholar] [CrossRef] [PubMed]
- Mesman, S.; von Oerthel, L.; Smidt, M.P. Mesodiencephalic Dopaminergic Neuronal Differentiation Does Not Involve GLI2A-Mediated SHH-Signaling and Is under the Direct Influence of Canonical WNT Signaling. PLoS ONE 2014, 9, e97926. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.-R.; Jeong, M.-H.; Leem, Y.-E.; Lee, S.-J.; Kim, H.-J.; Bae, G.-U.; Kang, J.-S. The Shh coreceptor Cdo is required for differentiation of midbrain dopaminergic neurons. Stem Cell Res. 2014, 13, 262–274. [Google Scholar] [CrossRef]
- Verwey, M.; Grant, A.; Meti, N.; Adye-White, L.; Torres-Berrío, A.; Rioux, V.; Lévesque, M.; Charron, F.; Flores, C. Mesocortical Dopamine Phenotypes in Mice Lacking the Sonic Hedgehog Receptor Cdon. eNeuro 2016, 3. [Google Scholar] [CrossRef]
- Feuerstein, M.; Chleilat, E.; Khakipoor, S.; Michailidis, K.; Ophoven, C.; Roussa, E. Expression patterns of key Sonic Hedgehog signaling pathway components in the developing and adult mouse midbrain and in the MN9D cell line. Cell Tissue Res. 2017. [Google Scholar] [CrossRef]
- Hammond, R.; Blaess, S.; Abeliovich, A. Sonic hedgehog is a chemoattractant for midbrain dopaminergic axons. PLoS ONE 2009, 4, e7007. [Google Scholar] [CrossRef] [PubMed]
- Kittappa, R.; Chang, W.W.; Awatramani, R.B.; McKay, R.D. The foxa2 gene controls the birth and spontaneous degeneration of dopamine neurons in old age. PLoS Biol. 2007, 5, e325. [Google Scholar] [CrossRef] [PubMed]
- Joksimovic, M.; Anderegg, A.; Roy, A.; Campochiaro, L.; Yun, B.; Kittappa, R.; McKay, R.; Awatramani, R. Spatiotemporally separable Shh domains in the midbrain define distinct dopaminergic progenitor pools. Proc. Natl. Acad. Sci. USA 2009, 106, 19185–19190. [Google Scholar] [CrossRef] [PubMed]
- Placzek, M.; Briscoe, J. The floor plate: Multiple cells, multiple signals. Nat. Rev. Neurosci. 2005, 6, 230–240. [Google Scholar] [CrossRef] [PubMed]
- Joksimovic, M.; Yun, B.A.; Kittappa, R.; Anderegg, A.M.; Chang, W.W.; Taketo, M.M.; McKay, R.D.; Awatramani, R.B. Wnt antagonism of Shh facilitates midbrain floor plate neurogenesis. Nat. Neurosci. 2009, 12, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Hayes, L.; Zhang, Z.; Albert, P.; Zervas, M.; Ahn, S. Timing of Sonic hedgehog and Gli1 expression segregates midbrain dopamine neurons. J. Comp. Neurol. 2011, 519, 3001–3018. [Google Scholar] [CrossRef] [PubMed]
- Bye, C.R.; Thompson, L.H.; Parish, C.L. Birth dating of midbrain dopamine neurons identifies A9 enriched tissue for transplantation into parkinsonian mice. Exp. Neurol. 2012, 236, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Panman, L.; Papathanou, M.; Laguna, A.; Oosterveen, T.; Volakakis, N.; Acampora, D.; Kurtsdotter, I.; Yoshitake, T.; Kehr, J.; Joodmardi, E.; et al. Sox6 and Otx2 control the specification of substantia nigra and ventral tegmental area dopamine neurons. Cell Rep. 2014, 8, 1018–1025. [Google Scholar] [CrossRef]
- Hynes, M.; Porter, J.A.; Chiang, C.; Chang, D.; Tessier-Lavigne, M.; Beachy, P.A.; Rosenthal, A. Induction of midbrain dopaminergic neurons by Sonic hedgehog. Neuron 1995, 15, 35–44. [Google Scholar] [CrossRef]
- Hynes, M.; Stone, D.M.; Dowd, M.; Pitts-Meek, S.; Goddard, A.; Gurney, A.; Rosenthal, A. Control of cell pattern in the neural tube by the zinc finger transcription factor and oncogene Gli-1. Neuron 1997, 19, 15–26. [Google Scholar] [CrossRef]
- Fedtsova, N.; Turner, E.E. Signals from the ventral midline and isthmus regulate the development of Brn3.0-expressing neurons in the midbrain. Mech. Dev. 2001, 105, 129–144. [Google Scholar] [CrossRef]
- Gazea, M.; Tasouri, E.; Tolve, M.; Bosch, V.; Kabanova, A.; Gojak, C.; Kurtulmus, B.; Novikov, O.; Spatz, J.; Pereira, G.; et al. Primary cilia are critical for Sonic hedgehog-mediated dopaminergic neurogenesis in the embryonic midbrain. Dev. Biol. 2016, 409, 55–71. [Google Scholar] [CrossRef] [PubMed]
- Kabanova, A.; Pabst, M.; Lorkowski, M.; Braganza, O.; Boehlen, A.; Nikbakht, N.; Pothmann, L.; Vaswani, A.R.; Musgrove, R.; Di Monte, D.A.; et al. Function and developmental origin of a mesocortical inhibitory circuit. Nat. Neurosci. 2015, 18, 872–882. [Google Scholar] [CrossRef] [PubMed]
