

Identifying Molecular Roadblocks for Transcription Factor-Induced Cellular Reprogramming In Vivo by Using C. elegans as a Model Organism

Abstract
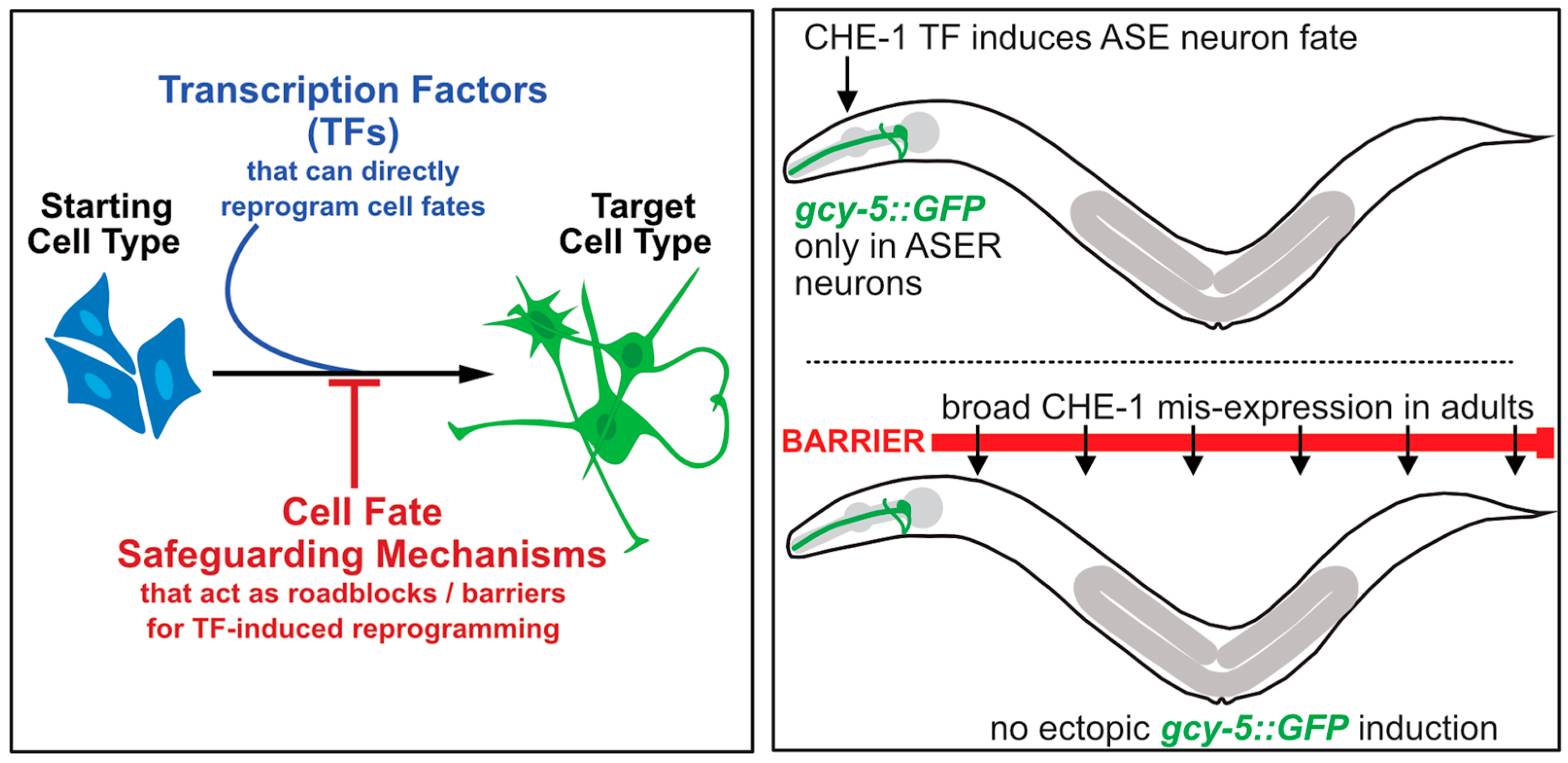
1. Introduction
2. Transcription Factor-Mediated Reprogramming of Cell Fates in C. elegans
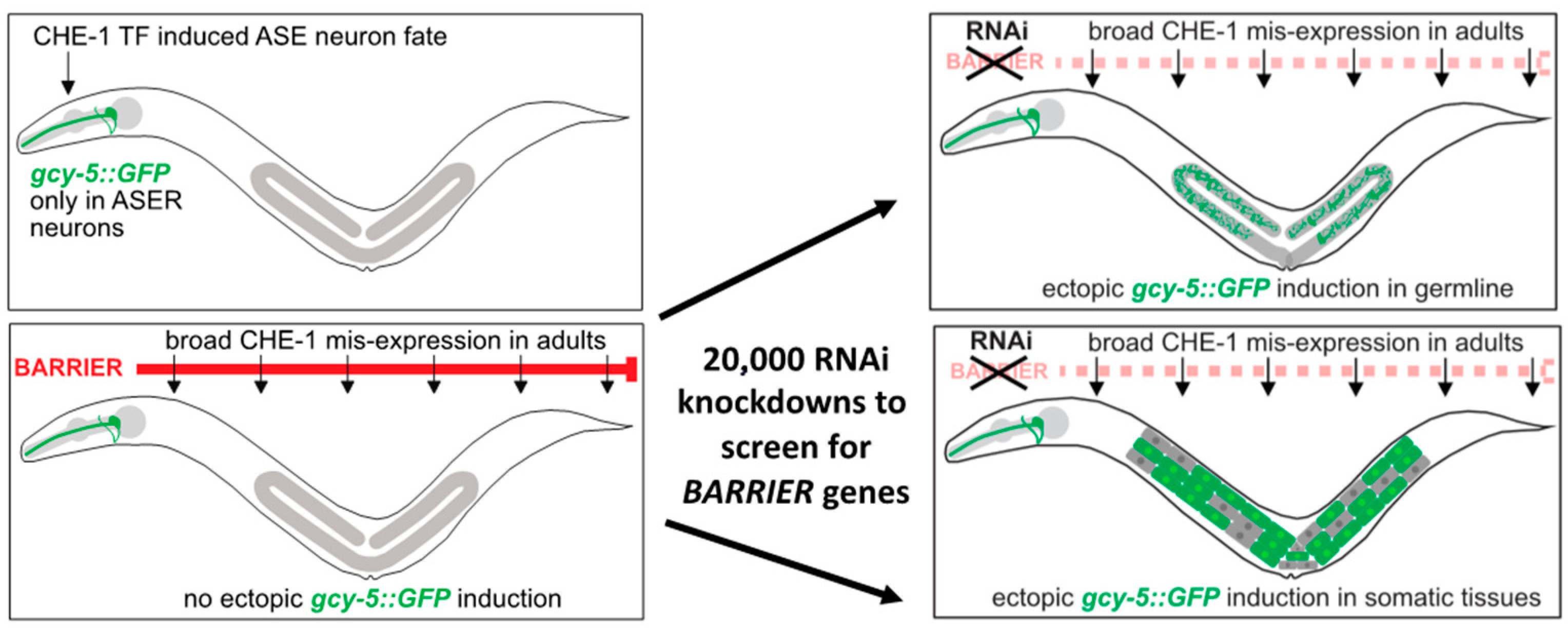
3. Identifying Reprogramming Barriers in C. elegans Using Neuronal Fate Induction
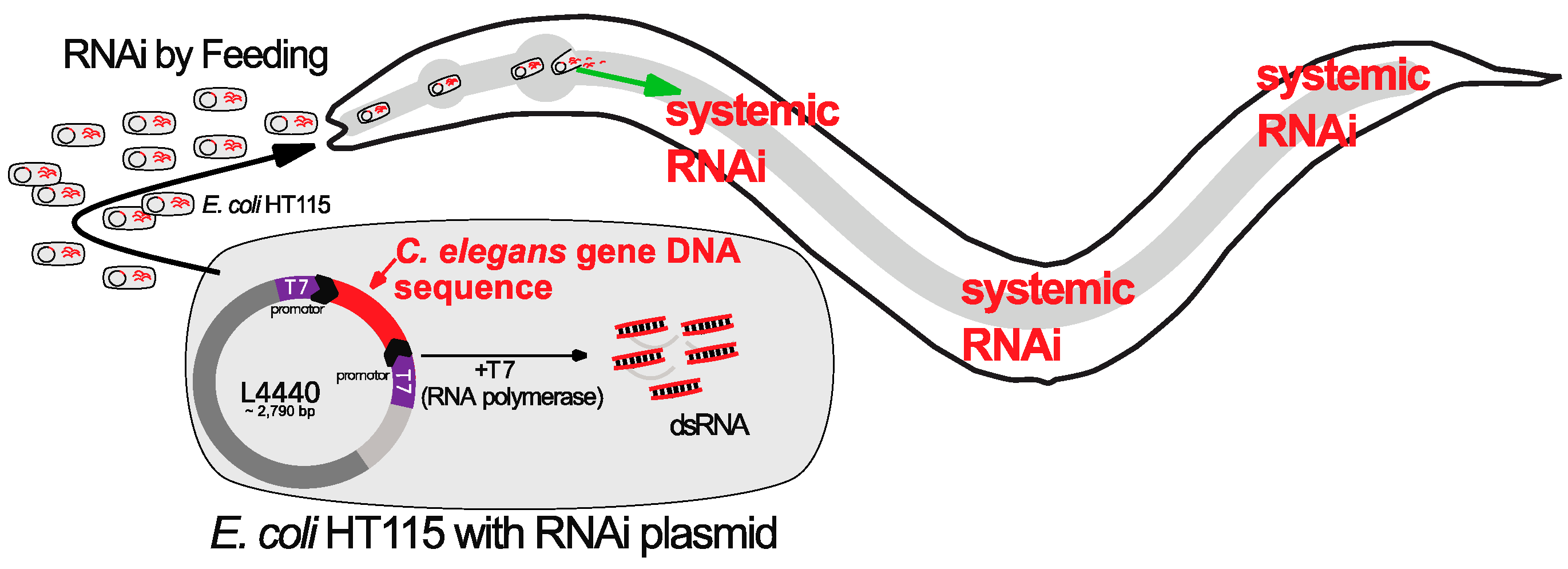
4. RNAi Screens in C. elegans to Identify Reprogramming Barriers
5. The Histone Chaperone LIN-53 Inhibits Germ Cell to Neuron Reprogramming
6. LIN-53 Cooperates with Polycomb Repressive Complex 2 to Prevent Germ Cell to Neuron Reprogramming in C. elegans
7. The FACT Complex Member HMG-3 Prevents TF-Mediated Germ Cell Reprogramming
8. The Chromodomain Protein MRG-1 Acts as a Reprogramming Barrier in the Germline
9. The Set1/MLL Methyltransferase Complex Member RBBP-5 Inhibits TF-Mediated Germ-Cell Reprogramming to GABAergic Neurons in C. elegans
10. Germ Granules (P granules) Safeguards Germ Cell Identity
11. Summary and Future Perspective
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sebban, S.; Buganim, Y. Nuclear Reprogramming by Defined Factors: Quantity versus Quality. Trends Cell Biol. 2016, 26, 65–75. [Google Scholar] [CrossRef]
- Guo, C.; Morris, S.A. Engineering cell identity: Establishing new gene regulatory and chromatin landscapes. Curr. Opin. Genet. Dev. 2017, 46, 50–57. [Google Scholar] [CrossRef]
- Schneuwly, S.; Klemenz, R.; Gehring, W.J. Redesigning the Body Plan of Drosophila by Ectopic Expression of the Homeotic Gene Antennapedia. Nature 1987, 325, 816–818. [Google Scholar] [CrossRef]
- Halder, G.; Callaerts, P.; Gehring, W.J. Induction of ectopic eyes by targeted expression of the eyeless gene in Drosophila. Science 1995, 267, 1788–1792. [Google Scholar] [CrossRef]
- Davis, R.L.; Weintraub, H.; Lassar, A.B. Expression of a single transfected cDNA converts fibroblasts to myoblasts. Cell 1987, 51, 987–1000. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- Reid, A.; Tursun, B. Transdifferentiation: Do transition states lie on the path of development? Curr. Opin. Syst. Biol. 2018, 11, 18–23. [Google Scholar] [CrossRef]
- Wang, H.; Yang, Y.; Liu, J.; Qian, L. Direct cell reprogramming: Approaches, mechanisms and progress. Nat. Rev. Mol. Cell Biol. 2021, 22, 410–424. [Google Scholar] [CrossRef]
