The Role of Hedgehog Signalling in the Formation of the Ventricular Septum


Abstract
1. Introduction
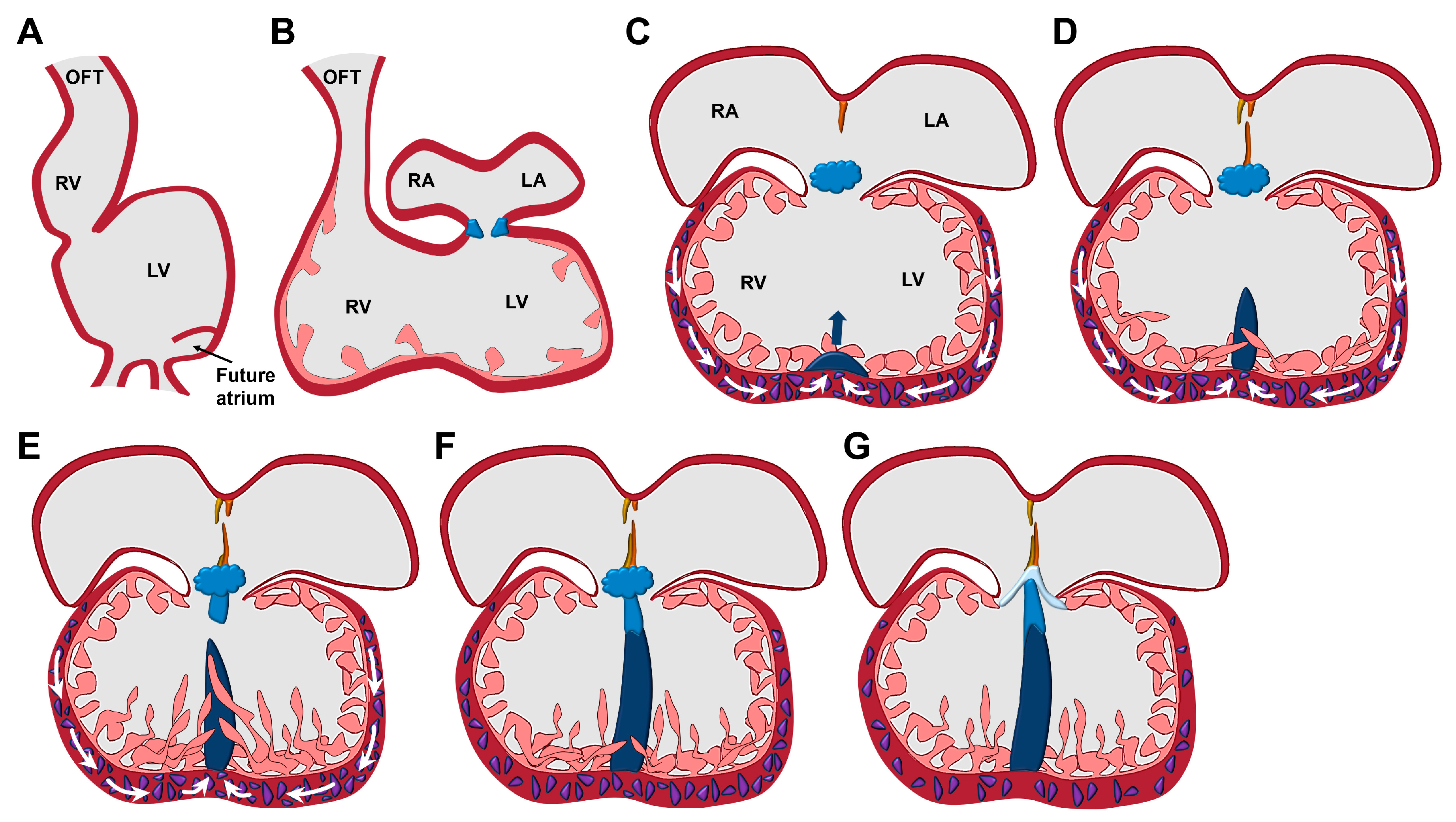
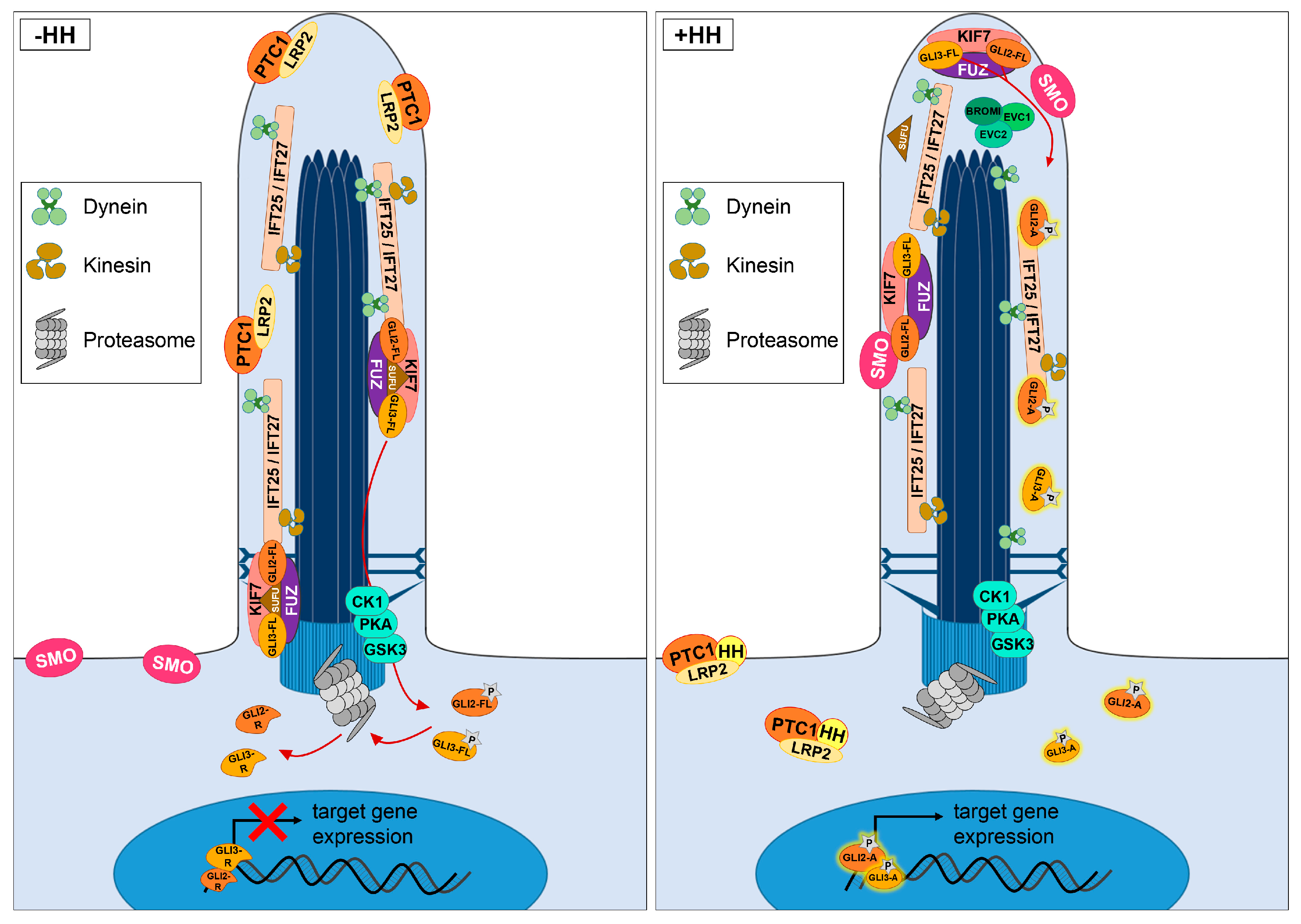
2. HH Signalling Plays an Essential Role in the Development of the VS
3. Is It Possible to Prevent the Development of VSDs by Targeting HH Signalling in Pregnancy?
Acknowledgments
Conflicts of Interest
References
- Waterston, D. The development of the heart in man. Trans. R. Soc. Edinb. 1918, 52, 257–302. [Google Scholar] [CrossRef]
- Frazer, J. A Manual of Embryology. In Development of the Heart, and Vessels of the Anterior Part of the Embryo, 2nd ed.; Bailliere, Tindall and Cox: London, UK, 1940; pp. 322–352. [Google Scholar]
- Goor, D.; Edwards, J.; Lillehei, C. The development of the interventricular septum of the human heart; Correlative morphogenetic study. Chest 1970, 58, 453–467. [Google Scholar] [CrossRef] [PubMed]
- Goor, D.; Lillehei, C.; Rees, R.; Edwards, J. Isolated ventricular septal defect. Development basis for various types and presentation of classification. Chest 1970, 58, 468–482. [Google Scholar] [CrossRef] [PubMed]
- Wenink, A. The conducting tissues in primitive ventricle with outlet chamber. Two different possibilities. J. Thorac. Cardiovasc. Surg. 1978, 75, 747–753. [Google Scholar] [PubMed]
- Wenink, A.; Oppenheimer-Dekker, A.; Moulaert, A. Muscular ventricular septal defects: A reappraisal of the anatomy. Am. J. Cardiol. 1979, 43, 259–264. [Google Scholar] [CrossRef]
- Anderson, R.; Spicer, D.; Brown, N.; Mohun, T. The development of septation in the four-chambered heart. Anat. Rec. 2014, 297, 1414–1429. [Google Scholar] [CrossRef] [PubMed]
- Moorman, A.; Christoffels, V. Cardiac chamber formation: Development, genes, and evolution. Physiol. Rev. 2003, 83, 1223–1267. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, L.; Markwald, R. Molecular regulation of atrioventricular valvuloseptal morphogenesis. Circ. Res. 1995, 77, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Steding, G.; Seidl, W. Contribution to the development of the heart. Part 1: Normal development. Thorac. Cardiovasc. Surg. 1980, 28, 386–409. [Google Scholar] [CrossRef] [PubMed]
- Franco, D.; Meilhac, S.; Christoffels, V.; Kispert, A.; Buckingham, M.; Kelly, R. Left and right ventricular contributions to the formation of the interventricular septum in the mouse heart. Dev. Biol. 2006, 294, 366–375. [Google Scholar] [CrossRef] [PubMed]
- Ben-Shachar, G.; Arcilla, R.; Lucas, R.; Manasek, F. Ventricular trabeculations in the chick embryo heart and their contribution to ventricular and muscular septal development. Circ. Res. 1985, 57, 759–766. [Google Scholar] [CrossRef] [PubMed]
- Contreras-Ramos, A.; Sánchez-Gómez, C.; García-Romero, H.; Cimarosti, L. Normal development of the muscular region of the interventricular septum—I. The significance of the ventricular trabeculations. Anat. Histol. Embryol. 2008, 37, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Hochstetter, F. Entwicklung des Venensystems. In Handbuch der Vergleichenden und Experimentellen Entwicklungslehre der Wirbeltiere; Gustav Fischer: Jena, Germany, 1906; Volume 3, pp. 141–145. [Google Scholar]
- Patten, B. Development of the chick during the third and fourth days of incubation. In Early Embryology of the Chick, 4th ed.; McGraw-Hill: New York, NY, USA, 1951; pp. 156–213. [Google Scholar]
- Goor, D.; Lillehei, C. The embryology of the heart. In Congenital Malformations of the Heart; Grune & Stratton: Orlando, FL, USA, 1975; pp. 38–88. [Google Scholar]
- Rychter, Z.; Rychterová, V.; Lemez, L. Formation of the heart loop and proliferation structure of its wall as a base for ventricular septation. Herz 1979, 4, 86–90. [Google Scholar] [PubMed]
- Van Mierop, L.; Kutsche, L. Development of the ventricular septum of the heart. Heart Vessels 1985, 1, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Meredith, M.; Hutchins, G.; Moore, G. Role of the left interventricular sulcus in formation of interventricular septum and crista supraventricularis in normal human cardiogenesis. Anat. Rec. 1979, 194, 417–428. [Google Scholar] [CrossRef] [PubMed]
- Sakata, Y.; Kamei, C.; Nakagami, H.; Bronson, R.; Liao, J.; Chin, M. Ventricular septal defect and cardiomyopathy in mice lacking the transcription factor CHF1/Hey2. Proc. Natl. Acad. Sci. USA 2002, 99, 16197–16202. [Google Scholar] [CrossRef] [PubMed]
