
Pax3 Hypomorphs Reveal Hidden Pax7 Functional Genetic Compensation in Utero



Abstract
:1. Introduction
2. Materials and Methods
2.1. Genetically Modified Mice
2.2. Histologic Analysis
2.3. Western Blot Analysis
2.4. Reverse Transcriptase-PCR Analysis
3. Results
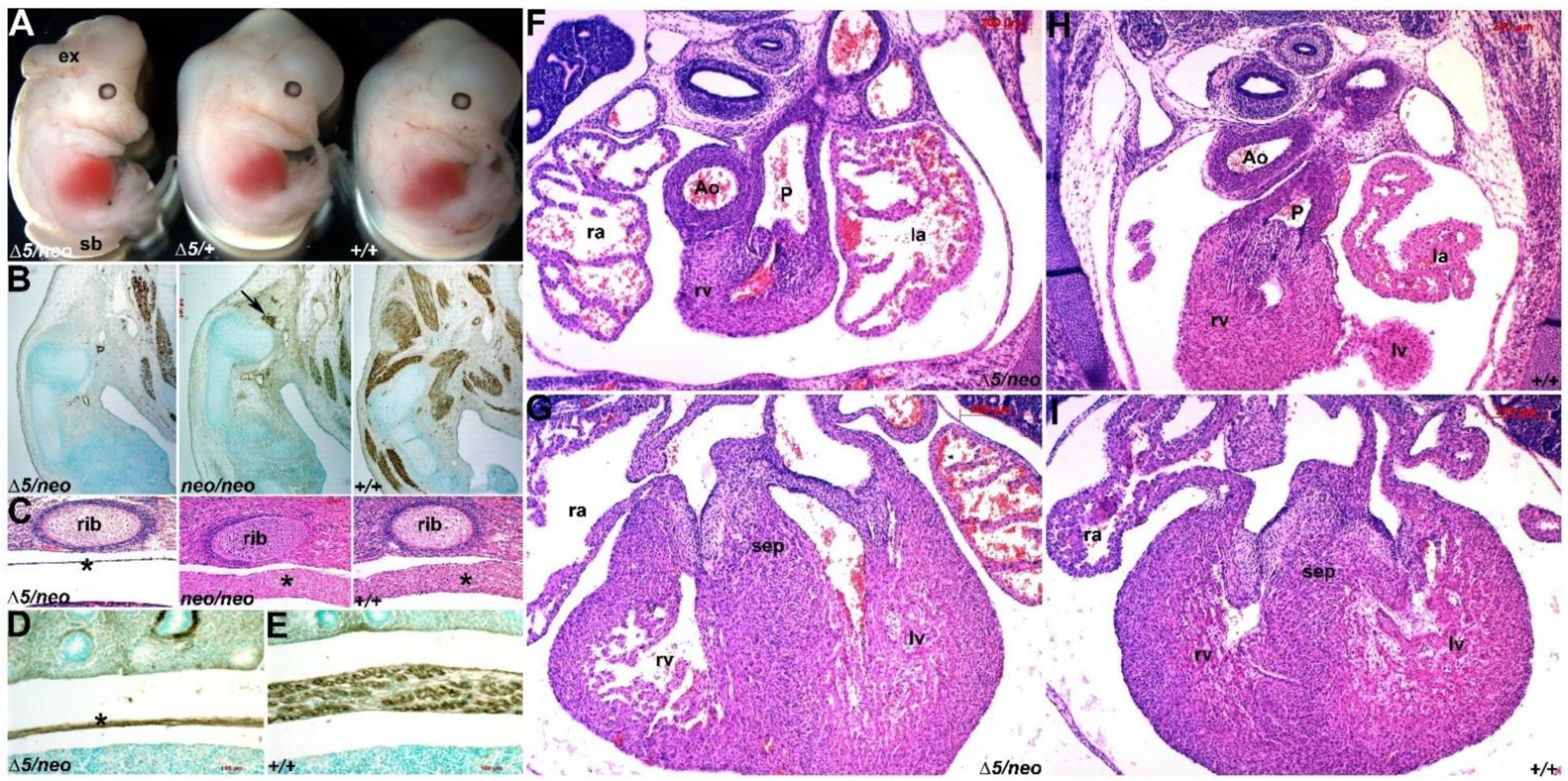
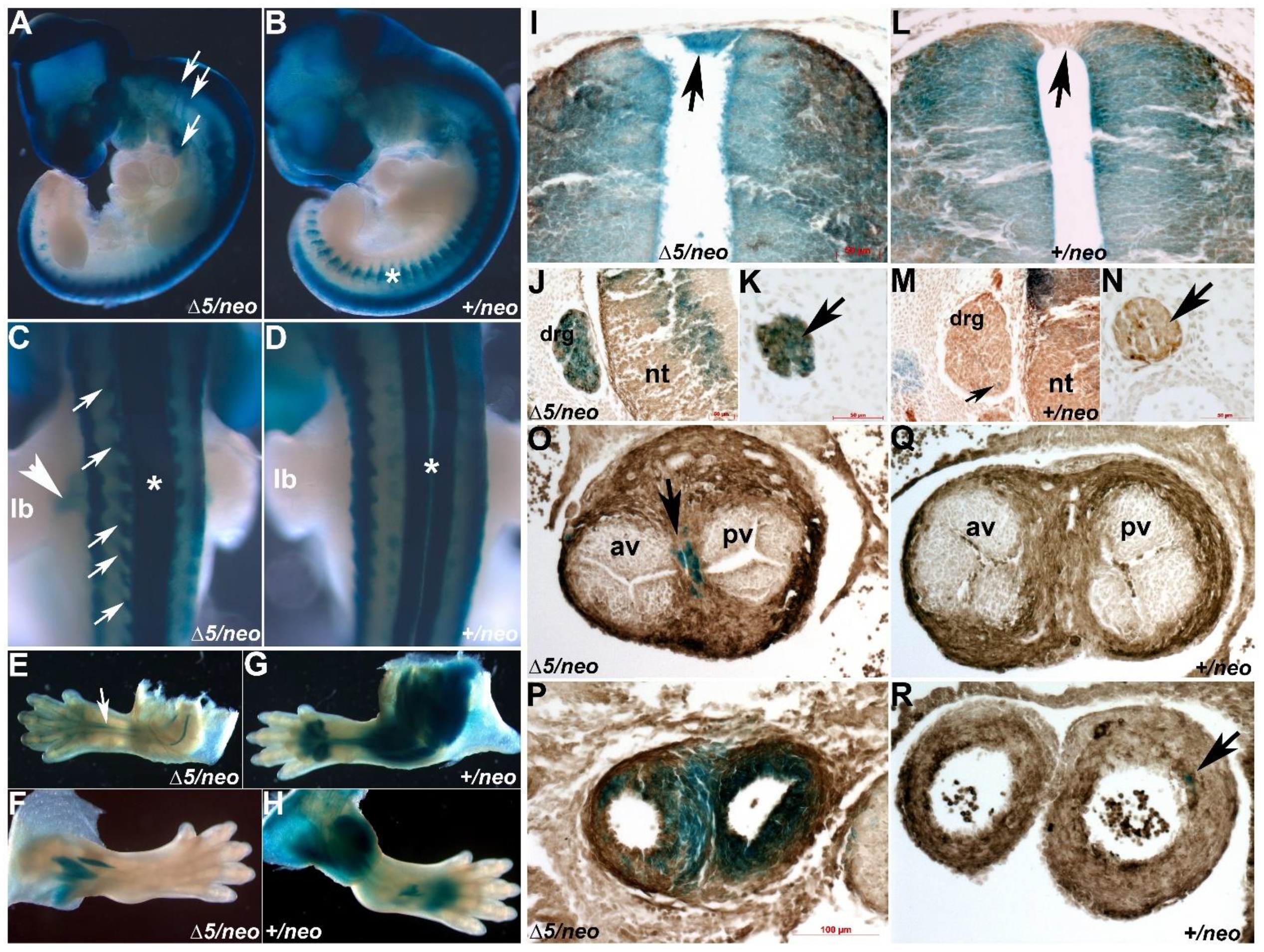
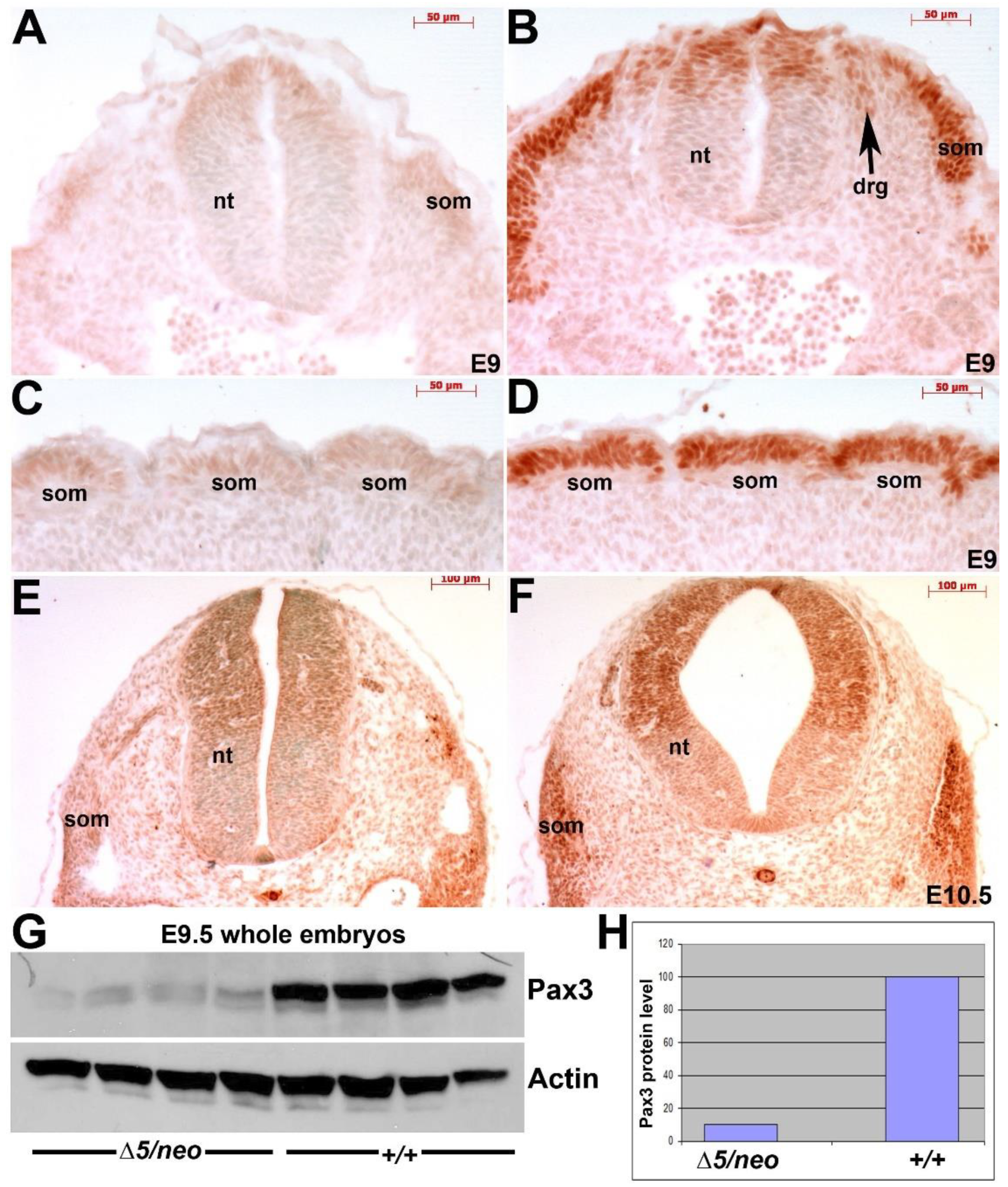
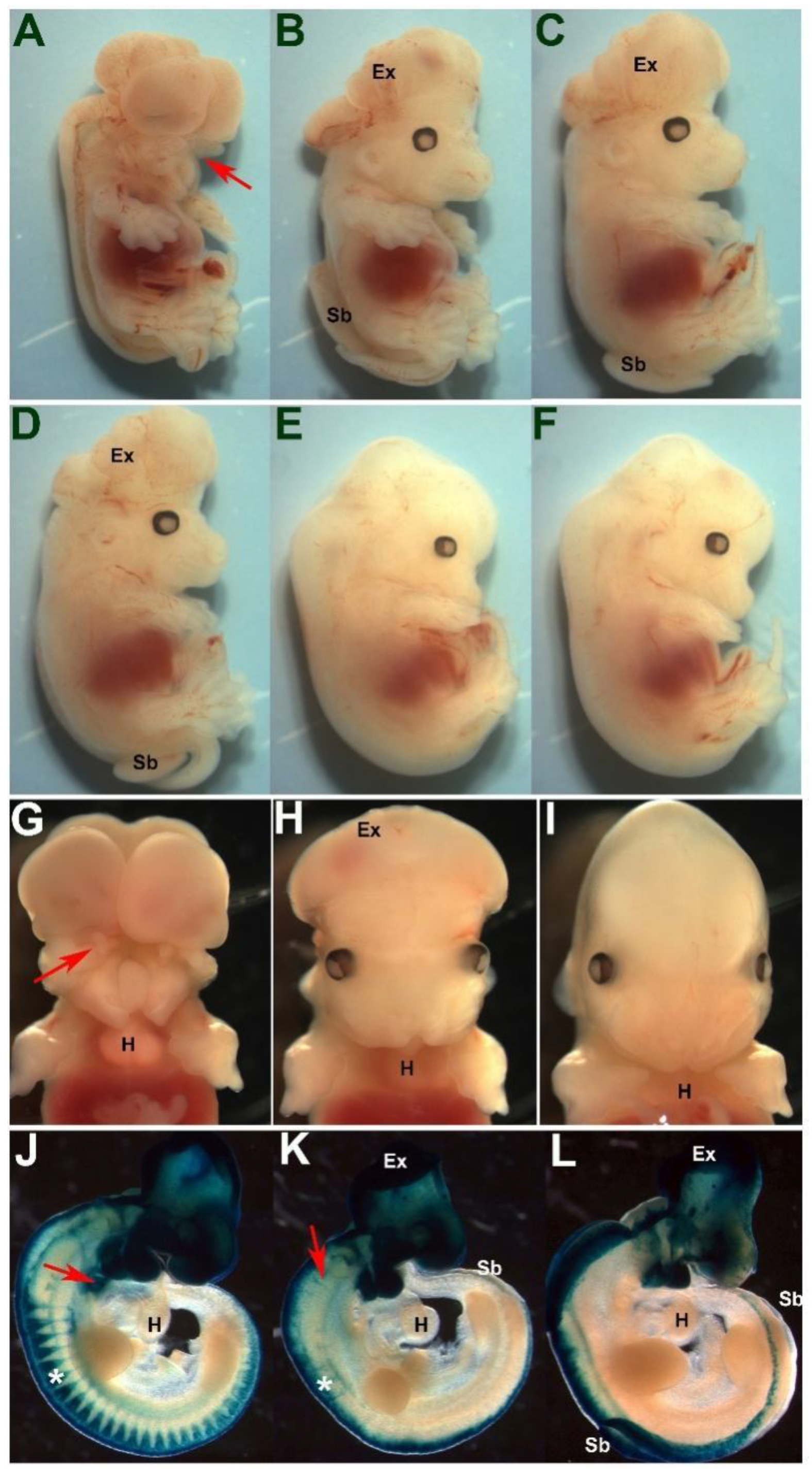
3.1. Select Neural Crest Lineages Are Resistant to a Drop in Pax3 Levels
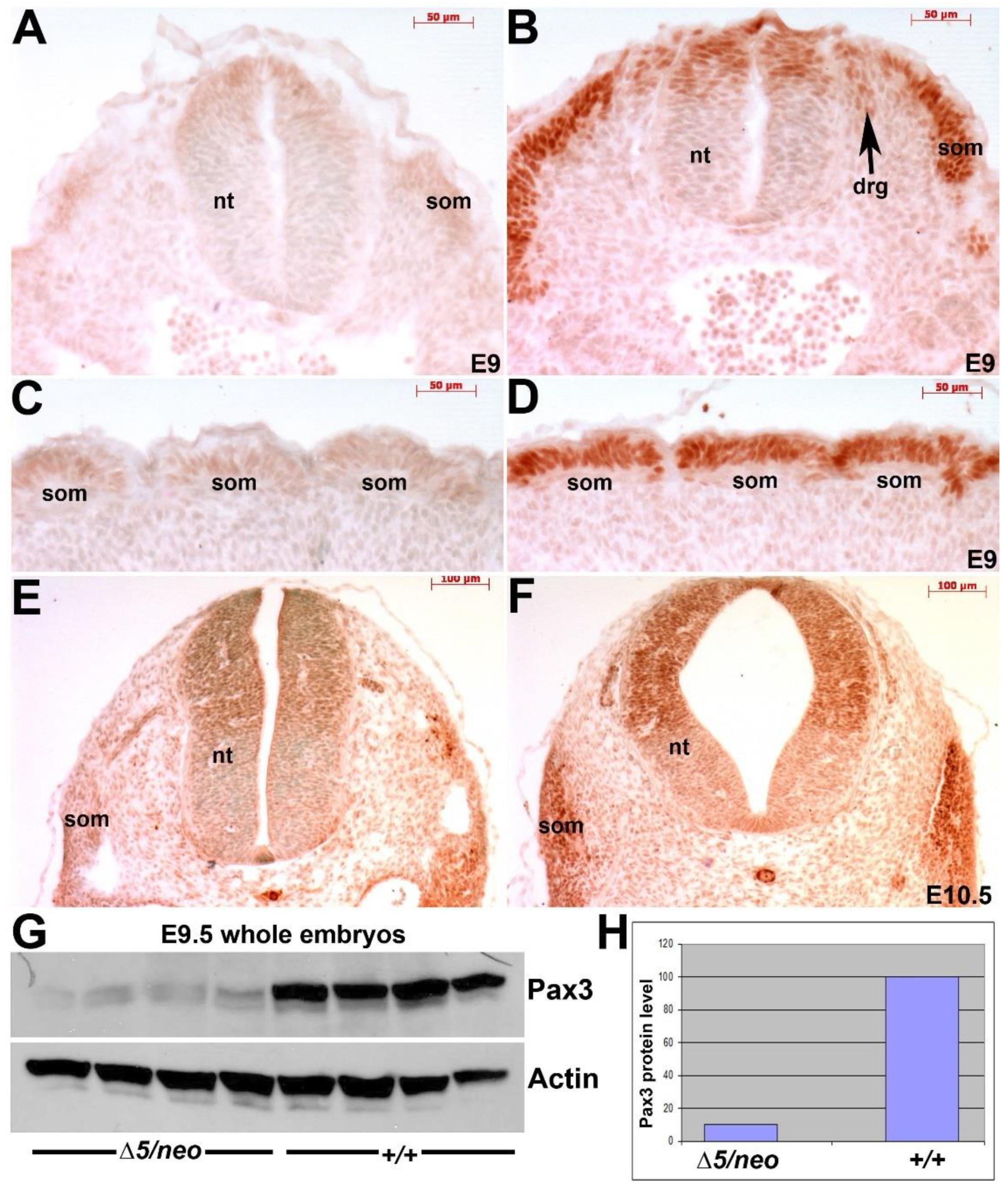
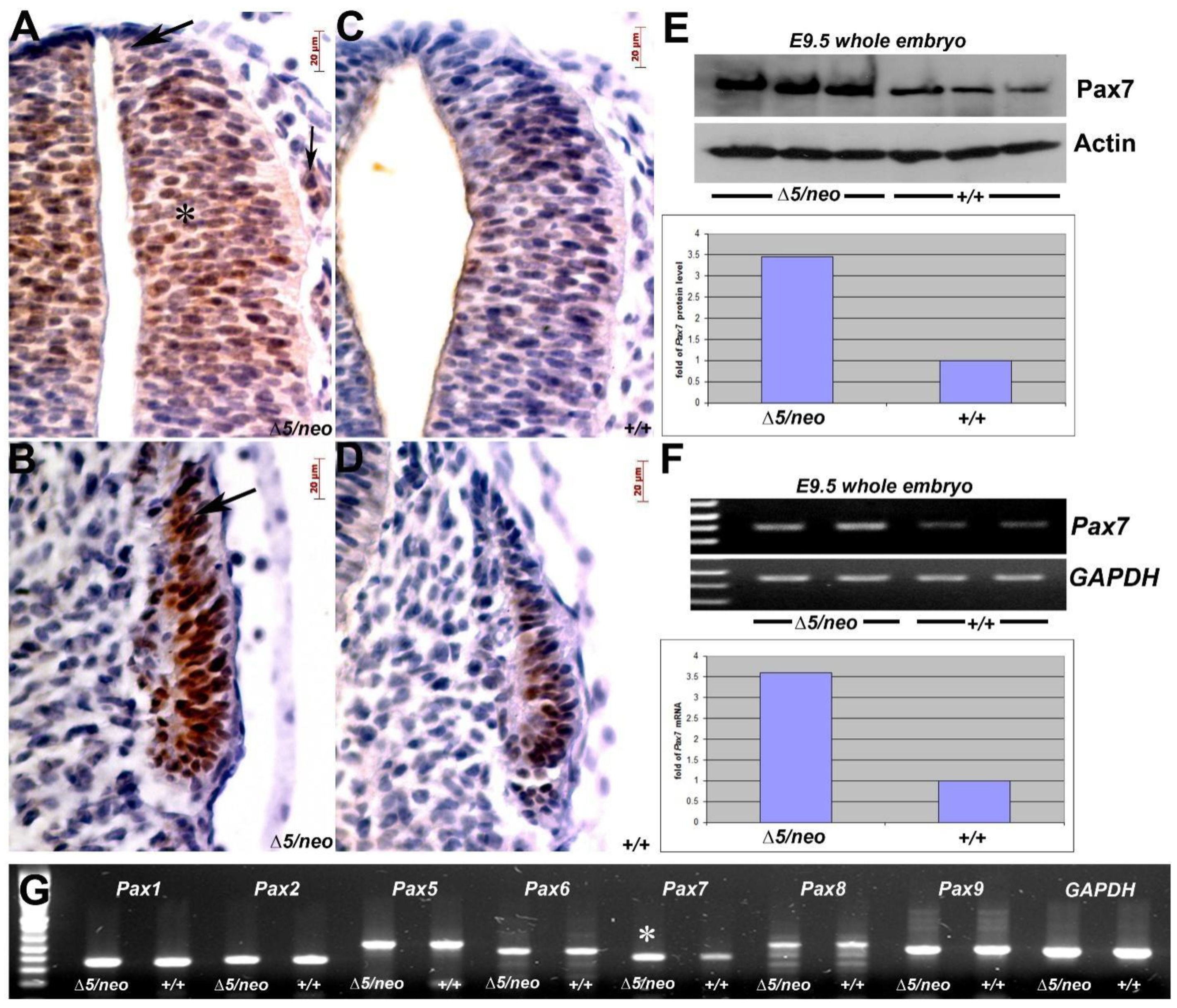
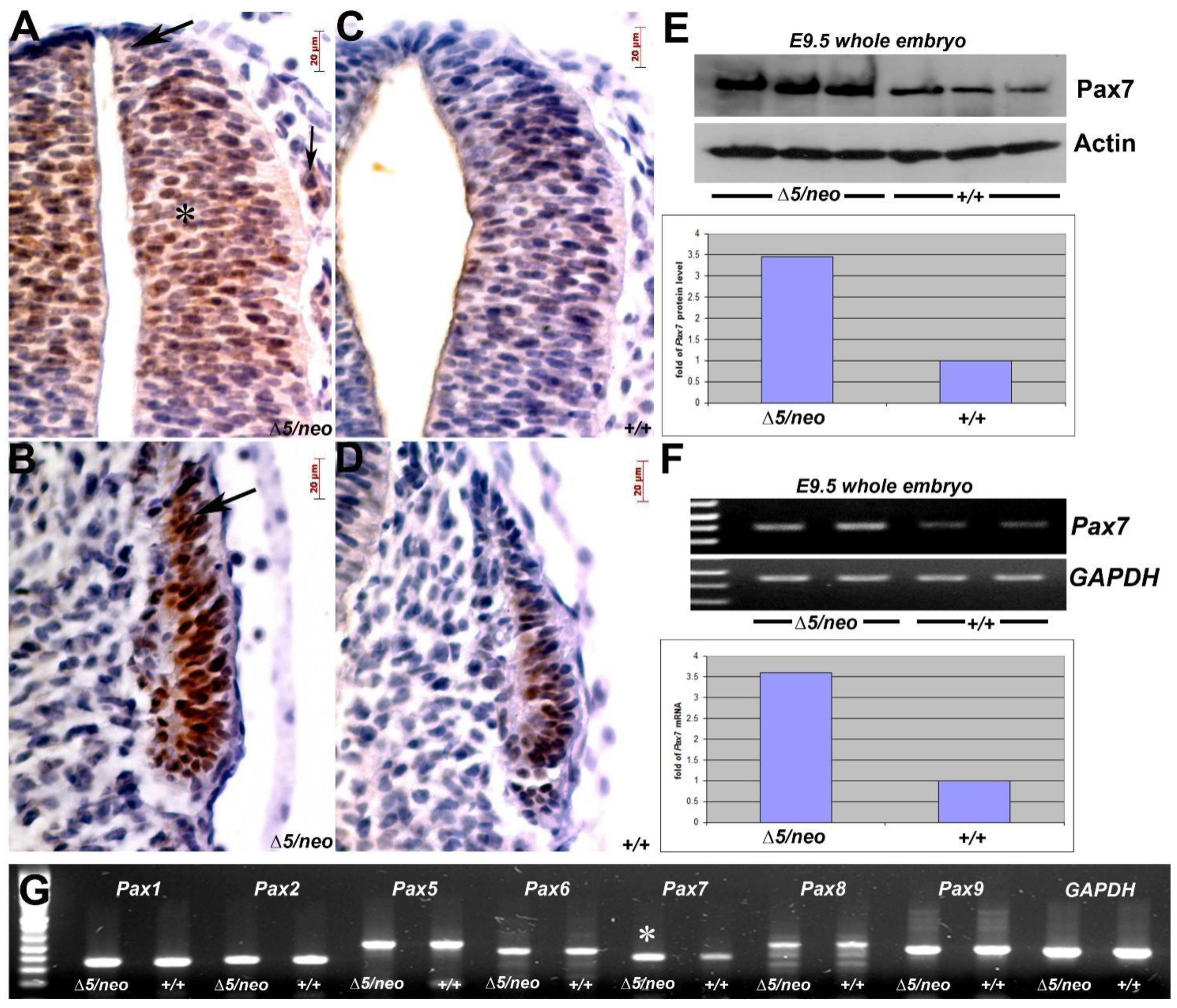
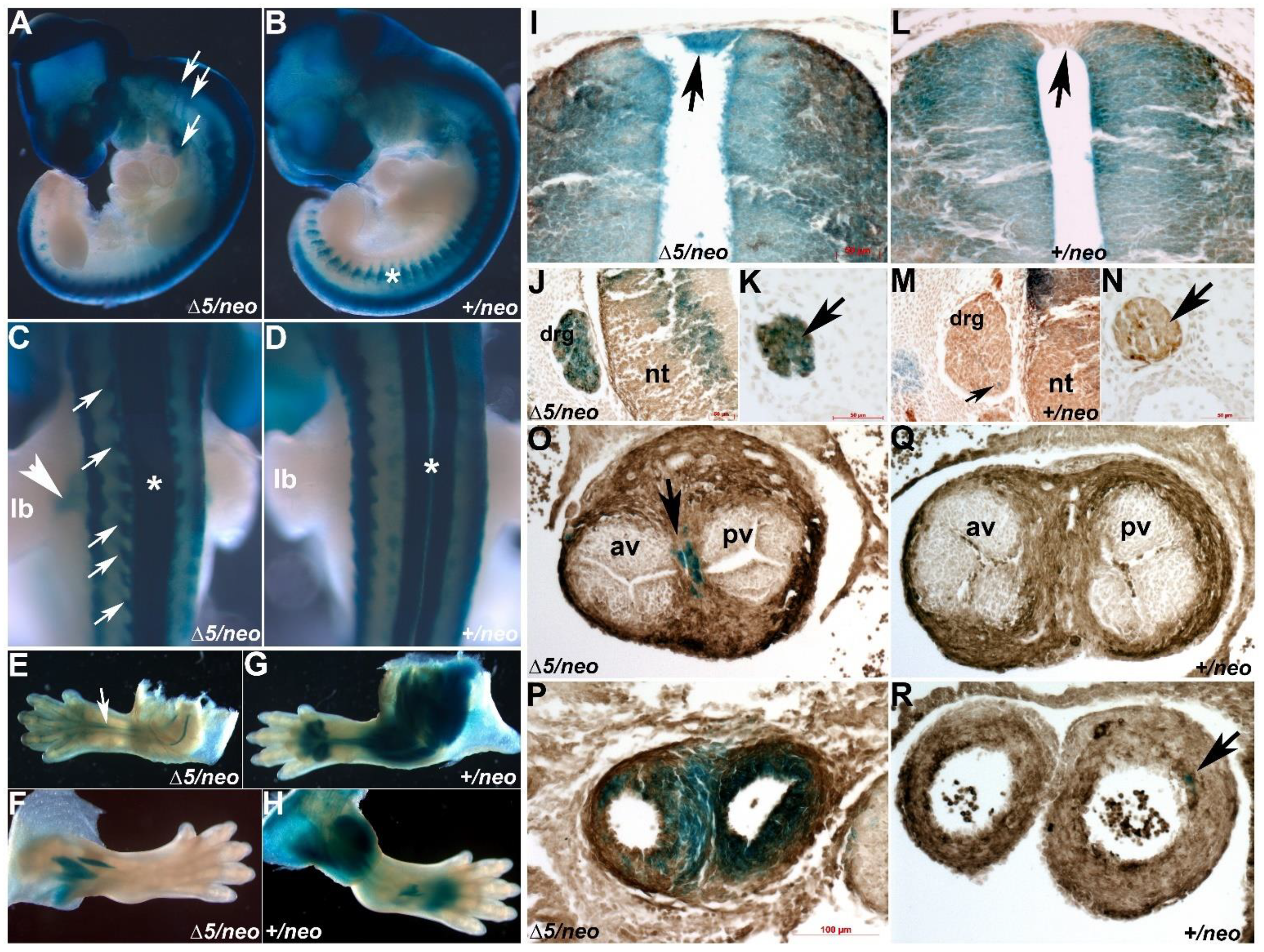
3.2. Reduction in Pax3 Expression Elicits Ectopic and Elevated Pax7 Expression
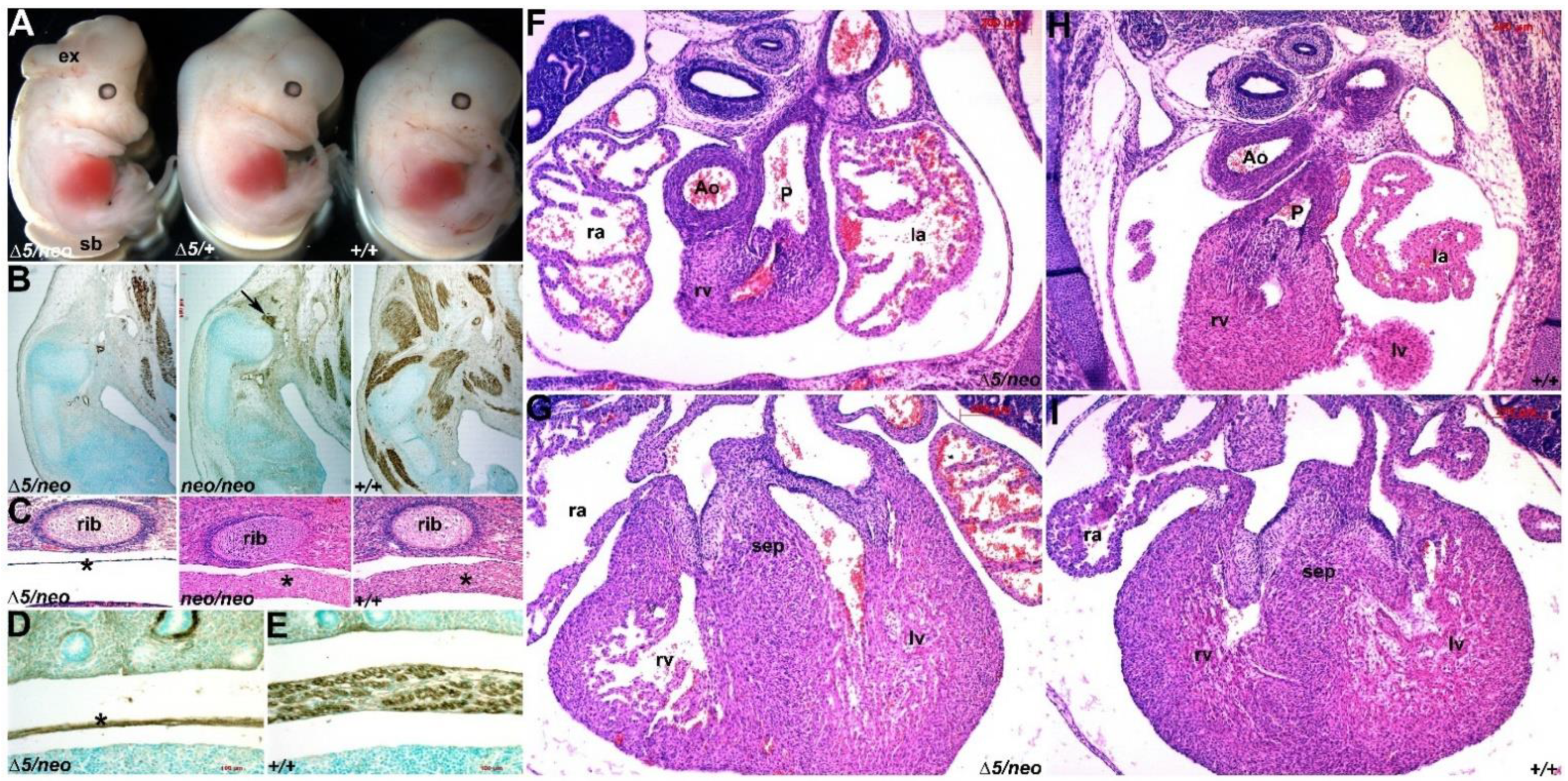
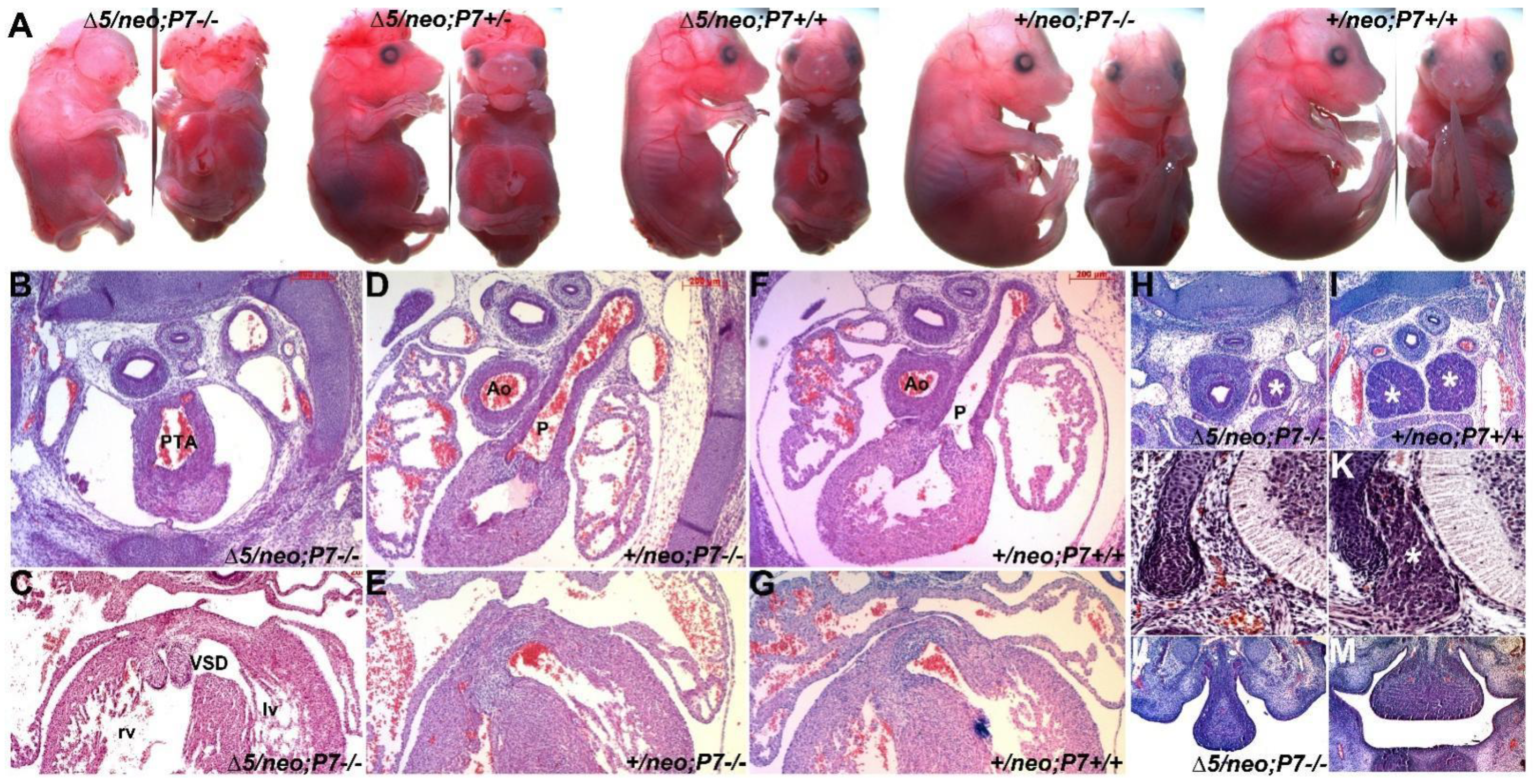
3.3. Pax7 Plays a Functional Genetic Compensation Role in Heart and Neural Tube Development
3.4. Pax7-Expressing Cell Lineage Is Ectopically Present in Neural Crest Derivative
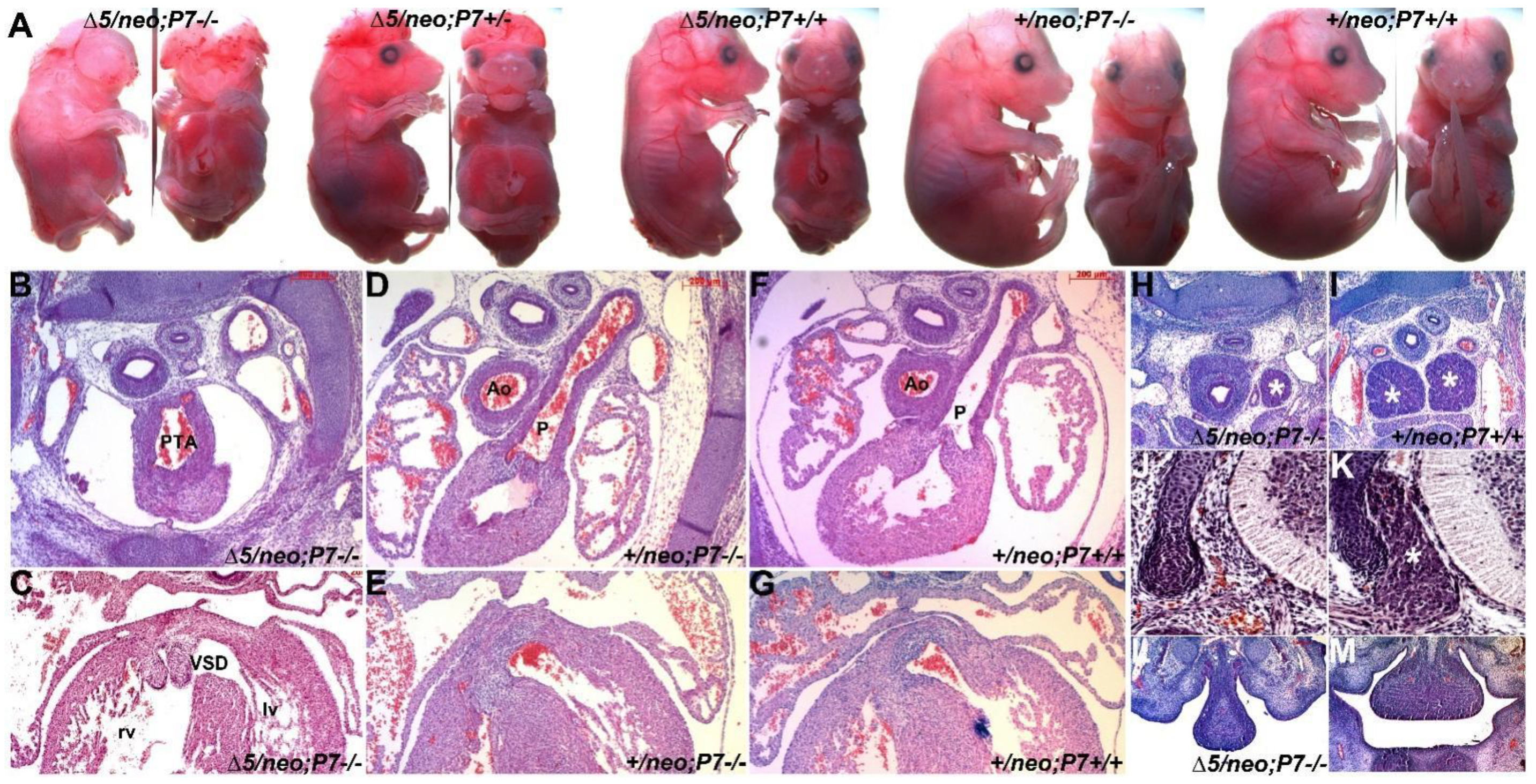
3.5. Pax3/7 Double Nulls Exhibit Exacerbated Defects
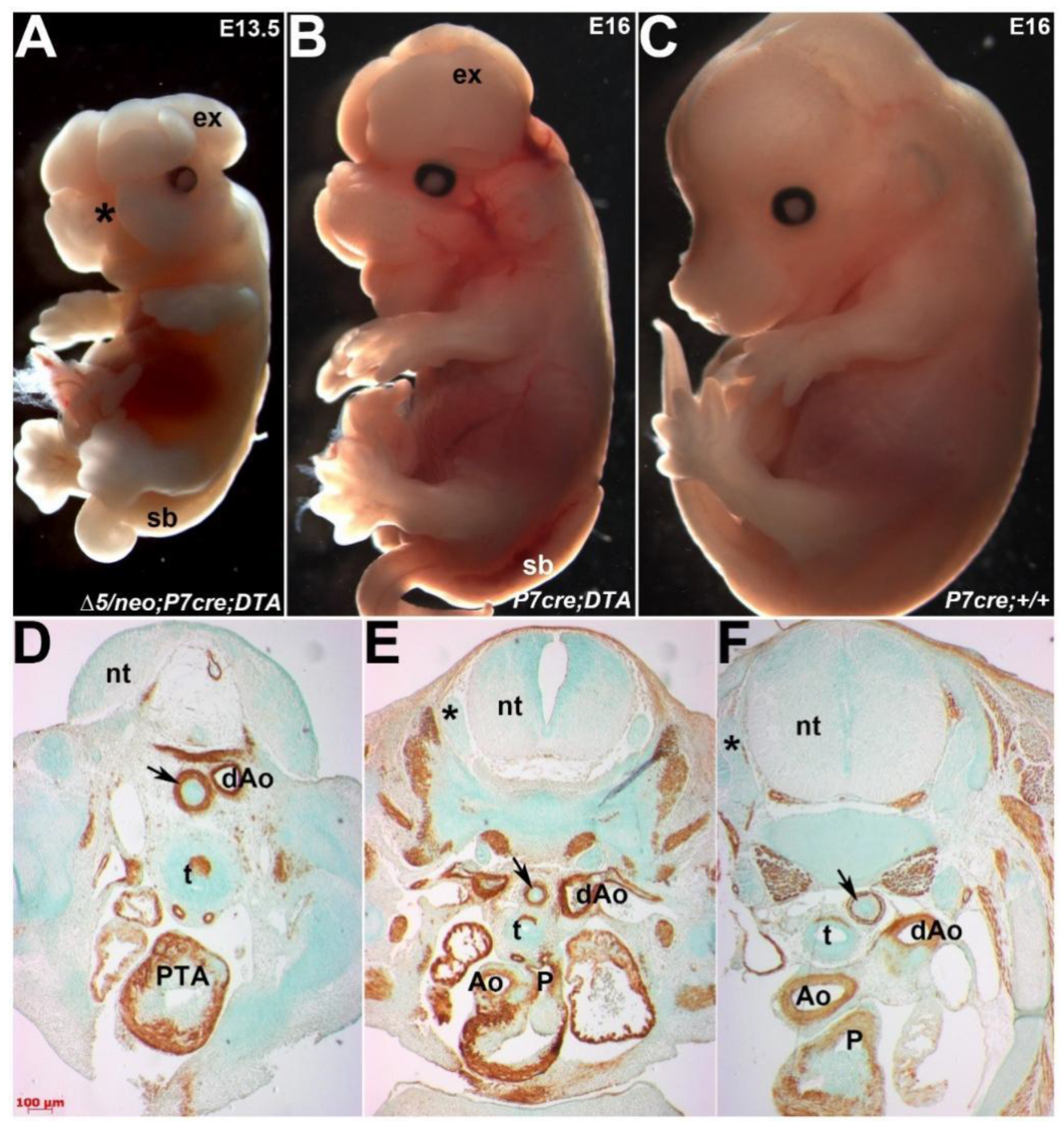
3.6. Ectopic Pax7-Expressing Lineage Is Essential for Heart and Craniofacial Morphogenesis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Chi, N.; Epstein, J.A. Getting your Pax straight: Pax proteins in development and disease. Trends Genet. 2002, 18, 41–47. [Google Scholar] [CrossRef]
- Zhou, H.M.; Wang, J.; Rogers, R.; Conway, S.J. Lineage-specific responses to reduced embryonic Pax3 expression levels. Dev. Biol. 2008, 315, 369–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goupille, O.; Pallafacchina, G.; Relaix, F.; Conway, S.J.; Cumano, A.; Robert, B.; Montarras, D.; Buckingham, M. Characterization of Pax3-expressing cells from adult blood vessels. J. Cell Sci. 2011, 124, 3980–3988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monsoro-Burq, A.H. PAX transcription factors in neural crest development. Semin. Cell Dev. Biol. 