- Vierbuchen, T.; Ostermeier, A.; Pang, Z.P.; Kokubu, Y.; Sudhof, T.C.; Wernig, M. Direct conversion of fibroblasts to functional neurons by defined factors. Nature 2010, 463, 1035–1041. [Google Scholar] [CrossRef]
- Marchal, I.; Tursun, B. Induced Neurons from Germ Cells in Caenorhabditis elegans. Front. Neurosci. 2021, 15, 771687. [Google Scholar] [CrossRef]
- Ofenbauer, A.; Tursun, B. Strategies for in vivo reprogramming. Curr. Opin. Cell Biol. 2019, 61, 9–15. [Google Scholar] [CrossRef]
- Zuryn, S.; Ahier, A.; Portoso, M.; White, E.R.; Morin, M.C.; Margueron, R.; Jarriault, S. Transdifferentiation. Sequential histone-modifying activities determine the robustness of transdifferentiation. Science 2014, 345, 826–829. [Google Scholar] [CrossRef]
- Jarriault, S.; Schwab, Y.; Greenwald, I. A Caenorhabditis elegans model for epithelial-neuronal transdifferentiation. Proc. Natl. Acad. Sci. USA 2008, 105, 3790–3795. [Google Scholar] [CrossRef]
- Lambert, J.; Lloret-Fernandez, C.; Laplane, L.; Poole, R.J.; Jarriault, S. On the origins and conceptual frameworks of natural plasticity—Lessons from single-cell models in C. elegans. Curr. Top. Dev. Biol. 2021, 144, 111–159. [Google Scholar] [CrossRef]
- Rothman, J.; Jarriault, S. Developmental Plasticity and Cellular Reprogramming in Caenorhabditis elegans. Genetics 2019, 213, 723–757. [Google Scholar] [CrossRef]
- Tursun, B.; Patel, T.; Kratsios, P.; Hobert, O. Direct conversion of C. elegans germ cells into specific neuron types. Science 2011, 331, 304–308. [Google Scholar] [CrossRef]
- Patel, T.; Tursun, B.; Rahe, D.P.; Hobert, O. Removal of Polycomb repressive complex 2 makes C. elegans germ cells susceptible to direct conversion into specific somatic cell types. Cell Rep. 2012, 2, 1178–1186. [Google Scholar] [CrossRef]
- Gaydos, L.J.; Rechtsteiner, A.; Egelhofer, T.A.; Carroll, C.R.; Strome, S. Antagonism between MES-4 and Polycomb repressive complex 2 promotes appropriate gene expression in C. elegans germ cells. Cell Rep. 2012, 2, 1169–1177. [Google Scholar] [CrossRef]
- Kolundzic, E.; Ofenbauer, A.; Bulut, S.I.; Uyar, B.; Baytek, G.; Sommermeier, A.; Seelk, S.; He, M.; Hirsekorn, A.; Vucicevic, D.; et al. FACT Sets a Barrier for Cell Fate Reprogramming in Caenorhabditis elegans and Human Cells. Dev. Cell 2018, 46, 611–626.e12. [Google Scholar] [CrossRef]