- Hui, C.-C.; Slusarski, D.; Platt, K.A.; Holmgren, R.; Joyner, A.L. Expression of three mouse homologs of the Drosophila segment polarity gene cubitus interruptus, Gli, Gli-2, and Gli-3, in ectoderm- and mesoderm-derived tissues suggests multiple roles during postimplantation development. Dev. Biol. 1994, 162, 402–413. [Google Scholar] [CrossRef] [PubMed]
- Szabó, N.-E.; Zhao, T.; Cankaya, M.; Theil, T.; Zhou, X.; Alvarez-Bolado, G. Role of neuroepithelial Sonic hedgehog in hypothalamic patterning. J. Neurosci. 2009, 29, 6989–7002. [Google Scholar] [CrossRef] [PubMed]
- Gazea, M.; Tasouri, E.; Heigl, T.; Bosch, V.; Tucker, K.L.; Blaess, S. Definition of a critical spatiotemporal window within which primary cilia control midbrain dopaminergic neurogenesis. Neurogenesis 2016, 3, e1248206. [Google Scholar] [CrossRef] [PubMed]
- Steinbeck, J.A.; Studer, L. Moving stem cells to the clinic: Potential and limitations for brain repair. Neuron 2015, 86, 187–206. [Google Scholar] [CrossRef] [PubMed]
- Ribes, V.; Balaskas, N.; Sasai, N.; Cruz, C.; Dessaud, E.; Cayuso, J.; Tozer, S.; Yang, L.L.; Novitch, B.; Marti, E.; et al. Distinct Sonic Hedgehog signaling dynamics specify floor plate and ventral neuronal progenitors in the vertebrate neural tube. Genes Dev. 2010, 24, 1186–1200. [Google Scholar] [CrossRef] [PubMed]
- Fasano, C.A.; Chambers, S.M.; Lee, G.; Tomishima, M.J.; Studer, L. Efficient derivation of functional floor plate tissue from human embryonic stem cells. Cell Stem Cell 2010, 6, 336–347. [Google Scholar] [CrossRef] [PubMed]
- Kirkeby, A.; Grealish, S.; Wolf, D.A.; Nelander, J.; Wood, J.; Lundblad, M.; Lindvall, O.; Parmar, M. Generation of Regionally Specified Neural Progenitors and Functional Neurons from Human Embryonic Stem Cells under Defined Conditions. Cell Rep. 2012, 1, 703–714. [Google Scholar] [CrossRef]
- Steinbeck, J.A.; Choi, S.J.; Mrejeru, A.; Ganat, Y.; Deisseroth, K.; Sulzer, D.; Mosharov, E.V.; Studer, L. Optogenetics enables functional analysis of human embryonic stem cell-derived grafts in a Parkinson’s disease model. Nat. Biotechnol. 2015, 33, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Castelo-Branco, G.; Wagner, J.; Rodriguez, F.J.; Kele, J.; Sousa, K.; Rawal, N.; Pasolli, H.A.; Fuchs, E.; Kitajewski, J.; Arenas, E. Differential regulation of midbrain dopaminergic neuron development by Wnt-1, Wnt-3a, and Wnt-5a. Proc. Natl. Acad. Sci. USA 2003, 100, 12747–12752. [Google Scholar] [CrossRef] [PubMed]
- Prakash, N.; Brodski, C.; Naserke, T.; Puelles, E.; Gogoi, R.; Hall, A.; Panhuysen, M.; Echevarria, D.; Sussel, L.; Weisenhorn, D.M.; et al. A Wnt1-regulated genetic network controls the identity and fate of midbrain-dopaminergic progenitors in vivo. Development 2006, 133, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Andersson, E.R.; Prakash, N.; Cajanek, L.; Minina, E.; Bryja, V.; Bryjova, L.; Yamaguchi, T.P.; Hall, A.C.; Wurst, W.; Arenas, E. Wnt5a regulates ventral midbrain morphogenesis and the development of A9-A10 dopaminergic cells in vivo. PLoS ONE 2008, 3, e3517. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Chen, S.; Li, J.-Y. Human induced pluripotent stem cells in Parkinson’s disease: A novel cell source of cell therapy and disease modeling. Prog. Neurobiol. 2015, 134, 161–177. [Google Scholar] [CrossRef] [PubMed]
- Tabar, V.; Studer, L. Pluripotent stem cells in regenerative medicine: Challenges and recent progress. Nat. Rev. Genet. 2014, 15, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Grainger, S.; Willert, K. Mechanisms of Wnt signaling and control. Wiley Interdiscip. Rev. Syst. Biol. Med. 2018, e1422. [Google Scholar] [CrossRef]
- Loh, K.M.; van Amerongen, R.; Nusse, R. Generating Cellular Diversity and Spatial Form: Wnt Signaling and the Evolution of Multicellular Animals. Dev. Cell 2016, 38, 643–655. [Google Scholar] [CrossRef]
- Nusse, R.; Clevers, H. Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef]
- Van Amerongen, R. Alternative Wnt pathways and receptors. Cold Spring Harb. Perspect. Biol. 2012, 4, a007914. [Google Scholar] [CrossRef]
