Engineering the Drosophila Genome for Developmental Biology


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
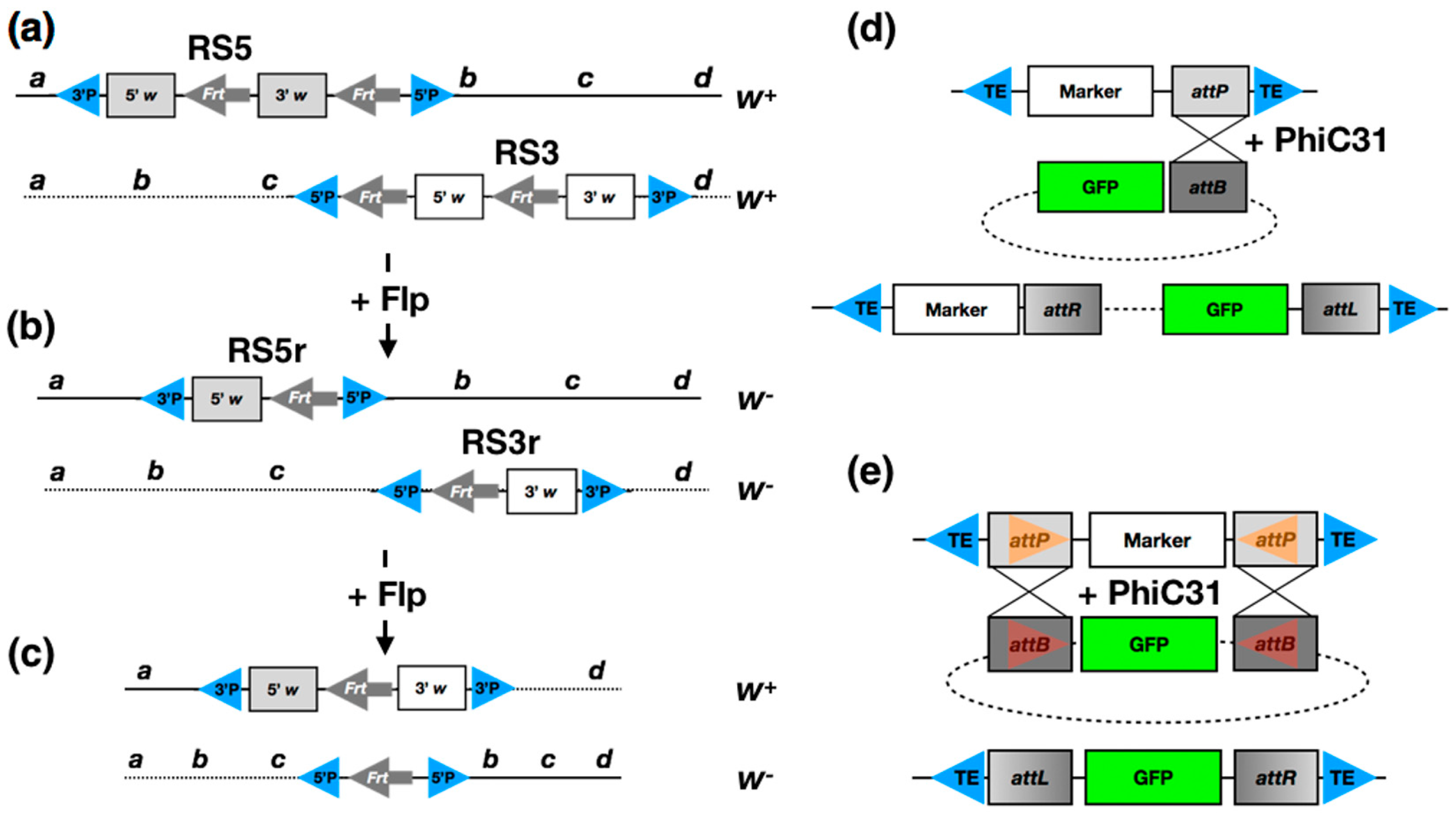
2. Classical Methods
3. CRISPR/Cas9
3.1. Overview
3.2. Mutagenesis with CRISPR/Cas9 Systems
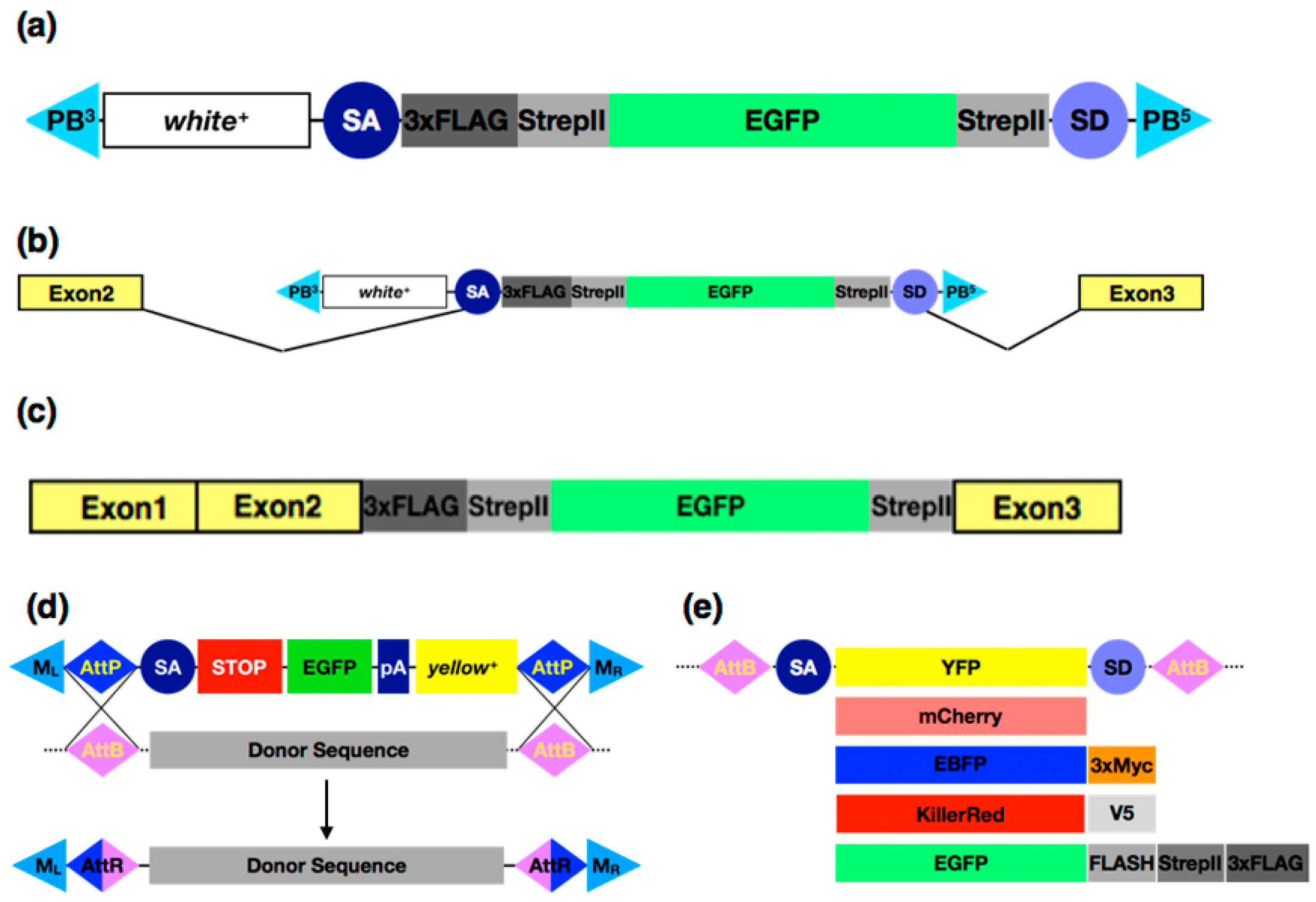
3.3. Protein Tagging with CRISPR/Cas9 Systems
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Rubin, G.M.; Lewis, E.B. A brief history of Drosophila’s contributions to genome research. Science 2000, 287, 2216–2218. [Google Scholar] [CrossRef] [PubMed]
- Arias, A.M. Drosophila melanogaster and the development of biology in the 20th century. Methods Mol. Biol. 2008, 420, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Adryan, B.; Russell, S. Genome mapping and genomics in Drosophila. In Genome Mapping and Genomics in Laboratory Animals; Denny, P., Kole, C., Eds.; Springer: Berilin, Germany, 2012; Volume 4, pp. 31–86. ISBN 978-3-642-31315-8. [Google Scholar]
- Russell, S. From sequence to function: The impact of the genome sequence on Drosophila biology. Brief. Funct. Genom. 2012, 11, 333–335. [Google Scholar] [CrossRef] [PubMed]
- Rong, Y.S.; Golic, K.G. Gene targeting by homologous recombination in Drosophila. Science 2000, 288, 2013–2018. [Google Scholar] [CrossRef] [PubMed]
- Gong, W.J.; Golic, K.G. Ends-out, or replacement, gene targeting in Drosophila. Proc. Natl. Acad. Sci. USA 2003, 100, 2556–2561. [Google Scholar] [CrossRef] [PubMed]
- Ryder, E.; Russell, S. Transposable elements as tools for genomics and genetics in Drosophila. Brief. Funct. Genom. Proteom. 2003, 2, 57–71. [Google Scholar] [CrossRef]
- Venken, K.J.; Bellen, H.J. Chemical mutagens, transposons, and transgenes to interrogate gene function in Drosophila melanogaster. Methods 2014, 68, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Rubin, G.M.; Spradling, A.C. Genetic transformation of Drosophila with transposable element vectors. Science 1982, 218, 348–353. [Google Scholar] [CrossRef] [PubMed]
- Brand, A.H.; Perrimon, N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 1993, 118, 401–415. [Google Scholar] [PubMed]
- O’Kane, C.J.; Gehring, W.J. Detection in situ of genomic regulatory elements in Drosophila. Proc. Natl. Acad. Sci. USA 1987, 84, 9123–9127. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.; Pearson, R.K.; Bellen, H.J.; O’Kane, C.J.; Grossniklaus, U.; Gehring, W.J. P-element-mediated enhancer detection: An efficient method for isolating and characterizing developmentally regulated genes in Drosophila. Genes. Dev. 1989, 3, 1301–1313. [Google Scholar] [CrossRef] [PubMed]
- Rørth, P. A modular misexpression screen in Drosophila detecting tissue-specific phenotypes. Proc. Natl. Acad. Sci. USA 1996, 93, 12418–12422. [Google Scholar] [CrossRef] [PubMed]
- Spradling, A.C.; Bellen, H.J.; Hoskins, R.A. Drosophila p elements preferentially transpose to replication origins. Proc. Natl. Acad. Sci. USA 2011, 108, 15948–15953. [Google Scholar] [CrossRef] [PubMed]
- Loukeris, T.G.; Arcà, B.; Livadaras, I.; Dialektaki, G.; Savakis, C. Introduction of the transposable element minos into the germ line of Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 1995, 92, 9485–9489. [Google Scholar] [CrossRef] [PubMed]
- Lobo, N.; Li, X.; Fraser, M.J. Transposition of the piggybac element in embryos of Drosophila melanogaster, aedes aegypti and trichoplusia ni. Mol. Gen. Genet. 1999, 261, 803–810. [Google Scholar] [CrossRef] [PubMed]
- Groth, A.C.; Fish, M.; Nusse, R.; Calos, M.P. Construction of transgenic Drosophila by using the site-specific integrase from phage phic31. Genetics 2004, 166, 1775–1782. [Google Scholar] [CrossRef] [PubMed]
- Venken, K.J.; Sarrion-Perdigones, A.; Vandeventer, P.J.; Abel, N.S.; Christiansen, A.E.; Hoffman, K.L. Genome engineering: Drosophila melanogaster and beyond. Wiley Interdiscip. Rev. Dev. Biol. 2016, 5, 233–267. [Google Scholar] [CrossRef] [PubMed]
- Golic, K.G.; Lindquist, S. The flp recombinase of yeast catalyzes site-specific recombination in the Drosophila genome. Cell 1989, 59, 499–509. [Google Scholar] [CrossRef]