- Lamers, W.; Moorman, A. Cardiac septation: A late contribution of the embryonic primary myocardium to heart morphogenesis. Circ. Res. 2002, 91, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, K.; Wakatsuki, S.; Yamada, S.; Yamamura, K.; Miyazaki, J.; Sehara-Fujisawa, A. Meltrin β expressed in cardiac neural crest cells is required for ventricular septum formation of the heart. Dev. Biol. 2007, 303, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Brickner, M.; Hillis, L.; Lange, R. Congenital heart disease in adults. First of two parts. N. Engl. J. Med. 2000, 342, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, A.; Shenoy, V.; Yamazato, Y.; Sriramula, S.; Francis, J.; Yuan, L.; Castellano, R.; Ostrov, D.; Oh, S.; Katovich, M.; et al. Evidence for angiotensin-converting enzyme 2 as a therapeutic target for the prevention of pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2009, 179, 1048–1054. [Google Scholar] [CrossRef] [PubMed]
- Selicorni, A.; Colli, A.; Passarini, A.; Milani, D.; Cereda, A.; Cerutti, M.; Maitz, S.; Alloni, V.; Salvini, L.; Galli, M.; et al. Analysis of congenital heart defects in 87 consecutive patients with Brachmann-de Lange syndrome. Am. J. Med. Genet. A 2009, 149A, 1268–1272. [Google Scholar] [CrossRef] [PubMed]
- Roger, H. Clinical researches on the congenital communication of the two sides of the heart by failure of occlusion of the interventricular septum. Bull. Acad. Med. 1879, 8, 1074–1094. [Google Scholar]
- Hoffman, J.; Kaplan, S. The incidence of congenital heart disease. J. Am. Coll. Cardiol. 2002, 39, 1890–1900. [Google Scholar] [CrossRef]
- Williams, D. Bicuspid aortic valve. J. Insur. Med. 2006, 38, 72–74. [Google Scholar] [PubMed]
- Benjamin, E.; Blaha, M.; Chiuve, S.; Cushman, M.; Das, S.; Deo, R.; de Ferranti, S.; Floyd, J.; Fornage, M.; Gillespie, C.; et al. Heart Disease and Stroke Statistics-2017 Update: A Report From the American Heart Association. Circulation 2017, 135, e146–e603. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, D. Heart disease: An ongoing genetic battle? Nature 2004, 429, 819–822. [Google Scholar] [CrossRef] [PubMed]
- Schipper, M.; Slieker, M.; Schoof, P.; Breur, J. Surgical Repair of Ventricular Septal Defect; Contemporary Results and Risk Factors for a Complicated Course. Pediatr. Cardiol. 2017, 38, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Scully, B.; Morales, D.; Zafar, F.; McKenzie, E.; Fraser, C.J.; Heinle, J. Current expectations for surgical repair of isolated ventricular septal defects. Ann. Thorac. Surg. 2010, 89, 544–549. [Google Scholar] [CrossRef] [PubMed]
- Heiberg, J.; Nyboe, C.; Hjortdal, V. Permanent chronotropic impairment after closure of atrial or ventricular septal defect. Scand. Cardiovasc. J. 2017, 51, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Klena, N.; Gabriel, G.; Liu, X.; Kim, A.; Lemke, K.; Chen, Y.; Chatterjee, B.; Devine, W.; Damerla, R.; et al. Global genetic analysis in mice unveils central role for cilia in congenital heart disease. Nature 2015, 521, 520–524. [Google Scholar] [CrossRef] [PubMed]
- Satir, P.; Pedersen, L.; Christensen, S. The primary cilium at a glance. J. Cell Sci. 2010, 123, 499–503. [Google Scholar] [CrossRef] [PubMed]
- Bisgrove, B.; Yost, H. The roles of cilia in developmental disorders and disease. Development 2006, 133, 4131–4143. [Google Scholar] [CrossRef] [PubMed]
- Basten, S.; Giles, R. Functional aspects of primary cilia in signaling, cell cycle and tumorigenesis. Cilia 2013, 2, 6. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.; Reiter, J. The primary cilium at the crossroads of mammalian hedgehog signaling. Curr. Top. Dev. Biol. 2008, 85, 225–260. [Google Scholar] [PubMed]
- Sasai, N.; Briscoe, J. Primary cilia and graded Sonic Hedgehog signaling. Wiley Interdiscip. Rev. Dev. Biol. 2012, 1, 753–772. [Google Scholar] [CrossRef] [PubMed]
- Bangs, F.; Anderson, K. Primary Cilia and Mammalian Hedgehog Signaling. Cold Spring Harb. Perspect. Biol. 2017, 9, a028175. [Google Scholar] [CrossRef] [PubMed]
- Tasouri, E.; Tucker, K. Primary cilia and organogenesis: Is Hedgehog the only sculptor? Cell Tissue Res. 2011, 345, 21–40. [Google Scholar] [CrossRef] [PubMed]
- Ingham, P.; Nakano, Y.; Seger, C. Mechanisms and functions of Hedgehog signalling across the metazoa. Nat. Rev. Genet. 2011, 12, 393–406. [Google Scholar] [CrossRef] [PubMed]