- Xiao, Q.; Chen, Z.; Jin, X.; Mao, R.; Chen, Z. The many postures of noncanonical Wnt signaling in development and diseases. Biomed. Pharmacother. 2017, 93, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Arenas, E. Wnt signaling in midbrain dopaminergic neuron development and regenerative medicine for Parkinson’s disease. J. Mol. Cell Biol. 2014, 6, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Brown, A.; Ellisor, D.; Paul, E.; Hagan, N.; Zervas, M. Dynamic temporal requirement of Wnt1 in midbrain dopamine neuron development. Development 2013, 140, 1342–1352. [Google Scholar] [CrossRef] [PubMed]
- Andersson, E.R.; Salto, C.; Villaescusa, J.C.; Cajanek, L.; Yang, S.; Bryjova, L.; Nagy, I.I.; Vainio, S.J.; Ramirez, C.; Bryja, V.; et al. Wnt5a cooperates with canonical Wnts to generate midbrain dopaminergic neurons in vivo and in stem cells. Proc. Natl. Acad. Sci. USA 2013, 110, 610. [Google Scholar] [CrossRef] [PubMed]
- Prakash, N.; Wurst, W. Development of dopaminergic neurons in the mammalian brain. Cell. Mol. Life Sci. 2006, 63, 187–206. [Google Scholar] [CrossRef] [PubMed]
- Bryja, V.; Schulte, G.; Rawal, N.; Grahn, A.; Arenas, E. Wnt-5a induces Dishevelled phosphorylation and dopaminergic differentiation via a CK1-dependent mechanism. J. Cell Sci. 2007, 120, 586–595. [Google Scholar] [CrossRef] [PubMed]
- Schulte, G.; Bryja, V.; Rawal, N.; Castelo-Branco, G.; Sousa, K.M.; Arenas, E. Purified Wnt-5a increases differentiation of midbrain dopaminergic cells and dishevelled phosphorylation. J. Neurochem. 2005, 92, 1550–1553. [Google Scholar] [CrossRef] [PubMed]
- Haegel, H.; Larue, L.; Ohsugi, M.; Fedorov, L.; Herrenknecht, K.; Kemler, R. Lack of beta-catenin affects mouse development at gastrulation. Development 1995, 121, 3529–3537. [Google Scholar]
- Chilov, D.; Sinjushina, N.; Saarimaki-Vire, J.; Taketo, M.M.; Partanen, J. β-Catenin regulates intercellular signalling networks and cell-type specific transcription in the developing mouse midbrain-rhombomere 1 region. PLoS ONE 2010, 5, e10881. [Google Scholar] [CrossRef]
- Joksimovic, M.; Patel, M.; Taketo, M.M.; Johnson, R.; Awatramani, R. Ectopic Wnt/β-catenin signaling induces neurogenesis in the spinal cord and hindbrain floor plate. PLoS ONE 2012, 7, e30266. [Google Scholar] [CrossRef]
- Nouri, N.; Patel, M.J.; Joksimovic, M.; Poulin, J.-F.; Anderegg, A.; Taketo, M.M.; Ma, Y.-C.; Awatramani, R. Excessive Wnt/β-catenin signaling promotes midbrain floor plate neurogenesis, but results in vacillating dopamine progenitors. Mol. Cell. Neurosci. 2015, 68, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Miyamoto, Y.; Huang, E.J. Multiple roles of β-catenin in controlling the neurogenic niche for midbrain dopamine neurons. Development 2009, 136, 2027–2038. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Villaescusa, J.C.; Luo, S.X.; Guitarte, C.; Lei, S.; Miyamoto, Y.; Taketo, M.M.; Arenas, E.; Huang, E.J. Interactions of Wnt/β-catenin signaling and sonic hedgehog regulate the neurogenesis of ventral midbrain dopamine neurons. J. Neurosci. 2010, 30, 9280–9291. [Google Scholar] [CrossRef] [PubMed]
- Chilov, D.; Sinjushina, N.; Rita, H.; Taketo, M.M.; Makela, T.P.; Partanen, J. Phosphorylated β-catenin localizes to centrosomes of neuronal progenitors and is required for cell polarity and neurogenesis in developing midbrain. Dev. Biol. 2011, 357, 259–268. [Google Scholar] [CrossRef]
- Castelo-Branco, G.; Andersson, E.R.; Minina, E.; Sousa, K.M.; Ribeiro, D.; Kokubu, C.; Imai, K.; Prakash, N.; Wurst, W.; Arenas, E. Delayed dopaminergic neuron differentiation in Lrp6 mutant mice. Dev. Dyn. 2010, 239, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Fernando, C.V.; Kele, J.; Bye, C.R.; Niclis, J.C.; Alsanie, W.; Blakely, B.D.; Stenman, J.; Turner, B.J.; Parish, C.L. Diverse roles for Wnt7a in ventral midbrain neurogenesis and dopaminergic axon morphogenesis. Stem Cells Dev. 2014, 23, 1991–2003. [Google Scholar] [CrossRef] [PubMed]
- Sousa, K.M.; Villaescusa, J.C.; Cajanek, L.; Ondr, J.K.; Castelo-Branco, G.; Hofstra, W.; Bryja, V.; Palmberg, C.; Bergman, T.; Wainwright, B.; et al. Wnt2 regulates progenitor proliferation in the developing ventral midbrain. J. Biol. Chem. 2010, 285, 7246–7253. [Google Scholar] [CrossRef] [PubMed]
- Fischer, T.; Guimera, J.; Wurst, W.; Prakash, N. Distinct but redundant expression of the Frizzled Wnt receptor genes at signaling centers of the developing mouse brain. Neuroscience 2007, 147, 693–711. [Google Scholar] [CrossRef] [PubMed]