- Nakazawa, N.; Taniguchi, K.; Okumura, T.; Maeda, R.; Matsuno, K. A novel cre/loxp system for mosaic gene expression in the Drosophila embryo. Dev. Dyn. 2012, 241, 965–974. [Google Scholar] [CrossRef] [PubMed]
- Del Valle Rodríguez, A.; Didiano, D.; Desplan, C. Power tools for gene expression and clonal analysis in Drosophila. Nat. Methods 2011, 9, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Potter, C.J.; Luo, L. Using the q system in Drosophila melanogaster. Nat. Protoc. 2011, 6, 1105–1120. [Google Scholar] [CrossRef] [PubMed]
- Boulina, M.; Samarajeewa, H.; Baker, J.D.; Kim, M.D.; Chiba, A. Live imaging of multicolor-labeled cells in Drosophila. Development 2013, 140, 1605–1613. [Google Scholar] [CrossRef] [PubMed]
- Griffin, R.; Binari, R.; Perrimon, N. Genetic odyssey to generate marked clones in Drosophila mosaics. Proc. Natl. Acad. Sci. USA 2014, 111, 4756–4763. [Google Scholar] [CrossRef] [PubMed]
- Pantazis, P.; Supatto, W. Advances in whole-embryo imaging: A quantitative transition is underway. Nat. Rev. Mol. Cell Biol. 2014, 15, 327–339. [Google Scholar] [CrossRef] [PubMed]
- Owald, D.; Lin, S.; Waddell, S. Light, heat, action: Neural control of fruit fly behaviour. Phil. Trans. R. Soc. Lond. B Biol. Sci. 2015, 370, 20140211. [Google Scholar] [CrossRef] [PubMed]
- Viktorin, G. Using marcm to study Drosophila brain development. Methods Mol. Biol. 2014, 1082, 79–96. [Google Scholar] [CrossRef] [PubMed]
- Parks, A.L.; Cook, K.R.; Belvin, M.; Dompe, N.A.; Fawcett, R.; Huppert, K.; Tan, L.R.; Winter, C.G.; Bogart, K.P.; Deal, J.E.; et al. Systematic generation of high-resolution deletion coverage of the Drosophila melanogaster genome. Nat. Genet. 2004, 36, 288–292. [Google Scholar] [CrossRef] [PubMed]
- Ryder, E.; Ashburner, M.; Bautista-Llacer, R.; Drummond, J.; Webster, J.; Johnson, G.; Morley, T.; Chan, Y.S.; Blows, F.; Coulson, D.; et al. The drosdel deletion collection: A Drosophila genomewide chromosomal deficiency resource. Genetics 2007, 177, 615–629. [Google Scholar] [CrossRef] [PubMed]
- Golic, K.G.; Golic, M.M. Engineering the Drosophila genome: Chromosome rearrangements by design. Genetics 1996, 144, 1693–1711. [Google Scholar] [PubMed]
- Bieli, D.; Kanca, O.; Gohl, D.; Denes, A.; Schedl, P.; Affolter, M.; Müller, M. The Drosophila melanogaster mutants apblot and apxasta affect an essential apterous wing enhancer. G3 Genes Genomes Genet. 2015, 5, 1129–1143. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Cho, D.Y.; Whitworth, C.; Eisman, R.; Phelps, M.; Roote, J.; Kaufman, T.; Cook, K.; Russell, S.; Przytycka, T.; et al. Effects of gene dose, chromatin, and network topology on expression in Drosophila melanogaster. PLoS Genet. 2016, 12, e1006295. [Google Scholar] [CrossRef] [PubMed]
- Meadows, L.A.; Chan, Y.S.; Roote, J.; Russell, S. Neighbourhood continuity is not required for correct testis gene expression in Drosophila. PLoS Biol. 2010, 8, e1000552. [Google Scholar] [CrossRef] [PubMed]
- Cook, R.K.; Christensen, S.J.; Deal, J.A.; Coburn, R.A.; Deal, M.E.; Gresens, J.M.; Kaufman, T.C.; Cook, K.R. The generation of chromosomal deletions to provide extensive coverage and subdivision of the Drosophila melanogaster genome. Genome Biol. 2012, 13, R21. [Google Scholar] [CrossRef] [PubMed]
- Bischof, J.; Maeda, R.K.; Hediger, M.; Karch, F.; Basler, K. An optimized transgenesis system for Drosophila using germ-line-specific phic31 integrases. Proc. Natl. Acad. Sci. USA 2007, 104, 3312–3317. [Google Scholar] [CrossRef] [PubMed]
- Markstein, M.; Pitsouli, C.; Villalta, C.; Celniker, S.E.; Perrimon, N. Exploiting position effects and the gypsy retrovirus insulator to engineer precisely expressed transgenes. Nat. Genet. 2008, 40, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Knapp, J.M.; Chung, P.; Simpson, J.H. Generating customized transgene landing sites and multi-transgene arrays in Drosophila using phic31 integrase. Genetics 2015, 199, 919–934. [Google Scholar] [CrossRef] [PubMed]
