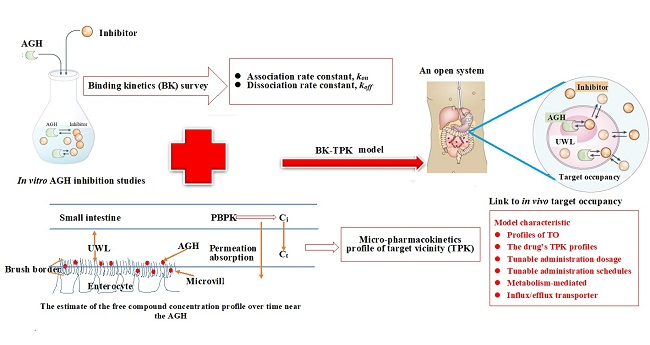
Utilizing the Combination of Binding Kinetics and Micro-Pharmacokinetics Link in Vitro α-Glucosidase Inhibition to in Vivo Target Occupancy

Abstract
:
1. Introduction
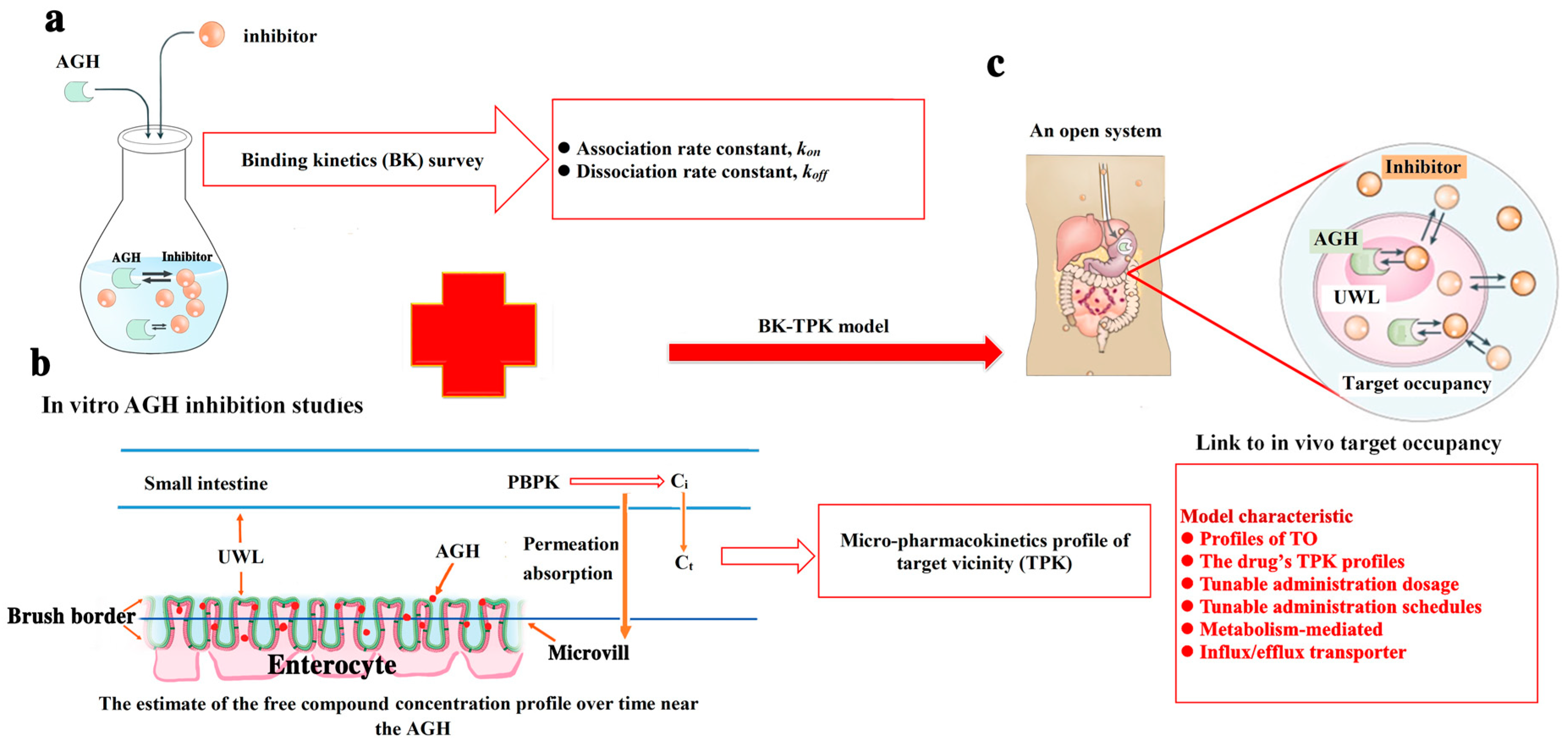
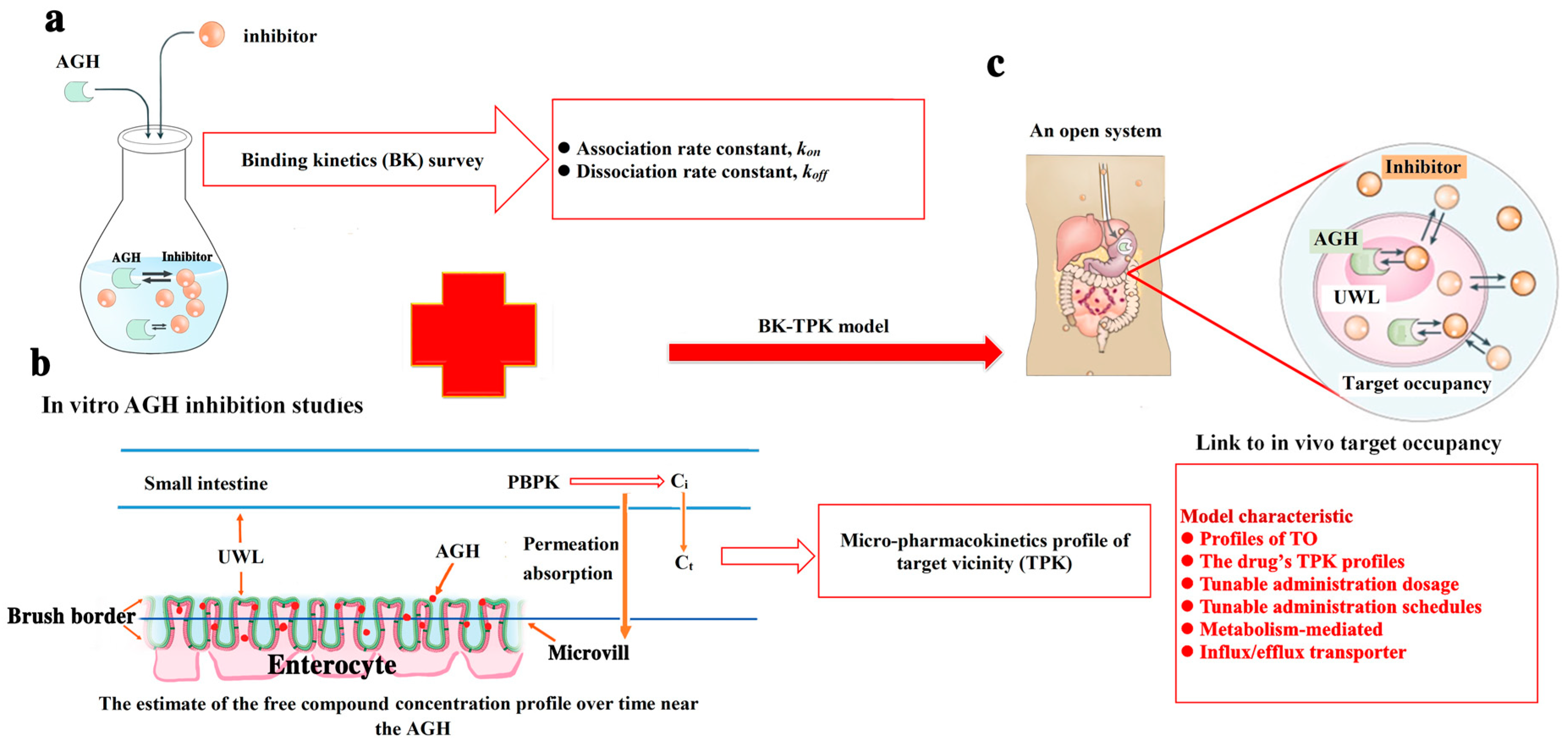
- Firstly, determining the values of kon and koff for two compounds investigated with AGH interaction.