- Rawal, N.; Castelo-Branco, G.; Sousa, K.M.; Kele, J.; Kobayashi, K.; Okano, H.; Arenas, E. Dynamic temporal and cell type-specific expression of Wnt signaling components in the developing midbrain. Exp. Cell Res. 2006, 312, 1626–1636. [Google Scholar] [CrossRef] [PubMed]
- Tissir, F.; Goffinet, A.M. Shaping the nervous system: Role of the core planar cell polarity genes. Nat. Rev. Neurosci. 2013, 14, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Stuebner, S.; Faus-Kessler, T.; Fischer, T.; Wurst, W.; Prakash, N. Fzd3 and Fzd6 deficiency results in a severe midbrain morphogenesis defect. Dev. Dyn. 2010, 239, 246–260. [Google Scholar] [CrossRef] [PubMed]
- Kele, J.; Andersson, E.R.; Villaescusa, J.C.; Cajanek, L.; Parish, C.L.; Bonilla, S.; Toledo, E.M.; Bryja, V.; Rubin, J.S.; Shimono, A.; et al. SFRP1 and 2 Dose-Dependently Regulate Midbrain Dopamine Neuron Development In vivo and in Embryonic Stem Cells. Stem Cells 2012, 30, 865–875. [Google Scholar] [CrossRef] [PubMed]
- Blakely, B.D.; Bye, C.R.; Fernando, C.V.; Prasad, A.A.; Pasterkamp, R.J.; Macheda, M.L.; Stacker, S.A.; Parish, C.L. Ryk, a Receptor Regulating Wnt5a-Mediated Neurogenesis and Axon Morphogenesis of Ventral Midbrain Dopaminergic Neurons. Stem Cells Dev. 2013, 22, 2132–2144. [Google Scholar] [CrossRef]
- Fukusumi, Y.; Meier, F.; Götz, S.; Matheus, F.; Irmler, M.; Beckervordersandforth, R.; Faus-Kessler, T.; Minina, E.; Rauser, B.; Zhang, J.; et al. Dickkopf 3 Promotes the Differentiation of a Rostrolateral Midbrain Dopaminergic Neuronal Subset In Vivo and from Pluripotent Stem Cells In Vitro in the Mouse. J. Neurosci. 2015, 35, 13385–13401. [Google Scholar] [CrossRef] [PubMed]
- Cruciat, C.-M.; Niehrs, C. Secreted and transmembrane wnt inhibitors and activators. Cold Spring Harb. Perspect. Biol. 2013, 5, a015081. [Google Scholar] [CrossRef] [PubMed]
- Vogt Weisenhorn, D.M.; Giesert, F.; Wurst, W. Diversity matters- heterogeneity of dopaminergic neurons in the ventral mesencephalon and its relation to Parkinson’s Disease. J. Neurochem. 2016, 139 (Suppl. 1), 8–26. [Google Scholar] [CrossRef] [PubMed]
- Omodei, D.; Acampora, D.; Mancuso, P.; Prakash, N.; Di Giovannantonio, L.G.; Wurst, W.; Simeone, A. Anterior-posterior graded response to Otx2 controls proliferation and differentiation of dopaminergic progenitors in the ventral mesencephalon. Development 2008, 135, 3459–3470. [Google Scholar] [CrossRef] [PubMed]
- Anderegg, A.; Lin, H.P.; Chen, J.A.; Caronia-Brown, G.; Cherepanova, N.; Yun, B.; Joksimovic, M.; Rock, J.; Harfe, B.D.; Johnson, R.; et al. An Lmx1b-miR135a2 Regulatory Circuit Modulates Wnt1/Wnt Signaling and Determines the Size of the Midbrain Dopaminergic Progenitor Pool. PLoS Genet. 2013, 9, e1003973. [Google Scholar] [CrossRef] [PubMed]
- Momcilovic, O.; Liu, Q.; Swistowski, A.; Russo-Tait, T.; Zhao, Y.; Rao, M.S.; Zeng, X. Genome wide profiling of dopaminergic neurons derived from human embryonic and induced pluripotent stem cells. Stem Cells Dev. 2014, 23, 406–420. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Ding, S. Small molecules that modulate embryonic stem cell fate and somatic cell reprogramming. Trends Pharmacol. Sci. 2010, 31, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Cajanek, L.; Ribeiro, D.; Liste, I.; Parish, C.L.; Bryja, V.; Arenas, E. Wnt/β-catenin signaling blockade promotes neuronal induction and dopaminergic differentiation in embryonic stem cells. Stem Cells 2009, 27, 2917–2927. [Google Scholar] [CrossRef] [PubMed]
- Cajanek, L.; Ganji, R.S.; Henriques-Oliveira, C.; Theofilopoulos, S.; Konik, P.; Bryja, V.; Arenas, E. Tiam1 regulates the Wnt/Dvl/Rac1 signaling pathway and the differentiation of midbrain dopaminergic neurons. Mol. Cell. Biol. 2013, 33, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Castelo-Branco, G.; Rawal, N.; Arenas, E. GSK-3β inhibition/beta-catenin stabilization in ventral midbrain precursors increases differentiation into dopamine neurons. J. Cell Sci. 2004, 117, 5731–5737. [Google Scholar] [CrossRef] [PubMed]
- Rhim, J.H.; Luo, X.; Xu, X.; Gao, D.; Zhou, T.; Li, F.; Qin, L.; Wang, P.; Xia, X.; Wong, S.T.C. A High-content screen identifies compounds promoting the neuronal differentiation and the midbrain dopamine neuron specification of human neural progenitor cells. Sci. Rep. 2015, 5, 16237. [Google Scholar] [CrossRef] [PubMed]