- Hajduskova, M.; Baytek, G.; Kolundzic, E.; Gosdschan, A.; Kazmierczak, M.; Ofenbauer, A.; Beato Del Rosal, M.L.; Herzog, S.; Ul Fatima, N.; Mertins, P.; et al. MRG-1/MRG15 Is a Barrier for Germ Cell to Neuron Reprogramming in Caenorhabditis elegans. Genetics 2019, 211, 121–139. [Google Scholar] [CrossRef]
- Kazmierczak, M.; Farre, I.D.C.; Ofenbauer, A.; Herzog, S.; Tursun, B. The CONJUDOR pipeline for multiplexed knockdown of gene pairs identifies RBBP-5 as a germ cell reprogramming barrier in C. elegans. Nucleic Acids Res. 2021, 49, e22. [Google Scholar] [CrossRef] [PubMed]
- Corsi, A.K.; Wightman, B.; Chalfie, M. A Transparent window into biology: A primer on Caenorhabditis elegans. WormBook 2015, 200, 1–31. [Google Scholar] [CrossRef]
- Fukushige, T.; Krause, M. The myogenic potency of HLH-1 reveals wide-spread developmental plasticity in early C. elegans embryos. Development 2005, 132, 1795–1805. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Fukushige, T.; McGhee, J.D.; Rothman, J.H. Reprogramming of early embryonic blastomeres into endodermal progenitors by a Caenorhabditis elegans GATA factor. Genes Dev. 1998, 12, 3809–3814. [Google Scholar] [CrossRef]
- Yuzyuk, T.; Fakhouri, T.H.; Kiefer, J.; Mango, S.E. The polycomb complex protein mes-2/E(z) promotes the transition from developmental plasticity to differentiation in C. elegans embryos. Dev. Cell 2009, 16, 699–710. [Google Scholar] [CrossRef]
- Strome, S.; Updike, D. Specifying and protecting germ cell fate. Nat. Rev. Mol. Cell Biol. 2015, 16, 406–416. [Google Scholar] [CrossRef]
- Jin, Y.; Hoskins, R.; Horvitz, H.R. Control of type-D GABAergic neuron differentiation by C. elegans UNC-30 homeodomain protein. Nature 1994, 372, 780–783. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.S.; Krause, M.; Sepanski, M.; Fire, A. The Caenorhabditis-Elegans Myod Homolog Hlh-1 Is Essential for Proper Muscle Function and Complete Morphogenesis. Development 1994, 120, 1631–1641. [Google Scholar] [CrossRef]
- Fukushige, T.; Hawkins, M.G.; McGhee, J.D. The GATA-factor elt-2 is essential for formation of the Caenorhabditis elegans intestine. Dev. Biol. 1998, 198, 286–302. [Google Scholar] [CrossRef]
- Riddle, M.R.; Weintraub, A.; Nguyen, K.C.; Hall, D.H.; Rothman, J.H. Transdifferentiation and remodeling of post-embryonic C. elegans cells by a single transcription factor. Development 2013, 140, 4844–4849. [Google Scholar] [CrossRef]