2015, 44, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Thompson, B.; Davidson, E.A.; Liu, W.; Nebert, D.W.; Bruford, E.A.; Zhao, H.; Dermitzakis, E.T.; Thompson, D.C.; Vasiliou, V. Overview of PAX gene family: Analysis of human tissue-specific variant expression and involvement in human disease. Hum. Genet. 2021, 140, 381–400. [Google Scholar] [CrossRef]
- Jun, S.; Desplan, C. Cooperative interactions between paired domain and homeodomain. Development 1996, 122, 2639–2650. [Google Scholar] [CrossRef]
- Schäfer, B.W.; Czerny, T.; Bernasconi, M.; Genini, M.; Busslinger, M. Molecular cloning and characterization of a human PAX-7 cDNA expressed in normal and neoplastic myocytes. Nucleic Acids Res. 1994, 22, 4574–4582. [Google Scholar] [CrossRef] [Green Version]
- Short, S.; Holland, L.Z. The evolution of alternative splicing in the Pax family: The view from the Basal chordate amphioxus. J. Mol. Evol. 2008, 66, 605–620. [Google Scholar] [CrossRef]
- Goulding, M.D.; Chalepakis, G.; Deutsch, U.; Erselius, J.R.; Gruss, P. Pax-3, a novel murine DNA binding protein expressed during early neurogenesis. EMBO J. 1991, 10, 1135–1147. [Google Scholar] [CrossRef]
- Walther, C.; Guenet, J.L.; Simon, D.; Deutsch, U.; Jostes, B.; Goulding, M.D.; Plachov, D.; Balling, R.; Gruss, P. Pax: A murine multigene family of paired box-containing genes. Genomics 1991, 11, 424–434. [Google Scholar] [CrossRef]
- Mansouri, A.; Stoykova, A.; Torres, M.; Gruss, P. Dysgenesis of cephalic neural crest derivatives in Pax7-/-mutant mice. Development 1996, 122, 831–838. [Google Scholar] [CrossRef]
- Conway, S.J.; Henderson, D.J.; Copp, A.J. Pax3 is required for cardiac neural crest migration in the mouse: Evidence from the splotch (Sp2H) mutant. Development 1997, 124, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Olaopa, M.; Zhou, H.M.; Snider, P.; Wang, J.; Schwartz, R.J.; Moon, A.M.; Conway, S.J. Pax3 is essential for normal cardiac neural crest morphogenesis but is not required during migration nor outflow tract septation. Dev. Biol. 2011, 356, 308–322. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Liu, K.C.; Jin, F.; Lu, M.M.; Epstein, J.A. Transgenic rescue of congenital heart disease and spina bifida in Splotch mice. Development 1999, 126, 2495–2503. [Google Scholar] [CrossRef]
- Snider, P.; Olaopa, M.; Firulli, A.B.; Conway, S.J. Cardiovascular development and the colonizing cardiac neural crest lineage. Sci. World J. 2007, 7, 1090–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borycki, A.G.; Li, J.; Jin, F.; Emerson, C.P.; Epstein, J.A. Pax3 functions in cell survival and in Pax7 regulation. Development 1999, 126, 1665–1674. [Google Scholar] [CrossRef]
- Williams, R.M.; Lukoseviciute, M.; Sauka-Spengler, T.; Bronner, M.E. Single-cell atlas of early chick development reveals gradual segregation of neural crest lineage from the neural plate border during neurulation. eLife 2022, 11, e74464. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, B.; DelConte, C.; García-Castro, M.I. Pax7 lineage contributions to the mammalian neural crest. PLoS ONE. 2012, 7, e41089. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.M.; Conway, S.J. Restricted Pax3 Deletion within the Neural Tube Results in Congenital Hydrocephalus. J. Dev. Biol. 2016, 4, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mansouri, A.; Gruss, P. Pax3 and Pax7 are expressed in commissural neurons and restrict ventral neuronal identity in the spinal cord. Mech Dev. 1998, 78, 171–178. [Google Scholar] [CrossRef]
- Relaix, F.; Rocancourt, D.; Mansouri, A.; Buckingham, M. Divergent functions of murine Pax3 and Pax7 in limb muscle development. Genes Dev. 2004, 18, 1088–1105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schubert, F.R.; Tremblay, P.; Mansouri, A.; Faisst, A.M.; Kammandel, B.; Lumsden, A.; Gruss, P.; Dietrich, S. Early mesodermal phenotypes in splotch suggest a role for Pax3 in the formation of epithelial somites. Dev. Dyn. 2001, 222, 