- Xi, J.; Liu, Y.; Liu, H.; Chen, H.; Emborg, M.E.; Zhang, S.C. Specification of midbrain dopamine neurons from primate pluripotent stem cells. Stem Cells 2012, 30, 1655–1663. [Google Scholar] [CrossRef] [PubMed]
- Okawa, S.; Salto, C.; Ravichandran, S.; Yang, S.; Toledo, E.M.; Arenas, E.; Del Sol, A. Transcriptional synergy as an emergent property defining cell subpopulation identity enables population shift. Nat. Commun. 2018, 9, 2595. [Google Scholar] [CrossRef]
- Zeng, X.-S.; Geng, W.-S.; Jia, J.-J.; Chen, L.; Zhang, P.-P. Cellular and Molecular Basis of Neurodegeneration in Parkinson Disease. Front. Aging Neurosci. 2018, 10, 109. [Google Scholar] [CrossRef]
- Awad, O.; Panicker, L.M.; Deranieh, R.M.; Srikanth, M.P.; Brown, R.A.; Voit, A.; Peesay, T.; Park, T.S.; Zambidis, E.T.; Feldman, R.A. Altered Differentiation Potential of Gaucher’s Disease iPSC Neuronal Progenitors due to Wnt/beta-Catenin Downregulation. Stem Cell Rep. 2017, 9, 1853–1867. [Google Scholar] [CrossRef]
- De Gregorio, R.; Pulcrano, S.; de Sanctis, C.; Volpicelli, F.; Guatteo, E.; von Oerthel, L.; Latagliata, E.C.; Esposito, R.; Piscitelli, R.M.; Perrone-Capano, C.; et al. miR-34b/c Regulates Wnt1 and Enhances Mesencephalic Dopaminergic Neuron Differentiation. Stem Cell Rep. 2018, 10, 1237–1250. [Google Scholar] [CrossRef]
- Kim, H.S.; Kim, J.; Jo, Y.; Jeon, D.; Cho, Y.S. Direct lineage reprogramming of mouse fibroblasts to functional midbrain dopaminergic neuronal progenitors. Stem Cell Res. 2013, 12, 60–68. [Google Scholar] [CrossRef]
- Di Rivetti Val Cervo, P.; Romanov, R.A.; Spigolon, G.; Masini, D.; Martin-Montanez, E.; Toledo, E.M.; La Manno, G.; Feyder, M.; Pifl, C.; Ng, Y.-H.; et al. Induction of functional dopamine neurons from human astrocytes in vitro and mouse astrocytes in a Parkinson’s disease model. Nat. Biotechnol. 2017, 35, 444–452. [Google Scholar] [CrossRef] [PubMed]
- Liss, B.; Franz, O.; Sewing, S.; Bruns, R.; Neuhoff, H.; Roeper, J. Tuning pacemaker frequency of individual dopaminergic neurons by Kv4.3L and KChip3.1 transcription. EMBO J. 2001, 20, 5715–5724. [Google Scholar] [CrossRef]
- Serodio, P.; Rudy, B. Differential expression of Kv4 K+ channel subunits mediating subthreshold transient K+ (A-type) currents in rat brain. J. Neurophysiol. 1998, 79, 1081–1091. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.L.; Sinton, C.M.; German, D.C. Midbrain dopaminergic neurons in the mouse: Co-localization with Calbindin-D28K and calretinin. Neuroscience 1996, 75, 523–533. [Google Scholar] [CrossRef]
- Katagiri, T.; Watabe, T. Bone Morphogenetic Proteins. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef]
- Chen, H.-L.; Panchision, D.M. Concise review: Bone morphogenetic protein pleiotropism in neural stem cells and their derivatives—Alternative pathways, convergent signals. Stem Cells 2007, 25, 63–68. [Google Scholar] [CrossRef]
- Bond, A.M.; Bhalala, O.G.; Kessler, J.A. The dynamic role of bone morphogenetic proteins in neural stem cell fate and maturation. Dev. Neurobiol. 2012, 72, 1068–1084. [Google Scholar] [CrossRef]
- Hegarty, S.V.; O’Keeffe, G.W.; Sullivan, A.M. BMP-Smad 1/5/8 signalling in the development of the nervous system. Prog. Neurobiol. 2013, 109, 28–41. [Google Scholar] [CrossRef]
- Furuta, Y.; Piston, D.W.; Hogan, B.L. Bone morphogenetic proteins (BMPs) as regulators of dorsal forebrain development. Development 1997, 124, 2203–2212. [Google Scholar]
- Liu, A.; Niswander, L.A. Bone morphogenetic protein signalling and vertebrate nervous system development. Nat. Rev. Neurosci. 2005, 6, 945–954. [Google Scholar] [CrossRef]
- Arnold, S.J.; Maretto, S.; Islam, A.; Bikoff, E.K.; Robertson, E.J. Dose-dependent Smad1, Smad5 and Smad8 signaling in the early mouse embryo. Dev. Biol. 2006, 296, 104–118. [Google Scholar] [CrossRef] [PubMed]