- Bateman, J.R.; Lee, A.M.; Wu, C.T. Site-specific transformation of Drosophila via phic31 integrase-mediated cassette exchange. Genetics 2006, 173, 769–777. [Google Scholar] [CrossRef] [PubMed]
- Venken, K.J.; He, Y.; Hoskins, R.A.; Bellen, H.J. P[acman]: A bac transgenic platform for targeted insertion of large DNA fragments in D. melanogaster. Science 2006, 314, 1747–1751. [Google Scholar] [CrossRef] [PubMed]
- Ejsmont, R.K.; Sarov, M.; Winkler, S.; Lipinski, K.A.; Tomancak, P. A toolkit for high-throughput, cross-species gene engineering in Drosophila. Nat. Methods 2009, 6, 435–437. [Google Scholar] [CrossRef] [PubMed]
- Sarov, M.; Barz, C.; Jambor, H.; Hein, M.Y.; Schmied, C.; Suchold, D.; Stender, B.; Janosch, S.; Vikas, V.K.J.; Krishnan, R.T.; et al. A genome-wide resource for the analysis of protein localisation in Drosophila. Elife 2016, 5, e12068. [Google Scholar] [CrossRef] [PubMed]
- Morin, X.; Daneman, R.; Zavortink, M.; Chia, W. A protein trap strategy to detect gfp-tagged proteins expressed from their endogenous loci in Drosophila. Proc. Natl. Acad. Sci. USA 2001, 98, 15050–15055. [Google Scholar] [CrossRef] [PubMed]
- Aleksic, J.; Lazic, R.; Müller, I.; Russell, S.R.; Adryan, B. Biases in Drosophila melanogaster protein trap screens. BMC Genom. 2009, 10, 249. [Google Scholar] [CrossRef] [PubMed]
- Buszczak, M.; Paterno, S.; Lighthouse, D.; Bachman, J.; Planck, J.; Owen, S.; Skora, A.D.; Nystul, T.G.; Ohlstein, B.; Allen, A.; et al. The carnegie protein trap library: A versatile tool for Drosophila developmental studies. Genetics 2007, 175, 1505–1531. [Google Scholar] [CrossRef] [PubMed]
- Quiñones-Coello, A.T.; Petrella, L.N.; Ayers, K.; Melillo, A.; Mazzalupo, S.; Hudson, A.M.; Wang, S.; Castiblanco, C.; Buszczak, M.; Hoskins, R.A.; et al. Exploring strategies for protein trapping in Drosophila. Genetics 2007, 175, 1089–1104. [Google Scholar] [CrossRef] [PubMed]
- Lowe, N.; Rees, J.S.; Roote, J.; Ryder, E.; Armean, I.M.; Johnson, G.; Drummond, E.; Spriggs, H.; Drummond, J.; Magbanua, J.P.; et al. Analysis of the expression patterns, subcellular localisations and interaction partners of Drosophila proteins using a pigp protein trap library. Development 2014, 141, 3994–4005. [Google Scholar] [CrossRef] [PubMed]
- Lye, C.M.; Naylor, H.W.; Sanson, B. Subcellular localisations of the cpti collection of yfp-tagged proteins in Drosophila embryos. Development 2014, 141, 4006–4017. [Google Scholar] [CrossRef] [PubMed]
- Rees, J.S.; Lowe, N.; Armean, I.M.; Roote, J.; Johnson, G.; Drummond, E.; Spriggs, H.; Ryder, E.; Russell, S.; St Johnston, D.; et al. In vivo analysis of proteomes and interactomes using parallel affinity capture (ipac) coupled to mass spectrometry. Mol. Cell. Proteom. 2011, 10. [Google Scholar] [CrossRef] [PubMed]
- Nashchekin, D.; Fernandes, A.R.; St Johnston, D. Patronin/shot cortical foci assemble the noncentrosomal microtubule array that specifies the Drosophila anterior-posterior axis. Dev. Cell 2016, 38, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Byri, S.; Misra, T.; Syed, Z.A.; Bätz, T.; Shah, J.; Boril, L.; Glashauser, J.; Aegerter-Wilmsen, T.; Matzat, T.; Moussian, B.; et al. The triple-repeat protein anakonda controls epithelial tricellular junction formation in Drosophila. Dev. Cell 2015, 33, 535–548. [Google Scholar] [CrossRef] [PubMed]
- Norman, M.; Vuilleumier, R.; Springhorn, A.; Gawlik, J.; Pyrowolakis, G. Pentagone internalises glypicans to fine-tune multiple signalling pathways. Elife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Choo, S.W.; White, R.; Russell, S. Genome-wide analysis of the binding of the hox protein ultrabithorax and the hox cofactor homothorax in Drosophila. PLoS ONE 2011, 6, e14778. [Google Scholar] [CrossRef] [PubMed]
- Venken, K.J.; Schulze, K.L.; Haelterman, N.A.; Pan, H.; He, Y.; Evans-Holm, M.; Carlson, J.W.; Levis, R.W.; Spradling, A.C.; Hoskins, R.A.; et al. Mimic: A highly versatile transposon insertion resource for engineering Drosophila melanogaster genes. Nat. Methods 2011, 8, 737–743. [Google Scholar] [CrossRef] [PubMed]