- Briscoe, J.; Thérond, P. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat. Rev. Mol. Cell Biol. 2013, 14, 416–429. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.; Zhao, Z.; Ingham, P. Hedgehog signalling. Development 2016, 143, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Christ, A.; Christa, A.; Kur, E.; Lioubinski, O.; Bachmann, S.; Willnow, T.; Hammes, A. LRP2 is an auxiliary SHH receptor required to condition the forebrain ventral midline for inductive signals. Dev. Cell. 2012, 22, 268–278. [Google Scholar] [CrossRef] [PubMed]
- Corbit, K.; Aanstad, P.; Singla, V.; Norman, A.; Stainier, D.; Reiter, J. Vertebrate Smoothened functions at the primary cilium. Nature 2005, 437, 1018–1021. [Google Scholar] [CrossRef] [PubMed]
- Rohatgi, R.; Milenkovic, L.; Scott, M. Patched1 regulates hedgehog signaling at the primary cilium. Science 2007, 317, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Wilson, C.; Li, Y.; Law, K.; Lu, C.; Gacayan, R.; Zhang, X.; Hui, C.; Chuang, P. Cilium-independent regulation of Gli protein function by Sufu in Hedgehog signaling is evolutionarily conserved. Genes Dev. 2009, 23, 1910–1928. [Google Scholar] [CrossRef] [PubMed]
- Humke, E.; Dorn, K.; Milenkovic, L.; Scott, M.; Rohatgi, R. The output of Hedgehog signaling is controlled by the dynamic association between Suppressor of Fused and the Gli proteins. Genes Dev. 2010, 24, 670–682. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Perez, V.; Blair, H.; Rodriguez-Andres, M.; Blanco, M.; Wilson, A.; Liu, Y.; Miles, C.; Peters, H.; Goodship, J. Evc is a positive mediator of Ihh-regulated bone growth that localises at the base of chondrocyte cilia. Development 2007, 134, 2903–2912. [Google Scholar] [CrossRef] [PubMed]
- Valencia, M.; Lapunzina, P.; Lim, D.; Zannolli, R.; Bartholdi, D.; Wollnik, B.; Al-Ajlouni, O.; Eid, S.; Cox, H.; Buoni, S.; et al. Widening the mutation spectrum of EVC and EVC2: Ectopic expression of Weyer variants in NIH 3T3 fibroblasts disrupts Hedgehog signaling. Hum. Mutat. 2009, 30, 1667–1675. [Google Scholar] [CrossRef] [PubMed]
- Blair, H.; Tompson, S.; Liu, Y.; Campbell, J.; MacArthur, K.; Ponting, C.; Ruiz-Perez, V.; Goodship, J. Evc2 is a positive modulator of Hedgehog signalling that interacts with Evc at the cilia membrane and is also found in the nucleus. BMC Biol. 2011, 9, 14. [Google Scholar] [CrossRef] [PubMed]
- Dorn, K.; Hughes, C.; Rohatgi, R. A Smoothened-Evc2 complex transduces the Hedgehog signal at primary cilia. Dev. Cell 2012, 23, 823–835. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Chen, W.; Chen, Y.; Jiang, J. Smoothened transduces Hedgehog signal by forming a complex with Evc/Evc2. Cell Res. 2012, 22, 1593–1604. [Google Scholar] [CrossRef] [PubMed]
- Caparrós-Martín, J.; Valencia, M.; Reytor, E.; Pacheco, M.; Fernandez, M.; Perez-Aytes, A.; Gean, E.; Lapunzina, P.; Peters, H.; Goodship, J.; et al. The ciliary Evc/Evc2 complex interacts with Smo and controls Hedgehog pathway activity in chondrocytes by regulating Sufu/Gli3 dissociation and Gli3 trafficking in primary cilia. Hum. Mol. Genet. 2013, 22, 124–139. [Google Scholar] [CrossRef] [PubMed]
- Ko, H.; Norman, R.; Tran, J.; Fuller, K.; Fukuda, M.; Eggenschwiler, J. Broad-minded links cell cycle-related kinase to cilia assembly and hedgehog signal transduction. Dev. Cell 2010, 18, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Keady, B.; Samtani, R.; Tobita, K.; Tsuchya, M.; San Agustin, J.; Follit, J.; Jonassen, J.; Subramanian, R.; Lo, C.; Pazour, G. IFT25 Links the Signal-Dependent Movement of Hedgehog Components to Intraflagellar Transport. Dev. Cell 2012, 22, 940–951. [Google Scholar] [CrossRef] [PubMed]
- Eguether, T.; San Agustin, J.; Keady, B.; Jonassen, J.; Liang, Y.; Francis, R.; Tobita, K.; Johnson, C.; Abdelhamed, Z.; Lo, C.; et al. IFT27 links the BBSome to IFT for maintenance of the ciliary signaling compartment. Dev. Cell 2014, 31, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Li, L.; Eguether, T.; Sundberg, J.; Pazour, G.; Chen, J. Intraflagellar transport 27 is essential for hedgehog signaling but dispensable for ciliogenesis during hair follicle morphogenesis. Development 2015, 142, 2194–2202. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Fallon, J.; Beachy, P. Hedgehog-regulated processing of Gli3 produces an anterior/posterior repressor gradient in the developing vertebrate limb. Cell 2000, 100, 423–434. [Google Scholar] [CrossRef]
- Gerhardt, C.; Lier, J.; Burmühl, S.; Struchtrup, A.; Deutschmann, K.; Vetter, M.; Leu, T.; Reeg, S.; Grune, T.; Rüther, U. The transition zone protein Rpgrip1l regulates proteasomal activity at the primary cilium. J. Cell Biol. 2015, 210, 115–133. [Google Scholar] [CrossRef] [PubMed]