- Rosenzweig, B.L.; Imamura, T.; Okadome, T.; Cox, G.N.; Yamashita, H.; ten Dijke, P.; Heldin, C.H.; Miyazono, K. Cloning and characterization of a human type II receptor for bone morphogenetic proteins. Proc. Natl. Acad. Sci. USA 1995, 92, 7632–7636. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.-H.; Derynck, R. Specificity and versatility in tgf-beta signaling through Smads. Annu. Rev. Cell Dev. Biol. 2005, 21, 659–693. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Jordan, J.; Böttner, M.; Schluesener, H.J.; Unsicker, K.; Krieglstein, K. Bone morphogenetic proteins: neurotrophic roles for midbrain dopaminergic neurons and implications of astroglial cells. Eur. J. Neurosci. 1997, 9, 1699–1709. [Google Scholar] [CrossRef] [PubMed]
- Hegarty, S.V.; Sullivan, A.M.; O’Keeffe, G.W. BMP2 and GDF5 induce neuronal differentiation through a Smad dependant pathway in a model of human midbrain dopaminergic neurons. Mol. Cell. Neurosci. 2013, 56, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Panchision, D.M.; Pickel, J.M.; Studer, L.; Lee, S.H.; Turner, P.A.; Hazel, T.G.; McKay, R.D. Sequential actions of BMP receptors control neural precursor cell production and fate. Genes Dev. 2001, 15, 2094–2110. [Google Scholar] [CrossRef]
- Tronche, F.; Kellendonk, C.; Kretz, O.; Gass, P.; Anlag, K.; Orban, P.C.; Bock, R.; Klein, R.; Schutz, G. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat. Genet. 1999, 23, 99–103. [Google Scholar] [CrossRef]
- Schulz, T.C.; Noggle, S.A.; Palmarini, G.M.; Weiler, D.A.; Lyons, I.G.; Pensa, K.A.; Meedeniya, A.C.B.; Davidson, B.P.; Lambert, N.A.; Condie, B.G. Differentiation of human embryonic stem cells to dopaminergic neurons in serum-free suspension culture. Stem Cells 2004, 22, 1218–1238. [Google Scholar] [CrossRef]
- Liu, Q.; Pedersen, O.Z.; Peng, J.; Couture, L.A.; Rao, M.S.; Zeng, X. Optimizing dopaminergic differentiation of pluripotent stem cells for the manufacture of dopaminergic neurons for transplantation. Cytotherapy 2013, 15, 999–1010. [Google Scholar] [CrossRef]
- Yang, H.; Wang, J.; Wang, F.; Liu, X.; Chen, H.; Duan, W.; Qu, T. Dopaminergic Neuronal Differentiation from the Forebrain-Derived Human Neural Stem Cells Induced in Cultures by Using a Combination of BMP-7 and Pramipexole with Growth Factors. Front. Neural Circuits 2016, 10, 29. [Google Scholar] [CrossRef]
- Thier, M.; Wörsdörfer, P.; Lakes, Y.B.; Gorris, R.; Herms, S.; Opitz, T.; Seiferling, D.; Quandel, T.; Hoffmann, P.; Nöthen, M.M.; et al. Direct conversion of fibroblasts into stably expandable neural stem cells. Cell Stem Cell 2012, 10, 473–479. [Google Scholar] [CrossRef] [PubMed]
- Kadari, A.; Lu, M.; Li, M.; Sekaran, T.; Thummer, R.P.; Guyette, N.; Chu, V.; Edenhofer, F. Excision of viral reprogramming cassettes by Cre protein transduction enables rapid, robust and efficient derivation of transgene-free human induced pluripotent stem cells. Stem Cell Res. Ther. 2014, 5, 47. [Google Scholar] [CrossRef] [PubMed]
- Meyer, S.; Wörsdörfer, P.; Günther, K.; Thier, M.; Edenhofer, F. Derivation of Adult Human Fibroblasts and their Direct Conversion into Expandable Neural Progenitor Cells. J. Vis. Exp. 2015, e52831. [Google Scholar] [CrossRef] [PubMed]
- Kwok, C.K.; Ueda, Y.; Kadari, A.; Günther, K.; Ergün, S.; Heron, A.; Schnitzler, A.C.; Rook, M.; Edenhofer, F. Scalable stirred suspension culture for the generation of billions of human induced pluripotent stem cells using single-use bioreactors. J. Tissue Eng. Regener. Med. 2018, 12, e1076–e1087. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, P.; Glatza, M.; Hemmer, K.; Tsytsyura, Y.; Thiel, C.S.; Hoing, S.; Moritz, S.; Parga, J.A.; Wagner, L.; Bruder, J.M.; et al. Derivation and expansion using only small molecules of human neural progenitors for neurodegenerative disease modeling. PLoS ONE 2013, 8, e59252. [Google Scholar] [CrossRef]
- Joyner, A.L.; Liu, A.; Millet, S. Otx2, Gbx2 and Fgf8 interact to position and maintain a mid-hindbrain organizer. Curr. Opin. Cell Biol. 2000, 12, 736–741. [Google Scholar] [CrossRef]
- Wurst, W.; Bally-Cuif, L. Neural plate patterning: Upstream and downstream of the isthmic organizer. Nat. Rev. Neurosci. 2001, 2, 99–108. [Google Scholar] [CrossRef]
- Olander, S.; Nordstrom, U.; Patthey, C.; Edlund, T. Convergent Wnt and FGF signaling at the gastrula stage induce the formation of the isthmic organizer. Mech. Dev. 2006, 123, 166–176. [Google Scholar] [CrossRef]
- Canning, C.A.; Lee, L.; Irving, C.; Mason, I.; Jones, C.M. Sustained interactive Wnt and FGF signaling is required to maintain isthmic identity. Dev. Biol. 2007, 305, 276–286. [Google Scholar] [CrossRef]