- Nagarkar-Jaiswal, S.; DeLuca, S.Z.; Lee, P.T.; Lin, W.W.; Pan, H.; Zuo, Z.; Lv, J.; Spradling, A.C.; Bellen, H.J. A genetic toolkit for tagging intronic mimic containing genes. Elife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Nagarkar-Jaiswal, S.; Lee, P.T.; Campbell, M.E.; Chen, K.; Anguiano-Zarate, S.; Gutierrez, M.C.; Busby, T.; Lin, W.W.; He, Y.; Schulze, K.L.; et al. A library of mimics allows tagging of genes and reversible, spatial and temporal knockdown of proteins in Drosophila. Elife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Caussinus, E.; Kanca, O.; Affolter, M. Fluorescent fusion protein knockout mediated by anti-gfp nanobody. Nat. Struct. Mol. Biol. 2011, 19, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Neumüller, R.A.; Wirtz-Peitz, F.; Lee, S.; Kwon, Y.; Buckner, M.; Hoskins, R.A.; Venken, K.J.; Bellen, H.J.; Mohr, S.E.; Perrimon, N. Stringent analysis of gene function and protein-protein interactions using fluorescently tagged genes. Genetics 2012, 190, 931–940. [Google Scholar] [CrossRef] [PubMed]
- Trost, M.; Blattner, A.C.; Lehner, C.F. Regulated protein depletion by the auxin-inducible degradation system in Drosophila melanogaster. Fly 2016, 10, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.D.; Lander, E.S.; Zhang, F. Development and applications of crispr-cas9 for genome engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef] [PubMed]
- Beumer, K.J.; Carroll, D. Targeted genome engineering techniques in Drosophila. Methods 2014, 68, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Chen, H.; Liu, J.; Zhang, H.; Yan, Y.; Zhu, N.; Guo, Y.; Yang, B.; Chang, Y.; Dai, F.; et al. Various applications of talen- and crispr/cas9-mediated homologous recombination to modify the Drosophila genome. Biol. Open 2014, 3, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Cebrian-Serrano, A.; Davies, B. Crispr-cas orthologues and variants: Optimizing the repertoire, specificity and delivery of genome engineering tools. Mamm. Genome 2017. [Google Scholar] [CrossRef] [PubMed]
- Gasiunas, G.; Barrangou, R.; Horvath, P.; Siksnys, V. Cas9-crrna ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. USA 2012, 109, E2579–E2586. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-rna-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Bétermier, M.; Bertrand, P.; Lopez, B.S. Is non-homologous end-joining really an inherently error-prone process? PLoS Genet. 2014, 10, e1004086. [Google Scholar] [CrossRef] [PubMed]
- Greene, E.C. DNA sequence alignment during homologous recombination. J. Biol. Chem. 2016, 291, 11572–11580. [Google Scholar] [CrossRef] [PubMed]
- Greene, E.C. On the influence of protein-DNA register during homologous recombination. Cell Cycle 2016, 15, 172–175. [Google Scholar] [CrossRef] [PubMed]
- Gratz, S.J.; Rubinstein, C.D.; Harrison, M.M.; Wildonger, J.; O’Connor-Giles, K.M. Crispr-cas9 genome editing in Drosophila. Curr. Protoc. Mol. Biol. 2015, 111. [Google Scholar] [CrossRef]
- Gratz, S.J.; Ukken, F.P.; Rubinstein, C.D.; Thiede, G.; Donohue, L.K.; Cummings, A.M.; O’Connor-Giles, K.M. Highly specific and efficient crispr/cas9-catalyzed homology-directed repair in Drosophila. Genetics 2014, 196, 961–971. [Google Scholar] [CrossRef] [PubMed]
- Port, F.; Chen, H.M.; Lee, T.; Bullock, S.L. Optimized crispr/cas tools for efficient germline and somatic genome engineering in Drosophila. Proc. Natl. Acad. Sci. USA 2014, 111, E2967–E2976. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, K.A.; Wu, W.H.; Colgan, D.F.; Tsang, S.H.; Bassuk, A.G.; Mahajan, V.B. Unexpected mutations after crispr-cas9 editing in vivo. Nat. Methods 2017, 14, 547–548. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.M.; Cradick, T.J.; Fine, E.J.; Bao, G. Nuclease target site selection for maximizing on-target activity and minimizing off-target effects in genome editing. Mol. Ther. 2016, 24, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Stella, S.; Montoya, G. The genome editing revolution: A crispr-cas tale off-target story. Bioessays 2016, 38 (Suppl. 1), S4–S13. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using crispr/cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Aach, J.; Stranges, P.B.; Esvelt, K.M.; Moosburner, M.; Kosuri, S.; Yang, L.; Church, G.M. Cas9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat. Biotechnol. 2013, 31, 833–838. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Lin, C.Y.; Gootenberg, J.S.; Konermann, S.; Trevino, A.E.; Scott, D.A.; Inoue, A.; Matoba, S.; Zhang, Y.; et al. Double nicking by rna-guided crispr cas9 for enhanced genome editing specificity. Cell 2013, 154, 1380–1389. [Google Scholar] [CrossRef] [PubMed]