- Gerhardt, C.; Wiegering, A.; Leu, T.; Rüther, U. Control of Hedgehog signalling by the cilia-regulated proteasome. J. Dev. Biol. 2016, 4, 27. [Google Scholar] [CrossRef]
- Tuson, M.; He, M.; Anderson, K. Protein kinase A acts at the basal body of the primary cilium to prevent Gli2 activation and ventralization of the mouse neural tube. Development 2011, 138, 4921–4930. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Li, Y. Evidence for the direct involvement of βTrCP in Gli3 protein processing. Proc. Natl. Acad. Sci. USA 2006, 103, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Bai, C.; Joyner, A.; Wang, B. Sonic hedgehog signaling regulates Gli2 transcriptional activity by suppressing its processing and degradation. Mol. Cell Biol. 2006, 26, 3365–3377. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Subramanian, R.; Bangs, F.; Omelchenko, T.; Liem, K.J.; Kapoor, T.; Anderson, K. The kinesin-4 protein Kif7 regulates mammalian Hedgehog signalling by organizing the cilium tip compartment. Nat. Cell Biol. 2014, 16, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Cheung, H.; Zhang, X.; Ribeiro, A.; Mo, R.; Makino, S.; Puviindran, V.; Law, K.; Briscoe, J.; Hui, C. The kinesin protein Kif7 is a critical regulator of Gli transcription factors in mammalian hedgehog signaling. Sci. Signal. 2009, 2, ra29. [Google Scholar] [CrossRef] [PubMed]
- Endoh-Yamagami, S.; Evangelista, M.; Wilson, D.; Wen, X.; Theunissen, J.; Phamluong, K.; Davis, M.; Scales, S.; Solloway, M.; de Sauvage, F.; et al. The mammalian Cos2 homolog Kif7 plays an essential role in modulating Hh signal transduction during development. Curr. Biol. 2009, 19, 1320–1326. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, L.; Akhmanova, A. Kif7 keeps cilia tips in shape. Nat. Cell Biol. 2014, 16, 623–625. [Google Scholar] [CrossRef] [PubMed]
- Heydeck, W.; Zeng, H.; Liu, A. Planar cell polarity effector gene Fuzzy regulates cilia formation and Hedgehog signal transduction in mouse. Dev. Dyn. 2009, 238, 3035–3042. [Google Scholar] [CrossRef] [PubMed]
- Washington Smoak, I.; Byrd, N.; Abu-Issa, R.; Goddeeris, M.; Anderson, R.; Morris, J.; Yamamura, K.; Klingensmith, J.; Meyers, E. Sonic hedgehog is required for cardiac outflow tract and neural crest cell development. Dev. Biol. 2005, 283, 357–372. [Google Scholar] [CrossRef] [PubMed]
- Kerkela, R.; Kockeritz, L.; Macaulay, K.; Zhou, J.; Doble, B.; Beahm, C.; Greytak, S.; Woulfe, K.; Trivedi, C.; Woodgett, J.; et al. Deletion of GSK-3beta in mice leads to hypertrophic cardiomyopathy secondary to cardiomyoblast hyperproliferation. J. Clin. Investig. 2008, 118, 3609–3618. [Google Scholar] [CrossRef] [PubMed]
- Coles, G.; Ackerman, K. Kif7 is required for the patterning and differentiation of the diaphragm in a model of syndromic congenital diaphragmatic hernia. Proc. Natl. Acad. Sci. USA. 2013, 110, E1898–E1905. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D. Extra-toes: A new mutant gene causing multiple abnormalities in the mouse. J. Embryol. Exp. Morphol. 1967, 17, 543–581. [Google Scholar] [PubMed]
- Patel, P.; Woodgett, J. Glycogen Synthase Kinase 3: A Kinase for All Pathways? Curr. Top. Dev. Biol. 2017, 123, 277–302. [Google Scholar] [PubMed]
- Nakaya, M.; Biris, K.; Tsukiyama, T.; Jaime, S.; Rawls, J.; Yamaguchi, T. Wnt3a links left-right determination with segmentation and anteroposterior axis elongation. Development 2005, 132, 5425–5436. [Google Scholar] [CrossRef] [PubMed]
- Bosada, F.; Devasthali, V.; Jones, K.; Stankunas, K. Wnt/β-catenin signaling enables developmental transitions during valvulogenesis. Development 2016, 143, 1041–1054. [Google Scholar] [CrossRef] [PubMed]
- Briggs, L.; Burns, T.; Lockhart, M.; Phelps, A.; Van den Hoff, M.; Wessels, A. Wnt/β-catenin and sonic hedgehog pathways interact in the regulation of the development of the dorsal mesenchymal protrusion. Dev. Dyn. 2016, 245, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Niessen, K.; Karsan, A. Notch signaling in cardiac development. Circ. Res. 2008, 102, 1169–1181. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Liu, J. Role of Notch signaling in the mammalian heart. Braz. J. Med. Biol. Res. 2014, 47, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wurdak, H.; Ittner, L.; Lang, K.; Leveen, P.; Suter, U.; Fischer, J.; Karlsson, S.; Born, W.; Sommer, L. Inactivation of TGFbeta signaling in neural crest stem cells leads to multiple defects reminiscent of DiGeorge syndrome. Genes Dev. 2005, 19, 530–535. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, B.; Ito, Y.; Makita, T.; Sasaki, T.; Chai, Y.; Sucov, H. Cardiovascular malformations with normal smooth muscle differentiation in neural crest-specific type II TGF β receptor (Tgfbr2) mutant mice. Dev. Biol. 2006, 289, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Chen, H.; Zheng, D.; Kuang, C.; Fang, H.; Zou, B.; Zhu, W.; Bu, G.; Jin, T.; Wang, Z.; et al. Smad7 is required for the development and function of the heart. J. Biol. Chem. 2009, 284, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Gray, R.; Abitua, P.; Wlodarczyk, B.; Szabo-Rogers, H.; Blanchard, O.; Lee, I.; Weiss, G.; Liu, K.; Marcotte, E.; Wallingford, J.; et al. The planar cell polarity effector Fuz is essential for targeted membrane trafficking, ciliogenesis and mouse embryonic development. Nat. Cell Biol. 2009, 11, 1225–1232. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.; Zhu, H.; Wlodarczyk, B.; Zhang, L.; Li, L.; Li, A.; Finnell, R.; Roop, D.; Chen, J. Fuz controls the morphogenesis and differentiation of hair follicles through the formation of primary cilia. J. Investig. Dermatol. 2011, 131, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Brooks, E.; Wallingford, J. Control of vertebrate intraflagellar transport by the planar cell polarity effector Fuz. J. Cell Biol. 2012, 198, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Zilber, Y.; Babayeva, S.; Seo, J.; Liu, J.; Mootin, S.; Torban, E. The PCP effector Fuzzy controls cilial assembly and signaling by recruiting Rab8 and Dishevelled to the primary cilium. Mol. Biol. Cell 2013, 24, 555–565. [Google Scholar] [CrossRef] [PubMed]
- Du Toit, A. Organelle dynamics. KIF7 organizes cilia. Nat. Rev. Mol. Cell Biol. 2014, 15, 498–499. [Google Scholar] [CrossRef] [PubMed]
- Digilio, M.; Marino, B.; Ammirati, A.; Borzaga, U.; Giannotti, A.; Dallapiccola, B. Cardiac malformations in patients with oral-facial-skeletal syndromes: Clinical similarities with heterotaxia. Am. J. Med. Genet. 1999, 84, 350–356. [Google Scholar] [CrossRef]
- Sund, K.; Roelker, S.; Ramachandran, V.; Durbin, L.; Benson, D. Analysis of Ellis van Creveld syndrome gene products: Implications for cardiovascular development and disease. Hum. Mol. Genet. 2009, 18, 1813–1824. [Google Scholar] [CrossRef] [PubMed]
- Calkoen, E.; Adriaanse, B.; Haak, M.; Bartelings, M.; Kolesnik, A.; Niszczota, C.; van Vugt, J.; Roest, A.; Blom, N.; Gittenberger-de Groot, A.; et al. How Normal is a ‘Normal’ Heart in Fetuses and Infants with Down Syndrome? Fetal Diagn. Ther. 2016, 39, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Goddeeris, M.; Schwartz, R.; Klingensmith, J.; Meyers, E. Independent requirements for Hedgehog signaling by both the anterior heart field and neural crest cells for outflow tract development. Development 2007, 134, 1593–1604. [Google Scholar] [CrossRef] [PubMed]
- Goddeeris, M.; Rho, S.; Petiet, A.; Davenport, C.; Johnson, G.; Meyers, E.; Klingensmith, J. Intracardiac septation requires hedgehog-dependent cellular contributions from outside the heart. Development 2008, 135, 1887–1895. [Google Scholar] [CrossRef] [PubMed]
- Dyer, L.; Kirby, M. Sonic hedgehog maintains proliferation in secondary heart field progenitors and is required for normal arterial pole formation. Dev. Biol. 2009, 330, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Vincent, S.; Buckingham, M. How to make a heart: The origin and regulation of cardiac progenitor cells. Curr. Top. Dev. Biol. 2010, 90, 1–41. [Google Scholar] [PubMed]
- Kelly, R. The second heart field. Curr. Top. Dev. Biol. 2012, 100, 33–65. [Google Scholar] [PubMed]
- Francou, A.; Saint-Michel, E.; Mesbah, K.; Théveniau-Ruissy, M.; Rana, M.; Christoffels, V.; Kelly, R. Second heart field cardiac progenitor cells in the early mouse embryo. Biochim. Biophys. Acta 2013, 1833, 795–798. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, A.; Peterson, M.; Friedland-Little, J.; Anderson, S.; Moskowitz, I. Sonic hedgehog is required in pulmonary endoderm for atrial septation. Development 2009, 136, 1761–1770. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Hoffmann, A.; Burnicka-Turek, O.; Friedland-Little, J.; Zhang, K.; Moskowitz, I. Tbx5-hedgehog molecular networks are essential in the second heart field for atrial septation. Dev. Cell 2012, 23, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, A.; Yang, X.; Burnicka-Turek, O.; Bosman, J.; Ren, X.; Steimle, J.; Vokes, S.; McMahon, A.; Kalinichenko, V.; Moskowitz, I. Foxf genes integrate tbx5 and hedgehog pathways in the second heart field for cardiac septation. PLoS Genet. 2014, 10, e1004604. [Google Scholar] [CrossRef] [PubMed]