- Riddle, M.R.; Spickard, E.A.; Jevince, A.; Nguyen, K.C.; Hall, D.H.; Joshi, P.M.; Rothman, J.H. Transorganogenesis and transdifferentiation in C. elegans are dependent on differentiated cell identity. Dev. Biol. 2016, 420, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Sallee, M.D.; Littleford, H.E.; Greenwald, I. A bHLH Code for Sexually Dimorphic Form and Function of the C. elegans Somatic Gonad. Curr. Biol. 2017, 27, 1853–1860.e5. [Google Scholar] [CrossRef]
- Tursun, B. Cellular reprogramming processes in Drosophila and C. elegans. Curr. Opin. Genet. Dev. 2012, 22, 475–484. [Google Scholar] [CrossRef]
- Etchberger, J.F.; Flowers, E.B.; Poole, R.J.; Bashllari, E.; Hobert, O. Cis-regulatory mechanisms of left/right asymmetric neuron-subtype specification in C. elegans. Development 2009, 136, 147–160. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.; Johnston, R.J., Jr.; Hobert, O. A transcriptional regulatory cascade that controls left/right asymmetry in chemosensory neurons of C. elegans. Genes Dev. 2003, 17, 2123–2137. [Google Scholar] [CrossRef]
- Ortiz, C.O.; Etchberger, J.F.; Posy, S.L.; Frokjaer-Jensen, C.; Lockery, S.; Honig, B.; Hobert, O. Searching for neuronal left/right asymmetry: Genomewide analysis of nematode receptor-type guanylyl cyclases. Genetics 2006, 173, 131–149. [Google Scholar] [CrossRef]
- Patel, T.; Hobert, O. Coordinated control of terminal differentiation and restriction of cellular plasticity. eLife 2017, 6, e24100. [Google Scholar] [CrossRef] [PubMed]
- Cheloufi, S.; Hochedlinger, K. Emerging roles of the histone chaperone CAF-1 in cellular plasticity. Curr. Opin. Genet. Dev. 2017, 46, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Cheloufi, S.; Elling, U.; Hopfgartner, B.; Jung, Y.L.; Murn, J.; Ninova, M.; Hubmann, M.; Badeaux, A.I.; Euong Ang, C.; Tenen, D.; et al. The histone chaperone CAF-1 safeguards somatic cell identity. Nature 2015, 528, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Fraser, A.G.; Kamath, R.S.; Zipperlen, P.; Martinez-Campos, M.; Sohrmann, M.; Ahringer, J. Functional genomic analysis of C. elegans chromosome I by systematic RNA interference. Nature 2000, 408, 325–330. [Google Scholar] [CrossRef]