- Overballe-Petersen, S.; Harms, K.; Orlando, L.A.; Mayar, J.V.; Rasmussen, S.; Dahl, T.W.; Rosing, M.T.; Poole, A.M.; Sicheritz-Ponten, T.; Brunak, S.; et al. Bacterial natural transformation by highly fragmented and damaged DNA. Proc. Natl. Acad. Sci. USA 2013, 110, 19860–19865. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.W.; Kim, S.; Kim, Y.; Kweon, J.; Kim, H.S.; Bae, S.; Kim, J.S. Analysis of off-target effects of crispr/cas-derived rna-guided endonucleases and nickases. Genome Res. 2014, 24, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Miyaoka, Y.; Berman, J.R.; Cooper, S.B.; Mayerl, S.J.; Chan, A.H.; Zhang, B.; Karlin-Neumann, G.A.; Conklin, B.R. Systematic quantification of hdr and nhej reveals effects of locus, nuclease, and cell type on genome-editing. Sci. Rep. 2016, 6, 23549. [Google Scholar] [CrossRef] [PubMed]
- Doudna, J.A.; Charpentier, E. Genome editing. The new frontier of genome engineering with crispr-cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef] [PubMed]
- Guilinger, J.P.; Thompson, D.B.; Liu, D.R. Fusion of catalytically inactive cas9 to foki nuclease improves the specificity of genome modification. Nat. Biotechnol. 2014, 32, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Kleinstiver, B.P.; Pattanayak, V.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.T.; Zheng, Z.; Joung, J.K. High-fidelity crispr-cas9 nucleases with no detectable genome-wide off-target effects. Nature 2016, 529, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Slaymaker, I.M.; Gao, L.; Zetsche, B.; Scott, D.A.; Yan, W.X.; Zhang, F. Rationally engineered cas9 nucleases with improved specificity. Science 2016, 351, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Kleinstiver, B.P.; Prew, M.S.; Tsai, S.Q.; Topkar, V.V.; Nguyen, N.T.; Zheng, Z.; Gonzales, A.P.; Li, Z.; Peterson, R.T.; Yeh, J.R.; et al. Engineered crispr-cas9 nucleases with altered pam specificities. Nature 2015, 523, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Zetsche, B.; Gootenberg, J.S.; Abudayyeh, O.O.; Slaymaker, I.M.; Makarova, K.S.; Essletzbichler, P.; Volz, S.E.; Joung, J.; van der Oost, J.; Regev, A.; et al. Cpf1 is a single rna-guided endonuclease of a class 2 crispr-cas system. Cell 2015, 163, 759–771. [Google Scholar] [CrossRef] [PubMed]
- Shmakov, S.; Abudayyeh, O.O.; Makarova, K.S.; Wolf, Y.I.; Gootenberg, J.S.; Semenova, E.; Minakhin, L.; Joung, J.; Konermann, S.; Severinov, K.; et al. Discovery and functional characterization of diverse class 2 crispr-cas systems. Mol. Cell. 2015, 60, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Gratz, S.J.; Cummings, A.M.; Nguyen, J.N.; Hamm, D.C.; Donohue, L.K.; Harrison, M.M.; Wildonger, J.; O’Connor-Giles, K.M. Genome engineering of Drosophila with the crispr rna-guided cas9 nuclease. Genetics 2013, 194, 1029–1035. [Google Scholar] [CrossRef] [PubMed]
- Hilton, I.B.; Gersbach, C.A. Enabling functional genomics with genome engineering. Genome Res. 2015, 25, 1442–1455. [Google Scholar] [CrossRef] [PubMed]
- Essletzbichler, P.; Konopka, T.; Santoro, F.; Chen, D.; Gapp, B.V.; Kralovics, R.; Brummelkamp, T.R.; Nijman, S.M.; Burckstummer, T. Megabase-scale deletion using crispr/cas9 to generate a fully haploid human cell line. Genome Res. 2014, 24, 2059–2065. [Google Scholar] [CrossRef] [PubMed]