- Francou, A.; De Bono, C.; Kelly, R. Epithelial tension in the second heart field promotes mouse heart tube elongation. Nat. Commun. 2017, 8, 14770. [Google Scholar] [CrossRef] [PubMed]
- Kathiriya, I.; Srivastava, D. Left-right asymmetry and cardiac looping: Implications for cardiac development and congenital heart disease. Am. J. Med. Genet. 2000, 97, 271–279. [Google Scholar] [CrossRef]
- Komatsu, Y.; Mishina, Y. Establishment of left-right asymmetry in vertebrate development: The node in mouse embryos. Cell. Mol. Life Sci. 2013, 70, 4659–4666. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Okada, Y.; Hirokawa, N. FGF-induced vesicular release of Sonic hedgehog and retinoic acid in leftward nodal flow is critical for left-right determination. Nature 2005, 435, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Tsiairis, C.; McMahon, A. An Hh-dependent pathway in lateral plate mesoderm enables the generation of left/right asymmetry. Curr. Biol. 2009, 19, 1912–1917. [Google Scholar] [CrossRef] [PubMed]
- Schilling, T.; Concordet, J.; Ingham, P. Regulation of left-right asymmetries in the zebrafish by Shh and BMP4. Dev. Biol. 1999, 210, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Meyers, E.; Martin, G. Differences in left-right axis pathways in mouse and chick: Functions of FGF8 and SHH. Science 1999, 285, 403–406. [Google Scholar] [CrossRef] [PubMed]
- Pagán-Westphal, S.; Tabin, C. The transfer of left-right positional information during chick embryogenesis. Cell 1998, 93, 25–35. [Google Scholar] [CrossRef]
- Levin, M.; Johnson, R.; Stern, C.; Kuehn, M.; Tabin, C. A molecular pathway determining left-right asymmetry in chick embryogenesis. Cell 1995, 82, 803–814. [Google Scholar] [CrossRef]
- Sutherland, M.; Ware, S. Disorders of left-right asymmetry: Heterotaxy and situs inversus. Am. J. Med. Genet. C. Semin. Med. Genet. 2009, 151C, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Ramsdell, A. Left-right asymmetry and congenital cardiac defects: Getting to the heart of the matter in vertebrate left-right axis determination. Dev. Biol. 2005, 288, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Liu, J.; Xiang, M.; Olson, P.; Guzzetta, A.; Zhang, K.; Moskowitz, I.; Xie, L. Gata4 potentiates second heart field proliferation and Hedgehog signaling for cardiac septation. Proc. Natl. Acad. Sci. USA 2017, 114, E1422–E1431. [Google Scholar] [CrossRef] [PubMed]
- Roux, M.; Laforest, B.; Capecchi, M.; Bertrand, N.; Zaffran, S. Hoxb1 regulates proliferation and differentiation of second heart field progenitors in pharyngeal mesoderm and genetically interacts with Hoxa1 during cardiac outflow tract development. Dev. Biol. 2015, 406, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Vincent, S.; Mayeuf-Louchart, A.; Watanabe, Y.; Brzezinski, J.T.; Miyagawa-Tomita, S.; Kelly, R.; Buckingham, M. Prdm1 functions in the mesoderm of the second heart field, where it interacts genetically with Tbx1, during outflow tract morphogenesis in the mouse embryo. Hum. Mol. Genet. 2014, 23, 5087–5101. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.; Brown, N.; Buckingham, M. The arterial pole of the mouse heart forms from Fgf10-expressing cells in pharyngeal mesoderm. Dev. Cell 2001, 1, 435–440. [Google Scholar] [CrossRef]
- Mjaatvedt, C.; Nakaoka, T.; Moreno-Rodriguez, R.; Norris, R.; Kern, M.; Eisenberg, C.; Turner, D.; Markwald, R. The outflow tract of the heart is recruited from a novel heart-forming field. Dev. Biol. 2001, 238, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Waldo, K.; Kumiski, D.; Wallis, K.; Stadt, H.; Hutson, M.; Platt, D.; Kirby, M. Conotruncal myocardium arises from a secondary heart field. Development 2001, 128, 3179–3188. [Google Scholar] [PubMed]
- Mostefa-Kara, M.; Bonnet, D.; Belli, E.; Fadel, E.; Houyel, L. Anatomy of the ventricular septal defect in outflow tract defects: Similarities and differences. J. Thorac. Cardiovasc. Surg. 2015, 149, 682–688.e1. [Google Scholar] [CrossRef] [PubMed]
- Gerhardt, C.; Lier, J.; Kuschel, S.; Rüther, U. The ciliary protein Ftm is required for ventricular wall and septal development. PLoS ONE 2013, 8, e57545. [Google Scholar] [CrossRef] [PubMed]