- Kamath, R.S.; Martinez-Campos, M.; Zipperlen, P.; Fraser, A.G.; Ahringer, J. Effectiveness of specific RNA-mediated interference through ingested double-stranded RNA in Caenorhabditis elegans. Genome Biol. 2001, 2, research0002.1. [Google Scholar] [CrossRef]
- Boutros, M.; Ahringer, J. The art and design of genetic screens: RNA interference. Nat. Rev. Genet. 2008, 9, 554–566. [Google Scholar] [CrossRef]
- Gouda, K.; Matsunaga, Y.; Iwasaki, T.; Kawano, T. An altered method of feeding RNAi that knocks down multiple genes simultaneously in the nematode Caenorhabditis elegans. Biosci. Biotechnol. Biochem. 2010, 74, 2361–2365. [Google Scholar] [CrossRef][Green Version]
- Min, K.; Kang, J.; Lee, J. A modified feeding RNAi method for simultaneous knock-down of more than one gene in Caenorhabditis elegans. Biotechniques 2010, 48, 229–232. [Google Scholar] [CrossRef]
- Ciosk, R.; DePalma, M.; Priess, J.R. Translational regulators maintain totipotency in the Caenorhabditis elegans germline. Science 2006, 311, 851–853. [Google Scholar] [CrossRef]
- Harrison, M.M.; Ceol, C.J.; Lu, X.; Horvitz, H.R. Some C. elegans class B synthetic multivulva proteins encode a conserved LIN-35 Rb-containing complex distinct from a NuRD-like complex. Proc. Natl. Acad. Sci. USA 2006, 103, 16782–16787. [Google Scholar] [CrossRef]
- Loyola, A.; Almouzni, G. Histone chaperones, a supporting role in the limelight. Biochim. Biophys. Acta 2004, 1677, 3–11. [Google Scholar] [CrossRef]
- Bender, L.B.; Cao, R.; Zhang, Y.; Strome, S. The MES-2/MES-3/MES-6 complex and regulation of histone H3 methylation in C. elegans. Curr. Biol. 2004, 14, 1639–1643. [Google Scholar] [CrossRef]
- Di Croce, L.; Helin, K. Transcriptional regulation by Polycomb group proteins. Nat. Struct. Mol. Biol. 2013, 20, 1147–1155. [Google Scholar] [CrossRef]
- Tursun, B. PcG Proteins in Caenorhabditis elegans. In Polycomb Group Proteins; Pirrotta, V., Tollefsbol, T.O., Eds.; Translational Epigenetics Series; Elsevier Academic Press: Cambridge, MA, USA, 2017; pp. 289–315. [Google Scholar]
- Fong, Y.; Bender, L.; Wang, W.; Strome, S. Regulation of the different chromatin states of autosomes and X chromosomes in the germ line of C. elegans. Science 2002, 296, 2235–2238. [Google Scholar] [CrossRef]
- Seelk, S.; Adrian-Kalchhauser, I.; Hargitai, B.; Hajduskova, M.; Gutnik, S.; Tursun, B.; Ciosk, R. Increasing Notch signaling antagonizes PRC2-mediated silencing to promote reprograming of germ cells into neurons. eLife 2016, 5, e15477. [Google Scholar] [CrossRef]