506–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crist, C.G.; Montarras, D.; Pallafacchina, G.; Rocancourt, D.; Cumano, A.; Conway, S.J.; Buckingham, M. Muscle stem cell behavior is modified by microRNA-27 regulation of Pax3 expression. Proc. Natl. Acad. Sci USA 2009, 106, 13383–13387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Epstein, J.A.; Li, J.; Lang, D.; Chen, F.; Brown, C.B.; Jin, F.; Lu, M.M.; Thomas, M.; Liu, E.; Wessels, A.; et al. Migration of cardiac neural crest cells in Splotch embryos. Development 2000, 127, 1869–1878. [Google Scholar] [CrossRef] [PubMed]
- Lang, D.; Chen, F.; Milewski, R.; Li, J.; Lu, M.M.; Epstein, J.A. Pax3 is required for enteric ganglia formation and functions with Sox10 to modulate expression of c-ret. J. Clin. Investig. 2000, 106, 963–971. [Google Scholar] [CrossRef] [Green Version]
- Koushik, S.V.; Chen, H.; Wang, J.; Conway, S.J. Generation of a conditional loxP allele of the Pax3 transcription factor that enables selective deletion of the homeodomain. Genesis 2002, 32, 114–117. [Google Scholar] [CrossRef]
- Lepper, C.; Conway, S.J.; Fan, C.M. Adult satellite cells and embryonic muscle progenitors have distinct genetic requirements. Nature 2009, 460, 627–631. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Rowitch, D.H.; Soriano, P.; McMahon, A.P.; Sucov, H.M. Fate of the mammalian cardiac neural crest. Development 2000, 127, 1607–1616. [Google Scholar] [CrossRef]
- Keller, C.; Hansen, M.S.; Coffin, C.M.; Capecchi, M.R. Pax3:Fkhr interferes with embryonic Pax3 and Pax7 function: Implications for alveolar rhabdomyosarcoma cell of origin. Genes Dev. 2004, 18, 2608–2613. [Google Scholar] [CrossRef] [Green Version]
- Ivanova, A.; Signore, M.; Caro, N.; Greene, N.D.; Copp, A.J.; Martinez-Barbera, J.P. In vivo genetic ablation by Cre-mediated expression of diphtheria toxin fragment A. Genesis 2005, 43, 129–135. [Google Scholar] [CrossRef] [Green Version]