- Auer, T.O.; Duroure, K.; De Cian, A.; Concordet, J.P.; Del Bene, F. Highly efficient crispr/cas9-mediated knock-in in zebrafish by homology-independent DNA repair. Genome Res. 2014, 24, 142–153. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Tan, C.; Wang, F.; Wang, Y.; Zhou, R.; Cui, D.; You, W.; Zhao, H.; Ren, J.; Feng, B. Knock-in of large reporter genes in human cells via crispr/cas9-induced homology-dependent and independent DNA repair. Nucleic Acids Res. 2016, 44, e85. [Google Scholar] [CrossRef] [PubMed]
- Böttcher, R.; Hollmann, M.; Merk, K.; Nitschko, V.; Obermaier, C.; Philippou-Massier, J.; Wieland, I.; Gaul, U.; Förstemann, K. Efficient chromosomal gene modification with crispr/cas9 and pcr-based homologous recombination donors in cultured Drosophila cells. Nucleic Acids Res. 2014, 42, e89. [Google Scholar] [CrossRef] [PubMed]
- Housden, B.E.; Lin, S.; Perrimon, N. Cas9-based genome editing in Drosophila. Methods Enzymol. 2014, 546, 415–439. [Google Scholar] [CrossRef] [PubMed]
- Salsman, J.; Dellaire, G. Precision genome editing in the crispr era. Biochem. Cell Biol. 2017, 95, 187–201. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Guell, M.; Byrne, S.; Yang, J.L.; De Los Angeles, A.; Mali, P.; Aach, J.; Kim-Kiselak, C.; Briggs, A.W.; Rios, X.; et al. Optimization of scarless human stem cell genome editing. Nucleic Acids Res. 2013, 41, 9049–9061. [Google Scholar] [CrossRef] [PubMed]
- Komor, A.C.; Badran, A.H.; Liu, D.R. Crispr-based technologies for the manipulation of eukaryotic genomes. Cell 2017, 169, 559. [Google Scholar] [CrossRef] [PubMed]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 2017. [Google Scholar] [CrossRef] [PubMed]
- Lamb, A.M.; Walker, E.A.; Wittkopp, P.J. Tools and strategies for scarless allele replacement in Drosophila using crispr/cas9. Fly 2017, 11, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Byrne, S.M.; Ortiz, L.; Mali, P.; Aach, J.; Church, G.M. Multi-kilobase homozygous targeted gene replacement in human induced pluripotent stem cells. Nucleic Acids Res. 2015, 43, e21. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Cai, X.; Tan, M.H.; Schaffert, S.; Arnold, C.P.; Gong, X.; Chen, C.Z.; Huang, S. Precise gene deletion and replacement using the crispr/cas9 system in human cells. Biotechniques 2014, 57, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhang, C.; Liu, W.; Gao, W.; Liu, C.; Song, G.; Li, W.X.; Mao, L.; Chen, B.; Xu, Y.; et al. An alternative strategy for targeted gene replacement in plants using a dual-sgrna/cas9 design. Sci. Rep. 2016, 6, 23890. [Google Scholar] [CrossRef] [PubMed]
- Adikusuma, F.; Pederick, D.; McAninch, D.; Hughes, J.; Thomas, P. Functional equivalence of the sox2 and sox3 transcription factors in the developing mouse brain and testes. Genetics 2017, 206, 1495–1503. [Google Scholar] [CrossRef] [PubMed]
- Evans, T.A. Crispr-based gene replacement reveals evolutionarily conserved axon guidance functions of Drosophila robo3 and tribolium robo2/3. Evodevo 2017, 8, 10. [Google Scholar] [CrossRef] [PubMed]
- Spitzweck, B.; Brankatschk, M.; Dickson, B.J. Distinct protein domains and expression patterns confer divergent axon guidance functions for Drosophila robo receptors. Cell 2010, 140, 409–420. [Google Scholar] [CrossRef] [PubMed]
- Ewen-Campen, B.; Mohr, S.E.; Hu, Y.; Perrimon, N. Accessing the phenotype gap: Enabling systematic investigation of paralog functional complexity with crispr. Dev. Cell 2017, 43, 6–9. [Google Scholar] [CrossRef] [PubMed]
- Overton, P.M.; Meadows, L.A.; Urban, J.; Russell, S. Evidence for differential and redundant function of the sox genes dichaete and soxn during cns development in Drosophila. Development 2002, 129, 4219–4228. [Google Scholar] [PubMed]
- Sánchez-Soriano, N.; Russell, S. Regulatory mutations of the Drosophila sox gene dichaete reveal new functions in embryonic brain and hindgut development. Dev. Biol. 2000, 220, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Yang, Z.; Xu, J.; Sun, J.; Mao, D.; Hu, Y.; Yang, S.J.; Qiao, H.H.; Wang, X.; Hu, Q.; et al. Enhanced specificity and efficiency of the crispr/cas9 system with optimized sgrna parameters in Drosophila. Cell Rep. 2014, 9, 1151–1162. [Google Scholar] [CrossRef] [PubMed]