- Belotserkovskaya, R.; Oh, S.; Bondarenko, V.A.; Orphanides, G.; Studitsky, V.M.; Reinberg, D. FACT facilitates transcription-dependent nucleosome alteration. Science 2003, 301, 1090–1093. [Google Scholar] [CrossRef]
- Orphanides, G.; Wu, W.H.; Lane, W.S.; Hampsey, M.; Reinberg, D. The chromatin-specific transcription elongation factor FACT comprises human SPT16 and SSRP1 proteins. Nature 1999, 400, 284–288. [Google Scholar] [CrossRef]
- Orphanides, G.; LeRoy, G.; Chang, C.H.; Luse, D.S.; Reinberg, D. FACT, a factor that facilitates transcript elongation through nucleosomes. Cell 1998, 92, 105–116. [Google Scholar] [CrossRef]
- Kolundzic, E.; Seelk, S.; Tursun, B. Application of RNAi and Heat-shock-induced Transcription Factor Expression to Reprogram Germ Cells to Neurons in C. elegans. J. Vis. Exp. 2018, 131, e56889. [Google Scholar] [CrossRef]
- Buganim, Y.; Faddah, D.A.; Jaenisch, R. Mechanisms and models of somatic cell reprogramming. Nat. Rev. Genet. 2013, 14, 427–439. [Google Scholar] [CrossRef]
- Ofenbauer, A.; Kraus, C.M.; Tursun, B. The C. elegans pseudogene sspt-16 (F55A3.7) is required to safeguard germ cells against reprogramming. MicroPubl. Biol. 2021, 2021, 1–2. [Google Scholar] [CrossRef]
- Beurton, F.; Stempor, P.; Caron, M.; Appert, A.; Dong, Y.; Chen, R.A.; Cluet, D.; Coute, Y.; Herbette, M.; Huang, N.; et al. Physical and functional interaction between SET1/COMPASS complex component CFP-1 and a Sin3S HDAC complex in C. elegans. Nucleic Acids Res. 2019, 47, 11164–11180. [Google Scholar] [CrossRef]
- Iwamori, N.; Tominaga, K.; Sato, T.; Riehle, K.; Iwamori, T.; Ohkawa, Y.; Coarfa, C.; Ono, E.; Matzuk, M.M. MRG15 is required for pre-mRNA splicing and spermatogenesis. Proc. Natl. Acad. Sci. USA 2016, 113, E5408–E5415. [Google Scholar] [CrossRef]
- Olgun, A.; Aleksenko, T.; Pereira-Smith, O.M.; Vassilatis, D.K. Functional analysis of MRG-1: The ortholog of human MRG15 in Caenorhabditis elegans. J. Gerontol. A Biol. Sci. Med. Sci. 2005, 60, 543–548. [Google Scholar] [CrossRef][Green Version]