- Dyer, C.; Blanc, E.; Hanisch, A.; Roehl, H.; Otto, G.W.; Yu, T.; Basson, M.A.; Knight, R. A bi-modal function of Wnt signalling directs an FGF activity gradient to spatially regulate neuronal differentiation in the midbrain. Development 2014, 141, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Wittmann, D.M.; Blochl, F.; Trumbach, D.; Wurst, W.; Prakash, N.; Theis, F.J. Spatial analysis of expression patterns predicts genetic interactions at the mid-hindbrain boundary. PLoS Comput. Biol. 2009, 5, e1000569. [Google Scholar] [CrossRef]
- Danielian, P.S.; McMahon, A.P. Engrailed-1 as a target of the Wnt-1 signalling pathway in vertebrate midbrain development. Nature 1996, 383, 332–334. [Google Scholar] [CrossRef] [PubMed]
- Gemel, J.; Jacobsen, C.; MacArthur, C.A. Fibroblast growth factor-8 expression is regulated by intronic engrailed and Pbx1-binding sites. J. Biol. Chem. 1999, 274, 6020–6026. [Google Scholar] [CrossRef] [PubMed]
- Ardayfio, P.A.; Leung, A.; Park, J.; Hwang, D.Y.; Moran-Gates, T.; Choi, Y.K.; Carlezon, W.A., Jr.; Tarazi, F.I.; Kim, K.S. Pitx3-deficient aphakia mice display unique behavioral responses to psychostimulant and antipsychotic drugs. Neuroscience 2010, 166, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Lee, J.S.; Hwang, H.S.; Lee, D.R.; Park, C.-Y.; Jung, S.J.; You, Y.R.; Kim, D.-S.; Kim, D.-W. Wnt signal activation induces midbrain specification through direct binding of the β-catenin/TCF4 complex to the EN1 promoter in human pluripotent stem cells. Exp. Mol. Med. 2018, 50, 24. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.M.; Kabotyanski, E.B.; Dou, Y.; Reineke, L.C.; Zhang, P.; Zhang, X.H.-F.; Malovannaya, A.; Jung, S.Y.; Mo, Q.; Roarty, K.P.; et al. FGFR1-Activated Translation of WNT Pathway Components with Structured 5′ UTRs Is Vulnerable to Inhibition of EIF4A-Dependent Translation Initiation. Cancer Res. 2018, 78, 4229–4240. [Google Scholar] [CrossRef]
- Aman, A.; Piotrowski, T. Wnt/β-catenin and Fgf signaling control collective cell migration by restricting chemokine receptor expression. Dev. Cell 2008, 15, 749–761. [Google Scholar] [CrossRef]
- Dailey, L.; Ambrosetti, D.; Mansukhani, A.; Basilico, C. Mechanisms underlying differential responses to FGF signaling. Cytokine Growth Factor Rev. 2005, 16, 233–247. [Google Scholar] [CrossRef]
- Ulloa, F.; Itasaki, N.; Briscoe, J. Inhibitory Gli3 activity negatively regulates Wnt/beta-catenin signaling. Curr. Biol. 2007, 17, 545–550. [Google Scholar] [CrossRef]
- Lei, Q.; Jeong, Y.; Misra, K.; Li, S.; Zelman, A.K.; Epstein, D.J.; Matise, M.P. Wnt signaling inhibitors regulate the transcriptional response to morphogenetic Shh-Gli signaling in the neural tube. Dev. Cell 2006, 11, 325–337. [Google Scholar] [CrossRef]
- Tasouri, E.; Tucker, K.L. Primary cilia and organogenesis: Is Hedgehog the only sculptor? Cell Tissue Res. 2011, 345, 21–40. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Schleidt, S.; Pelta-Heller, J.; Hutchings, D.; Cannarsa, G.; Iacovitti, L. BMP and TGF-beta pathway mediators are critical upstream regulators of Wnt signaling during midbrain dopamine differentiation in human pluripotent stem cells. Dev. Biol. 2013, 376, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Falk, S.; Wurdak, H.; Ittner, L.M.; Ille, F.; Sumara, G.; Schmid, M.-T.; Draganova, K.; Lang, K.S.; Paratore, C.; Leveen, P.; et al. Brain area-specific effect of TGF-β signaling on Wnt-dependent neural stem cell expansion. Cell Stem Cell 2008, 2, 472–483. [Google Scholar] [CrossRef] [PubMed]
- Veeck, J.; Dahl, E. Targeting the Wnt pathway in cancer: The emerging role of Dickkopf-3. Biochim. Biophys. Acta 2012, 1825, 18–28. [Google Scholar] [CrossRef]
- Zhang, J.; Götz, S.; Vogt Weisenhorn, D.M.; Simeone, A.; Wurst, W.; Prakash, N. A WNT1-regulated developmental gene cascade prevents dopaminergic neurodegeneration in adult En1(+/−) mice. Neurobiol. Dis. 2015, 82, 32–45. [Google Scholar] [CrossRef]
- Rekaik, H.; Blaudin de The, F.-X.; Prochiantz, A.; Fuchs, J.; Joshi, R.L. Dissecting the role of Engrailed in adult dopaminergic neurons--Insights into Parkinson disease pathogenesis. FEBS Lett. 2015, 589, 3786–3794. [Google Scholar] [CrossRef]