- Port, F.; Bullock, S.L. Augmenting crispr applications in Drosophila with trna-flanked sgrnas. Nat. Methods 2016, 13, 852–854. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, H.; Shivalila, C.S.; Cheng, A.W.; Shi, L.; Jaenisch, R. One-step generation of mice carrying reporter and conditional alleles by crispr/cas-mediated genome engineering. Cell 2013, 154, 1370–1379. [Google Scholar] [CrossRef] [PubMed]
- Arribere, J.A.; Bell, R.T.; Fu, B.X.; Artiles, K.L.; Hartman, P.S.; Fire, A.Z. Efficient marker-free recovery of custom genetic modifications with crispr/cas9 in caenorhabditis elegans. Genetics 2014, 198, 837–846. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.D. Rapid and precise engineering of the caenorhabditis elegans genome with lethal mutation co-conversion and inactivation of nhej repair. Genetics 2015, 199, 363–377. [Google Scholar] [CrossRef] [PubMed]
- Kane, N.S.; Vora, M.; Varre, K.J.; Padgett, R.W. Efficient screening of crispr/cas9-induced events in Drosophila using a co-crispr strategy. G3 Genes Genomes Genet. 2017, 7, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Ge, D.T.; Tipping, C.; Brodsky, M.H.; Zamore, P.D. Rapid screening for crispr-directed editing of the Drosophila genome using white coconversion. G3 Genes Genomes Genet. 2016, 6, 3197–3206. [Google Scholar] [CrossRef]
- Kondo, S.; Ueda, R. Highly improved gene targeting by germline-specific cas9 expression in Drosophila. Genetics 2013, 195, 715–721. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Sun, J.; Housden, B.E.; Hu, Y.; Roesel, C.; Lin, S.; Liu, L.P.; Yang, Z.; Mao, D.; Sun, L.; et al. Optimized gene editing technology for Drosophila melanogaster using germ line-specific cas9. Proc. Natl. Acad. Sci. USA 2013, 110, 19012–19017. [Google Scholar] [CrossRef] [PubMed]
- Xue, Z.; Wu, M.; Wen, K.; Ren, M.; Long, L.; Zhang, X.; Gao, G. Crispr/cas9 mediates efficient conditional mutagenesis in Drosophila. G3 Genes Genomes Genet. 2014, 4, 2167–2173. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Koolhaas, W.H.; Schnorrer, F. A versatile two-step crispr- and rmce-based strategy for efficient genome engineering in Drosophila. G3 Genes Genomes Genet. 2014, 4, 2409–2418. [Google Scholar] [CrossRef] [PubMed]
- Stern, D.L.; Frankel, N. The structure and evolution of cis-regulatory regions: The shavenbaby story. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2013, 368, 20130028. [Google Scholar] [CrossRef] [PubMed]
- Wilczynski, B.; Furlong, E.E. Challenges for modeling global gene regulatory networks during development: Insights from Drosophila. Dev. Biol. 2010, 340, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Mendoza-Garcia, P.; Hugosson, F.; Fallah, M.; Higgins, M.L.; Iwasaki, Y.; Pfeifer, K.; Wolfstetter, G.; Varshney, G.; Popichenko, D.; Gergen, J.P.; et al. The zic family homologue odd-paired regulates alk expression in Drosophila. PLoS Genet. 2017, 13, e1006617. [Google Scholar] [CrossRef] [PubMed]
- Zandvakili, A.; Gebelein, B. Mechanisms of specificity for hox factor activity. J. Dev. Biol. 2016, 4, 16. [Google Scholar] [CrossRef] [PubMed]
- Rogers, W.A.; Goyal, Y.; Yamaya, K.; Shvartsman, S.Y.; Levine, M.S. Uncoupling neurogenic gene networks in the Drosophila embryo. Genes Dev. 2017, 31, 634–638. [Google Scholar] [CrossRef] [PubMed]
- Lagha, M.; Bothma, J.P.; Levine, M. Mechanisms of transcriptional precision in animal development. Trends Genet. 2012, 28, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Consortium, E.P. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef]
- Lee, Y.; Rio, D.C. Mechanisms and regulation of alternative pre-mrna splicing. Annu. Rev. Biochem. 2015, 84, 291–323. [Google Scholar] [CrossRef] [PubMed]
- Beagan, J.A.; Phillips-Cremins, J.E. Crispr/cas9 genome editing throws descriptive 3-D genome folding studies for a loop. Wiley Interdiscip. Rev. Syst. Biol. Med. 2016, 8, 286–299. [Google Scholar] [CrossRef] [PubMed]
- Mir, M.; Reimer, A.; Haines, J.E.; Li, X.Y.; Stadler, M.; Garcia, H.; Eisen, M.B.; Darzacq, X. Dense bicoid hubs accentuate binding along the morphogen gradient. Genes Dev. 2017, 31, 1784–1794. [Google Scholar] [CrossRef] [PubMed]