- Bax, N.; Bleyl, S.; Gallini, R.; Wisse, L.; Hunter, J.; Van Oorschot, A.; Mahtab, E.; Lie-Venema, H.; Goumans, M.; Betsholtz, C.; et al. Cardiac malformations in Pdgfralpha mutant embryos are associated with increased expression of WT1 and Nkx2.5 in the second heart field. Dev. Dyn. 2010, 239, 2307–2317. [Google Scholar] [CrossRef] [PubMed]
- Schatteman, G.; Motley, S.; Effmann, E.; Bowen-Pope, D. Platelet-derived growth factor receptor alpha subunit deleted Patch mouse exhibits severe cardiovascular dysmorphogenesis. Teratology 1995, 51, 351–366. [Google Scholar] [CrossRef] [PubMed]
- Obican, S.; Finnell, R.; Mills, J.; Shaw, G.; Scialli, A. Folic acid in early pregnancy: A public health success story. FASEB J. 2010, 24, 4167–4174. [Google Scholar] [CrossRef] [PubMed]
- Ruat, M.; Hoch, L.; Faure, H.; Rognan, D. Targeting of Smoothened for therapeutic gain. Trends Pharmacol. Sci. 2014, 35, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Taipale, J.; Young, K.; Maiti, T.; Beachy, P. Small molecule modulation of Smoothened activity. Proc. Natl. Acad. Sci. USA 2002, 99, 14071–14076. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Chen, J. Purmorphamine activates the Hedgehog pathway by targeting Smoothened. Nat. Chem. Biol. 2006, 2, 29–30. [Google Scholar] [CrossRef] [PubMed]
- Nachtergaele, S.; Mydock, L.; Krishnan, K.; Rammohan, J.; Schlesinger, P.; Covey, D.; Rohatgi, R. Oxysterols are allosteric activators of the oncoprotein Smoothened. Nat. Chem. Biol. 2012, 8, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Gorojankina, T.; Hoch, L.; Faure, H.; Roudaut, H.; Traiffort, E.; Schoenfelder, A.; Girard, N.; Mann, A.; Manetti, F.; Solinas, A.; et al. Discovery, molecular and pharmacological characterization of GSA-10, a novel small-molecule positive modulator of Smoothened. Mol. Pharmacol. 2013, 83, 1020–1029. [Google Scholar] [CrossRef] [PubMed]
- Watkins, D.; Berman, D.; Baylin, S. Hedgehog signaling: Progenitor phenotype in small-cell lung cancer. Cell Cycle 2003, 2, 196–198. [Google Scholar] [CrossRef] [PubMed]
- Xie, J. Hedgehog signaling in prostate cancer. Future Oncol. 2005, 1, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Athar, M.; Tang, X.; Lee, J.; Kopelovich, L.; Kim, A. Hedgehog signalling in skin development and cancer. Exp. Dermatol. 2006, 15, 667–677. [Google Scholar] [CrossRef] [PubMed]
- Daniel, V.; Peacock, C.; Watkins, D. Developmental signalling pathways in lung cancer. Respirology 2006, 11, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Douard, R.; Moutereau, S.; Pernet, P.; Chimingqi, M.; Allory, Y.; Manivet, P.; Conti, M.; Vaubourdolle, M.; Cugnenc, P.; Loric, S. Sonic Hedgehog-dependent proliferation in a series of patients with colorectal cancer. Surgery 2006, 139, 665–670. [Google Scholar] [CrossRef] [PubMed]
- Evangelista, M.; Tian, H.; de Sauvage, F. The hedgehog signaling pathway in cancer. Clin. Cancer Res. 2006, 12, 5924–5928. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Sheng, T.; Zhang, Y.; Zhang, X.; He, J.; Huang, S.; Chen, K.; Sultz, J.; Adegboyega, P.; Zhang, H.; et al. Hedgehog signaling is activated in subsets of esophageal cancers. Int. J. Cancer 2006, 118, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Clement, V.; Sanchez, P.; de Tribolet, N.; Radovanovic, I.; Ruiz i Altaba, A. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr. Biol. 2007, 17, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Fukaya, M.; Isohata, N.; Ohta, H.; Aoyagi, K.; Ochiya, T.; Saeki, N.; Yanagihara, K.; Nakanishi, Y.; Taniguchi, H.; Sakamoto, H.; et al. Hedgehog signal activation in gastric pit cell and in diffuse-type gastric cancer. Gastroenterology 2006, 131, 14–29. [Google Scholar] [CrossRef] [PubMed]
- Barakat, M.; Humke, E.; Scott, M. Learning from Jekyll to control Hyde: Hedgehog signaling in development and cancer. Trends Mol. Med. 2010, 16, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Fish, E.; Parnell, S.; Sulik, K.; Baker, L.; Murdaugh, L.; Lamson, D.; Williams, K. Preaxial polydactyly following early gestational exposure to the smoothened agonist, SAG, in C57BL/6J mice. Birth Defects Res. 2017, 109, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, L.; Inglés-Esteve, J.; Aguilera, C.; Bigas, A. Phosphorylation by glycogen synthase kinase-3β down-regulates Notch activity, a link for Notch and Wnt pathways. J. Biol. Chem. 2003, 278, 32227–32235. [Google Scholar] [CrossRef] [PubMed]