- Fujita, M.; Takasaki, T.; Nakajima, N.; Kawano, T.; Shimura, Y.; Sakamoto, H. MRG-1, a mortality factor-related chromodomain protein, is required maternally for primordial germ cells to initiate mitotic proliferation in C. elegans. Mech. Dev. 2002, 114, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.; Leahul, L.; Wang, X.; Wang, C.; Bakos, B.; Jasper, K.; Hansen, D. Proteasome regulation of the chromodomain protein MRG-1 controls the balance between proliferative fate and differentiation in the C. elegans germ line. Development 2015, 142, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Baytek, G.; Blume, A.; Demirel, F.G.; Bulut, S.; Popp, O.; Mertins, P.; Tursun, B. SUMOylation of the chromodomain factor MRG-1 in C. elegans affects chromatin-regulatory dynamics. Biotechniques 2022, 73, 5–17. [Google Scholar] [CrossRef]
- Baytek, G.; Popp, O.; Mertins, P.; Tursun, B. Robust co-immunoprecipitation with mass spectrometry for Caenorhabditis elegans using solid-phase enhanced sample preparation. Biotechniques 2022, 72, 175–184. [Google Scholar] [CrossRef]
- Borkent, M.; Bennett, B.D.; Lackford, B.; Bar-Nur, O.; Brumbaugh, J.; Wang, L.; Du, Y.; Fargo, D.C.; Apostolou, E.; Cheloufi, S.; et al. A Serial shRNA Screen for Roadblocks to Reprogramming Identifies the Protein Modifier SUMO2. Stem Cell Rep. 2016, 6, 704–716. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Kelly, W.G. A role for Set1/MLL-related components in epigenetic regulation of the Caenorhabditis elegans germ line. PLoS Genet. 2011, 7, e1001349. [Google Scholar] [CrossRef]
- Updike, D.; Strome, S. P granule assembly and function in Caenorhabditis elegans germ cells. J. Androl. 2010, 31, 53–60. [Google Scholar] [CrossRef]
- Thomas, L.; Putnam, A.; Folkmann, A. Germ granules in development. Development 2023, 150, dev201037. [Google Scholar] [CrossRef]
- Updike, D.L.; Strome, S. A genomewide RNAi screen for genes that affect the stability, distribution and function of P granules in Caenorhabditis elegans. Genetics 2009, 183, 1397–1419. [Google Scholar] [CrossRef]
- Knutson, A.K.; Egelhofer, T.; Rechtsteiner, A.; Strome, S. Germ Granules Prevent Accumulation of Somatic Transcripts in the Adult Caenorhabditis elegans Germline. Genetics 2017, 206, 163–178. [Google Scholar] [CrossRef]
- Updike, D.L.; Knutson, A.K.; Egelhofer, T.A.; Campbell, A.C.; Strome, S. Germ-granule components prevent somatic development in the C. elegans germline. Curr. Biol. 2014, 24, 970–975. [Google Scholar] [CrossRef]
- Rahe, D.P.; Hobert, O. Restriction of Cellular Plasticity of Differentiated Cells Mediated by Chromatin Modifiers, Transcription Factors and Protein Kinases. G3 Genes Genomes Genet. 2019, 9, 2287–2302. [Google Scholar] [CrossRef]