- Crews, S. Creating cell type-specific mutants by enhancer mutagenesis. Genes Dev. 2017, 31, 629–631. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Zhang, S.; Shang, S.; Zhang, B.; Li, S.; Wang, X.; Wang, F.; Su, J.; Wu, Q.; Liu, H.; et al. Sea: A super-enhancer archive. Nucleic Acids Res. 2016, 44, D172–D179. [Google Scholar] [CrossRef] [PubMed]
- Thorn, K. Genetically encoded fluorescent tags. Mol. Biol. Cell 2017, 28, 848–857. [Google Scholar] [CrossRef] [PubMed]
- Rees, J.S.; Lilley, K.S.; Jackson, A.P. Silac-ipac: A quantitative method for distinguishing genuine from non-specific components of protein complexes by parallel affinity capture. J. Proteom. 2015, 115, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Cabantous, S.; Terwilliger, T.C.; Waldo, G.S. Protein tagging and detection with engineered self-assembling fragments of green fluorescent protein. Nat. Biotechnol. 2005, 23, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Kamiyama, D.; Sekine, S.; Barsi-Rhyne, B.; Hu, J.; Chen, B.; Gilbert, L.A.; Ishikawa, H.; Leonetti, M.D.; Marshall, W.F.; Weissman, J.S.; et al. Versatile protein tagging in cells with split fluorescent protein. Nat. Commun. 2016, 7, 11046. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Barish, S.; Okuwa, S.; Volkan, P.C. Examination of endogenous rotund expression and function in developing Drosophila olfactory system using crispr-cas9-mediated protein tagging. G3 Genes Genomes Genet. 2015, 5, 2809–2816. [Google Scholar] [CrossRef] [PubMed]
- Van Nostrand, E.L.; Gelboin-Burkhart, C.; Wang, R.; Pratt, G.A.; Blue, S.M.; Yeo, G.W. Crispr/cas9-mediated integration enables tag-eclip of endogenously tagged rna binding proteins. Methods 2017, 118–119, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Savic, D.; Partridge, E.C.; Newberry, K.M.; Smith, S.B.; Meadows, S.K.; Roberts, B.S.; Mackiewicz, M.; Mendenhall, E.M.; Myers, R.M. Cetch-seq: Crispr epitope tagging chip-seq of DNA-binding proteins. Genome Res. 2015, 25, 1581–1589. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.; Zhang, Y.; Yan, J.; Jain, S.; Chee, S.; Ren, B.; Zhao, H. A scalable epitope tagging approach for high throughput chip-seq analysis. ACS Synth. Biol. 2017, 6, 1034–1042. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.M.; McVey, M. Synthesis-dependent microhomology-mediated end joining accounts for multiple types of repair junctions. Nucleic Acids Res. 2010, 38, 5706–5717. [Google Scholar] [CrossRef] [PubMed]
- Koles, K.; Yeh, A.R.; Rodal, A.A. Tissue-specific tagging of endogenous loci in Drosophila melanogaster. Biol. Open 2015, 5, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.M.; Huang, Y.; Pfeiffer, B.D.; Yao, X.; Lee, T. An enhanced gene targeting toolkit for Drosophila: Golic+. Genetics 2015, 199, 683–694. [Google Scholar] [CrossRef] [PubMed]
- Kunzelmann, S.; Böttcher, R.; Schmidts, I.; Förstemann, K. A comprehensive toolbox for genome editing in cultured Drosophila melanogaster cells. G3 Genes Genomes Genet. 2016, 6, 1777–1785. [Google Scholar] [CrossRef] [PubMed]
- Kunzelmann, S.; Förstemann, K. Reversible perturbations of gene regulation after genome editing in Drosophila cells. PLoS ONE 2017, 12, e0180135. [Google Scholar] [CrossRef] [PubMed]



© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Korona, D.; Koestler, S.A.; Russell, S. Engineering the Drosophila Genome for Developmental Biology. J. Dev. Biol. 2017, 5, 16. https://doi.org/10.3390/jdb5040016
Korona D, Koestler SA, Russell S. Engineering the Drosophila Genome for Developmental Biology. Journal of Developmental Biology. 2017; 5(4):16. https://doi.org/10.3390/jdb5040016
Chicago/Turabian StyleKorona, Dagmara, Stefan A. Koestler, and Steven Russell. 2017. "Engineering the Drosophila Genome for Developmental Biology" Journal of Developmental Biology 5, no. 4: 16. https://doi.org/10.3390/jdb5040016
APA StyleKorona, D., Koestler, S. A., & Russell, S. (2017). Engineering the Drosophila Genome for Developmental Biology. Journal of Developmental Biology, 5(4), 16. https://doi.org/10.3390/jdb5040016