- Snider, P.; Tang, S.; Lin, G.; Wang, J.; Conway, S.J. Generation of Smad7(-Cre) recombinase mice: A useful tool for the study of epithelial-mesenchymal transformation within the embryonic heart. Genesis 2009, 47, 469–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engleka, K.A.; Gitler, A.D.; Zhang, M.; Zhou, D.D.; High, F.A.; Epstein, J.A. Insertion of Cre into the Pax3 locus creates a new allele of Splotch and identifies unexpected Pax3 derivatives. Dev. Biol. 2005, 280, 396–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conway, S.J.; Bundy, J.; Chen, J.; Dickman, E.; Rogers, R.; Will, B.M. Decreased neural crest stem cell expansion is responsible for the conotruncal heart defects within the splotch (Sp(2H))/Pax3 mouse mutant. Cardiovasc. Res. 2000, 47, 314–328. [Google Scholar] [CrossRef] [Green Version]
- Hunt, P.; Wilkinson, D.; Krumlauf, R. Patterning the vertebrate head: Murine Hox 2 genes mark distinct subpopulations of premigratory and migrating cranial neural crest. Development 1991, 112, 43–50. [Google Scholar] [CrossRef]
- Nikolopoulou, E.; Galea, G.L.; Rolo, A.; Greene, N.D.; Copp, A.J. Neural tube closure: Cellular, molecular and biomechanical mechanisms. Development 2017, 144, 552–566. [Google Scholar] [CrossRef] [Green Version]
- Monsoro-Burq, A.H.; Wang, E.; Harland, R. Msx1 and Pax3 cooperate to mediate FGF8 and WNT signals during Xenopus neural crest induction. Dev. Cell 2005, 8, 167–178. [Google Scholar] [CrossRef] [Green Version]
- Basch, M.L.; Bronner-Fraser, M.; García-Castro, M.I. Specification of the neural crest occurs during gastrulation and requires Pax7. Nature 2006, 441, 218–222. [Google Scholar] [CrossRef]
- Pani, L.; Horal, M.; Loeken, M.R. Rescue of neural tube defects in Pax-3-deficient embryos by p53 loss of function: Implications for Pax-3- dependent development and tumorigenesis. Genes Dev. 2002, 16, 676–680. [Google Scholar] [CrossRef] [Green Version]
- Boudjadi, S.; Chatterjee, B.; Sun, W.; Vemu, P.; Barr, F.G. The expression and function of PAX3 in development and disease. Gene 2018, 666, 145–157. [Google Scholar] [CrossRef]