- Caraci, F.; Gili, E.; Calafiore, M.; Failla, M.; La Rosa, C.; Crimi, N.; Sortino, M.; Nicoletti, F.; Copani, A.; Vancheri, C. TGF-beta1 targets the GSK-3beta/beta-catenin pathway via ERK activation in the transition of human lung fibroblasts into myofibroblasts. Pharmacol. Res. 2008, 57, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Strickland, S.; Smith, K.; Marotti, K. Hormonal induction of differentiation in teratocarcinoma stem cells: Generation of parietal endoderm by retinoic acid and dibutyryl cAMP. Cell 1980, 21, 347–355. [Google Scholar] [CrossRef]
- Deol, G.; Cuthbert, T.; Gatie, M.; Spice, D.; Hilton, L.; Kelly, G. Wnt and Hedgehog Signaling Regulate the Differentiation of F9 Cells into Extraembryonic Endoderm. Front. Cell Dev. Biol. 2017, 5, 93. [Google Scholar] [CrossRef] [PubMed]
- Vis, J.; Duffels, M.; Winter, M.; Weijerman, M.; Cobben, J.; Huisman, S.; Mulder, B. Down syndrome: A cardiovascular perspective. J. Intellect. Disabil. Res. 2009, 53, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Frid, C.; Drott, P.; Lundell, B.; Rasmussen, F.; Annerén, G. Mortality in Down’s syndrome in relation to congenital malformations. J. Intellect. Disabil. Res. 1999, 43, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Freeman, S.; Bean, L.; Allen, E.; Tinker, S.; Locke, A.; Druschel, C.; Hobbs, C.; Romitti, P.; Royle, M.; Torfs, C.; et al. Ethnicity, sex, and the incidence of congenital heart defects: A report from the National Down Syndrome Project. Genet. Med. 2008, 10, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Laursen, H. Congenital heart disease in Down’s syndrome. Br. Heart J. 1976, 38, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Currier, D.; Polk, R.; Reeves, R. A Sonic hedgehog (Shh) response deficit in trisomic cells may be a common denominator for multiple features of Down syndrome. Prog. Brain Res. 2012, 197, 223–236. [Google Scholar] [PubMed]
- Blom, N.; Ottenkamp, J.; Wenink, A.; Gittenberger-de Groot, A. Deficiency of the vestibular spine in atrioventricular septal defects in human fetuses with down syndrome. Am. J. Cardiol. 2003, 91, 180–184. [Google Scholar] [CrossRef]
- Snarr, B.; O’Neal, J.; Chintalapudi, M.; Wirrig, E.; Phelps, A.; Kubalak, S.; Wessels, A. Isl1 expression at the venous pole identifies a novel role for the second heart field in cardiac development. Circ. Res. 2007, 101, 971–974. [Google Scholar] [CrossRef] [PubMed]
- Webb, S.; Anderson, R.; Lamers, W.; Brown, N. Mechanisms of deficient cardiac septation in the mouse with trisomy 16. Circ. Res. 1999, 84, 897–905. [Google Scholar] [CrossRef] [PubMed]
- Roper, R.; Baxter, L.; Saran, N.; Klinedinst, D.; Beachy, P.; Reeves, R. Defective cerebellar response to mitogenic Hedgehog signaling in Down [corrected] syndrome mice. Proc. Natl. Acad. Sci. USA 2006, 103, 1452–1456. [Google Scholar] [CrossRef] [PubMed]
- Das, I.; Park, J.; Shin, J.; Jeon, S.; Lorenzi, H.; Linden, D.; Worley, P.; Reeves, R. Hedgehog agonist therapy corrects structural and cognitive deficits in a Down syndrome mouse model. Sci. Transl. Med. 2013, 5, 201ra120. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Castellanos, N.; Winkelman, B.; Tolosa-Rodriguez, L.; Devenney, B.; Reeves, R.; De Zeeuw, C. Size does not always matter: Ts65Dn Down syndrome mice show cerebellum-dependent motor learning deficits that cannot be rescued by postnatal SAG treatment. J. Neurosci. 2013, 33, 15408–15413. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Gene Symbol | Cardiac Phenotype | Literature |
|---|---|---|
| Shh |
| [71] |
| Lrp2 |
| [34] |
| Sufu |
| [34] |
| Bromi (Tbc1d32) |
| [34] |
| Ift25 |
| [57,58] |
| Ift27 |
| [57,58] |
| EVC1 |
| [86,87] |
| EVC2 |
| [86,87] |
| Kif7 |
| [34,73] |
| Fuz |
| [34,73] |
| Gsk3-β |
| [72] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wiegering, A.; Rüther, U.; Gerhardt, C. The Role of Hedgehog Signalling in the Formation of the Ventricular Septum. J. Dev. Biol. 2017, 5, 17. https://doi.org/10.3390/jdb5040017
Wiegering A, Rüther U, Gerhardt C. The Role of Hedgehog Signalling in the Formation of the Ventricular Septum. Journal of Developmental Biology. 2017; 5(4):17. https://doi.org/10.3390/jdb5040017
Chicago/Turabian StyleWiegering, Antonia, Ulrich Rüther, and Christoph Gerhardt. 2017. "The Role of Hedgehog Signalling in the Formation of the Ventricular Septum" Journal of Developmental Biology 5, no. 4: 17. https://doi.org/10.3390/jdb5040017
APA StyleWiegering, A., Rüther, U., & Gerhardt, C. (2017). The Role of Hedgehog Signalling in the Formation of the Ventricular Septum. Journal of Developmental Biology, 5(4), 17. https://doi.org/10.3390/jdb5040017