- Chung, S.; Leung, A.; Han, B.S.; Chang, M.Y.; Moon, J.I.; Kim, C.H.; Hong, S.; Pruszak, J.; Isacson, O.; Kim, K.S. Wnt1-lmx1a forms a novel autoregulatory loop and controls midbrain dopaminergic differentiation synergistically with the SHH-FoxA2 pathway. Cell Stem Cell 2009, 5, 646–658. [Google Scholar] [CrossRef] [PubMed]
- Matthes, M.; Preusse, M.; Zhang, J.; Schechter, J.; Mayer, D.; Lentes, B.; Theis, F.; Prakash, N.; Wurst, W.; Trumbach, D. Mouse IDGenes: A reference database for genetic interactions in the developing mouse brain. Database 2014, 2014, bau083. [Google Scholar] [CrossRef]
- Peng, C.; Aron, L.; Klein, R.; Li, M.; Wurst, W.; Prakash, N.; Le, W. Pitx3 is a critical mediator of GDNF-induced BDNF expression in nigrostriatal dopaminergic neurons. J. Neurosci. 2011, 31, 12802–12815. [Google Scholar] [CrossRef] [PubMed]
- Prakash, N.; Wurst, W. A Wnt signal regulates stem cell fate and differentiation in vivo. Neurodegener. Dis. 2007, 4, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Hegarty, S.V.; Sullivan, A.M.; O’Keeffe, G.W. Roles for the TGFβ superfamily in the development and survival of midbrain dopaminergic neurons. Mol. Neurobiol. 2014, 50, 559–573. [Google Scholar] [CrossRef]
- Pinho, S.; Niehrs, C. Dkk3 is required for TGF-beta signaling during Xenopus mesoderm induction. Differentiation 2007, 75, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Hsu, R.J.; Lin, C.C.; Su, Y.F.; Tsai, H.J. dickkopf-3-related gene regulates the expression of zebrafish myf5 gene through phosphorylated p38a-dependent Smad4 activity. J. Biol. Chem. 2011, 286, 6855–6864. [Google Scholar] [CrossRef] [PubMed]
- Romero, D.; Kawano, Y.; Bengoa, N.; Walker, M.M.; Maltry, N.; Niehrs, C.; Waxman, J.; Kypta, R. Downregulation of Dickkopf-3 disrupts prostate acinar morphogenesis through TGF-β/Smad signalling. J. Cell Sci. 2013, 126, 1858–1867. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, H.; Liang, Y.; Peng, P.; Ma, X.; Zhang, X. DKK3 regulates cell proliferation, apoptosis and collagen synthesis in keloid fibroblasts via TGF-β1/Smad signaling pathway. Biomed. Pharmacother. 2017, 91, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Itasaki, N.; Hoppler, S. Crosstalk between Wnt and bone morphogenic protein signaling: A turbulent relationship. Dev. Dyn. 2010, 239, 16–33. [Google Scholar] [CrossRef] [PubMed]
- Arkell, R.; Beddington, R.S. BMP-7 influences pattern and growth of the developing hindbrain of mouse embryos. Development 1997, 124, 1–12. [Google Scholar] [PubMed]



{kind=link}
{kind=link}
| FGFs | SHH | WNTs | BMPs | |
|---|---|---|---|---|
| VM patterning | Yes | Yes | Yes | No 1 |
| Progenitor proliferation | Yes | Yes | Yes | Yes |
| Subtype specification | Yes? 2 | Yes | Yes? 2 | Yes |
| Differentiation | Yes | No | Yes | Yes |
| Survival | Yes | No | Yes | N.D. 3 |
| In vitro stem/somatic cell differentiation | Mouse and human ESCs, iPSCs, iDA | Mouse and human ESCs, iPSCs, iNSC | Mouse and human ESCs, iPSCs, iDA | Human iPSCs, Human iNSC |
| Known pathway interaction | Promotes Wnt1 expression | Regulates Wnt expression | Represses Shh, promotes Fgf8 expression | Represses SHH expression |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brodski, C.; Blaess, S.; Partanen, J.; Prakash, N. Crosstalk of Intercellular Signaling Pathways in the Generation of Midbrain Dopaminergic Neurons In Vivo and from Stem Cells. J. Dev. Biol. 2019, 7, 3. https://doi.org/10.3390/jdb7010003
Brodski C, Blaess S, Partanen J, Prakash N. Crosstalk of Intercellular Signaling Pathways in the Generation of Midbrain Dopaminergic Neurons In Vivo and from Stem Cells. Journal of Developmental Biology. 2019; 7(1):3. https://doi.org/10.3390/jdb7010003
Chicago/Turabian StyleBrodski, Claude, Sandra Blaess, Juha Partanen, and Nilima Prakash. 2019. "Crosstalk of Intercellular Signaling Pathways in the Generation of Midbrain Dopaminergic Neurons In Vivo and from Stem Cells" Journal of Developmental Biology 7, no. 1: 3. https://doi.org/10.3390/jdb7010003
APA StyleBrodski, C., Blaess, S., Partanen, J., & Prakash, N. (2019). Crosstalk of Intercellular Signaling Pathways in the Generation of Midbrain Dopaminergic Neurons In Vivo and from Stem Cells. Journal of Developmental Biology, 7(1), 3. https://doi.org/10.3390/jdb7010003