- White, R.B.; Ziman, M.R. Genome-wide discovery of Pax7 target genes during development. Physiol. Genom. 2008, 33, 41–49. [Google Scholar] [CrossRef] [Green Version]
- Bennicelli, J.L.; Advani, S.; Schäfer, B.W.; Barr, F.G. PAX3 and PAX7 exhibit conserved cis-acting transcription repression domains and utilize a common gain of function mechanism in alveolar rhabdomyosarcoma. Oncogene 1999, 18, 4348–4356. [Google Scholar] [CrossRef] [Green Version]
- Soleimani, V.D.; Punch, V.G.; Kawabe, Y.; Jones, A.E.; Palidwor, G.A.; Porter, C.J.; Cross, J.W.; Carvajal, J.J.; Kockx, C.E.; van IJcken, W.F.; et al. Transcriptional dominance of Pax7 in adult myogenesis is due to high-affinity recognition of homeodomain motifs. Dev. Cell 2012, 22, 1208–1220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gard, C.; Gonzalez Curto, G.; Frarma, Y.E.; Chollet, E.; Duval, N.; Auzié, V.; Auradé, F.; Vigier, L.; Relaix, F.; Pierani, A.; et al. Pax3- and Pax7-mediated Dbx1 regulation orchestrates the patterning of intermediate spinal interneurons. Dev. Biol. 2017, 432, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Gan, Q.; Stokes, A.; Lassiter, R.N.; Wang, Y.; Chan, J.; Han, J.X.; Pleasure, D.E.; Epstein, J.A.; Zhou, C.J. β-catenin regulates Pax3 and Cdx2 for caudal neural tube closure and elongation. Development 2014, 141, 148–157. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, L.; Hulin, J.A.; Gromova, A.; Tran Nguyen, T.D.; Yu, R.T.; Liddle, C.; Downes, M.; Evans, R.M.; Makarenkova, H.P.; Meech, R. Barx2 and Pax7 have antagonistic functions in regulation of wnt signaling and satellite cell differentiation. Stem Cells 2014, 32, 1661–1673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwang, S.J.; Brugger, S.M.; Lazik, A.; Merrill, A.E.; Wu, L.Y.; Liu, Y.H.; Ishii, M.; Sangiorgi, F.O.; Rauchman, M.; Sucov, H.M.; et al. Msx2 is an immediate downstream effector of Pax3 in the development of the murine cardiac neural crest. Development 2002, 129, 527–538. [Google Scholar] [CrossRef]