- Seydoux, G. The P Granules of C. elegans: A Genetic Model for the Study of RNA-Protein Condensates. J. Mol. Biol. 2018, 430, 4702–4710. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Reprogramming Observed | Transcription Factor (TF) | Reprogramming Barrier | Function/Role | Human Counterpart | Reference |
|---|---|---|---|---|---|
| Germ cell to neuron | Znf TF CHE-1 ZnF TF UNC-3 Homeobox UNC-30 | LIN-53 | Histone Chaperone | RBBP4/7 | Tursun et al., 2011 [16] |
| Germ cell to neuron | ZnF TF CHE-1 | PRC-2 (MES-2/-3) | H3K27 Methylation | EZH2 | Patel et al., 2012 [17] |
| Germ cell to muscle | bHLH TF HLH-1 | PRC-2 (MES-6) | H3K27 Methylation | EED, MyoD, MYF5,6 | Patel et al., 2012 [17] |
| Germ cell to neuron | ZnF TF CHE-1 | MES-4 | Histone Chaperone H3K36 Methylation | NSD proteins | Gaydos et al., 2012 [18] |
| Germ cell and intestine to neuron | ZnF TF CHE-1 | FACT (HMG-3/-4, SPT-16) | Histone Chaperone | SSRP1, SUPT16H | Kolundzic et al., 2018 [19] |
| Germ cell to neuron | ZnF TF CHE-1 | MRG-1 | Part of NuA4 histone acetyltransferase complex | MRG-15 | Hadjuskova et al., 2019 [20] |
| Germ cell to neuron | ZnF TF CHE-1 Homeobox UNC-30 | RBBP-5 | Set1/MLL methyltransferase complex member | RBBP5 | Kazmierczak et al., 2021 [21] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Özcan, I.; Tursun, B. Identifying Molecular Roadblocks for Transcription Factor-Induced Cellular Reprogramming In Vivo by Using C. elegans as a Model Organism. J. Dev. Biol. 2023, 11, 37. https://doi.org/10.3390/jdb11030037
Özcan I, Tursun B. Identifying Molecular Roadblocks for Transcription Factor-Induced Cellular Reprogramming In Vivo by Using C. elegans as a Model Organism. Journal of Developmental Biology. 2023; 11(3):37. https://doi.org/10.3390/jdb11030037
Chicago/Turabian StyleÖzcan, Ismail, and Baris Tursun. 2023. "Identifying Molecular Roadblocks for Transcription Factor-Induced Cellular Reprogramming In Vivo by Using C. elegans as a Model Organism" Journal of Developmental Biology 11, no. 3: 37. https://doi.org/10.3390/jdb11030037
APA StyleÖzcan, I., & Tursun, B. (2023). Identifying Molecular Roadblocks for Transcription Factor-Induced Cellular Reprogramming In Vivo by Using C. elegans as a Model Organism. Journal of Developmental Biology, 11(3), 37. https://doi.org/10.3390/jdb11030037