- Duval, N.; Daubas, P.; Bourcier de Carbon, C.; St Cloment, C.; Tinevez, J.Y.; Lopes, M.; Ribes, V.; Robert, B. Msx1 and Msx2 act as essential activators of Atoh1 expression in the murine spinal cord. Development 2014, 141, 1726–1736. [Google Scholar] [CrossRef] [Green Version]
- Zalc, A.; Rattenbach, R.; Auradé, F.; Cadot, B.; Relaix, F. Pax3 and Pax7 play essential safeguard functions against environmental stress-induced birth defects. Dev. Cell 2015, 33, 56–66. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Li, J.; Engleka, K.A.; Zhou, B.; Lu, M.M.; Plotkin, J.B.; Epstein, J.A. Persistent expression of Pax3 in the neural crest causes cleft palate and defective osteogenesis in mice. J. Clin. Investig. 2008, 118, 2076–2087. [Google Scholar] [CrossRef] [Green Version]
- Leslie, E.J.; Marazita, M.L. Genetics of cleft lip and cleft palate. Am. J. Med. Genet. C Semin Med. Genet. 2013, 163C, 246–258. [Google Scholar] [CrossRef] [Green Version]
- van Raamsdonk, C.D.; Tilghman, S.M. Dosage requirement and allelic expression of PAX6 during lens placode formation. Development 2000, 127, 5439–5448. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, S.; Rocancourt, D.; Buckingham, M.; Relaix, F. Lack of in vivo functional compensation between Pax family groups II and III in rodents. Mol. Biol. Evol. 2011, 28, 2787–2798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corry, G.N.; Raghuram, N.; Missiaen, K.K.; Hu, N.; Hendzel, M.J.; Underhill, D.A. The PAX3 paired domain and homeodomain function as a single binding module in vivo to regulate subnuclear localization and mobility by a mechanism that requires base-specific recognition. J. Mol. Biol. 2010, 402, 178–193. [Google Scholar] [CrossRef] [PubMed]
- Prelich, G. Gene overexpression: Uses, mechanisms, and interpretation. Genetics 2012, 190, 841–854. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Trelles, F.; Tarrío, R.; Ayala, F.J. Is ectopic expression caused by deregulatory mutations or due to gene-regulation leaks with evolutionary potential? Bioessays 2005, 27, 592–601. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer | Reverse Primer | Size (bp) | AT (oC) | Cycle # |
|---|---|---|---|---|---|
| Pax1 | GACTGGGCGGGTGTGAACCG | GTGCATCCGTGGCCTTGCCT | 361 | 58 | 33 |
| Pax2 | CGGATGGGGCAGGGACAGGA | CTGCCCAGCTCAGGGTTGGC | 430 | 68 | 36 |
| Pax3 5′ | GTCTCGCCTTCACCTGGATA | GTTGTCACCTGCTTGGGTTT | 379 | 58 | 32 |
| Pax3 3′ | GATGGAGGAAACAAGCTGGA | GATGGAGGCACAAAGCTGTC | 304 | 58 | 32 |
| Pax3 exon5-6 | TTACCGCTGAAGAGGAAG | GTGTACAGTGCTCGGAGGAAG | 383 | 58 | 32 |
| Pax4 | GGCTCCCAGTGTGTCCTCTA | CCCATTTCAGCTTCTCTTGC | 350 | 64 | 36 |
| Pax5 | CAGGGACCGGCTGTTGGCAG | GGACTGTGGGCCTGGAACGC | 553 | 58 | 31 |
| Pax6 | CCGCAGCACTCGAGCACCAA | GAGCTGGTTGGCAGCCTCCG | 471 | 58 | 33 |
| Pax7 | AAGTGTCCACCCCTCTTGG | TGATTCTGAGCACTCGGCTA | 399 | 64 | 36 |
| Pax8 | AGGCCTATGCCTCCCCCAGC | GGAAGCGCCAGGCCTCACTG | 537 | 66 | 33 |
| Pax9 | GGGCCGGGTTGGTGTACTGC | GCGGGCTGGTGCTGCTTGTA | 474 | 66 | 31 |
| GAPDH | ACCACAGTCCATGCCATCAC | TCCACCACCCTGTTGCTGTA | 452 | 58 | 25 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, H.-M.; Conway, S.J. Pax3 Hypomorphs Reveal Hidden Pax7 Functional Genetic Compensation in Utero. J. Dev. Biol. 2022, 10, 19. https://doi.org/10.3390/jdb10020019
Zhou H-M, Conway SJ. Pax3 Hypomorphs Reveal Hidden Pax7 Functional Genetic Compensation in Utero. Journal of Developmental Biology. 2022; 10(2):19. https://doi.org/10.3390/jdb10020019
Chicago/Turabian StyleZhou, Hong-Ming, and Simon J. Conway. 2022. "Pax3 Hypomorphs Reveal Hidden Pax7 Functional Genetic Compensation in Utero" Journal of Developmental Biology 10, no. 2: 19. https://doi.org/10.3390/jdb10020019
APA StyleZhou, H.-M., & Conway, S. J. (2022). Pax3 Hypomorphs Reveal Hidden Pax7 Functional Genetic Compensation in Utero. Journal of Developmental Biology, 10(2), 19. https://doi.org/10.3390/jdb10020019