- Secondly, micro-pharmacokinetics models of free compounds in target vicinity (TPK) were established to assess the compound concentrations changes over time in the UWL (i.e., target vicinity).
- Finally, TO simulation modelling was used to evaluate complex potential pharmacological effect of tested compounds by kon, koff in in vivo and the compound concentration versus time change in the biological target vicinity.
2. Materials and Methods
2.1. Chemicals and Materials
2.2. Assay for Steady-State AGH activity
2.3. IC50 Determination
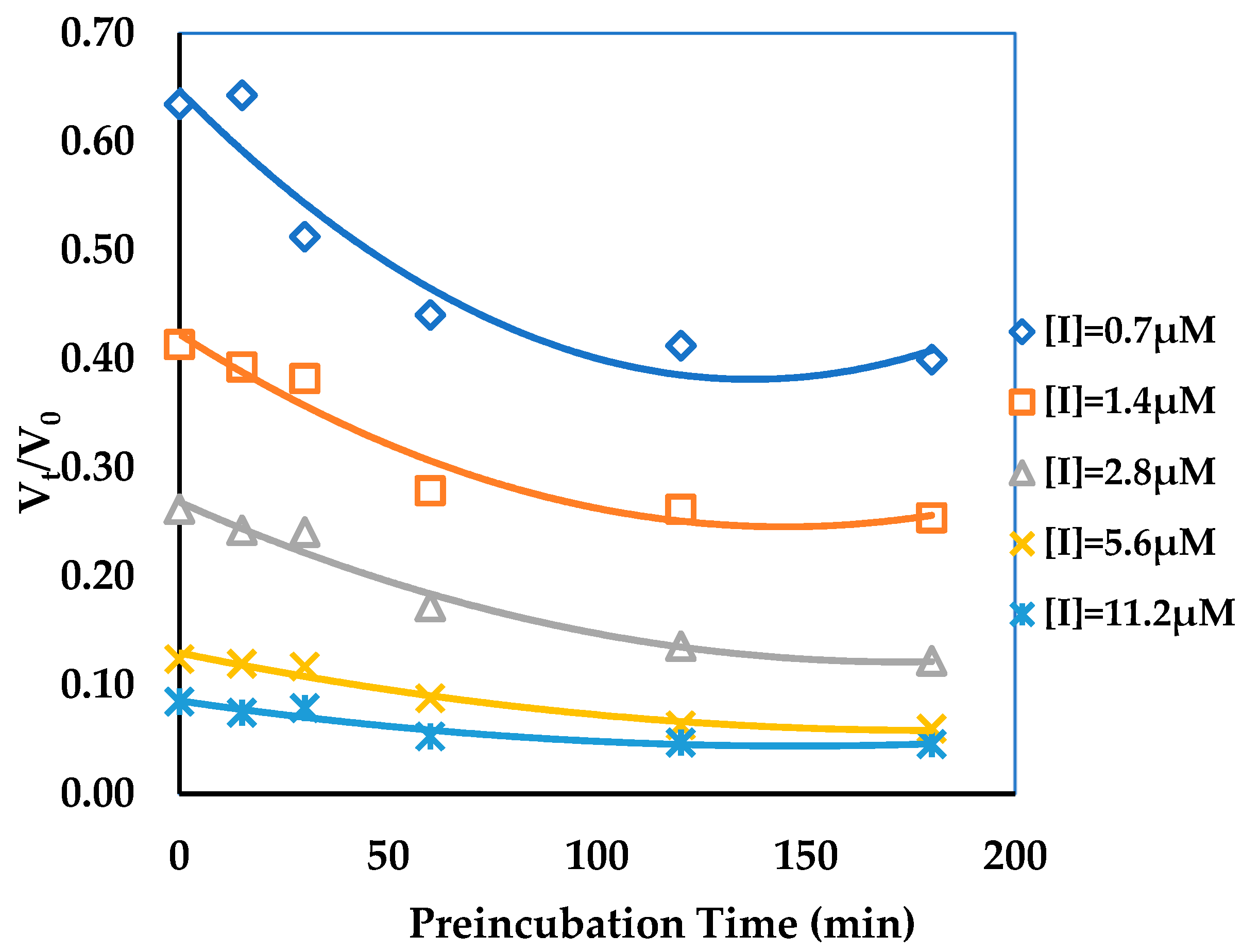
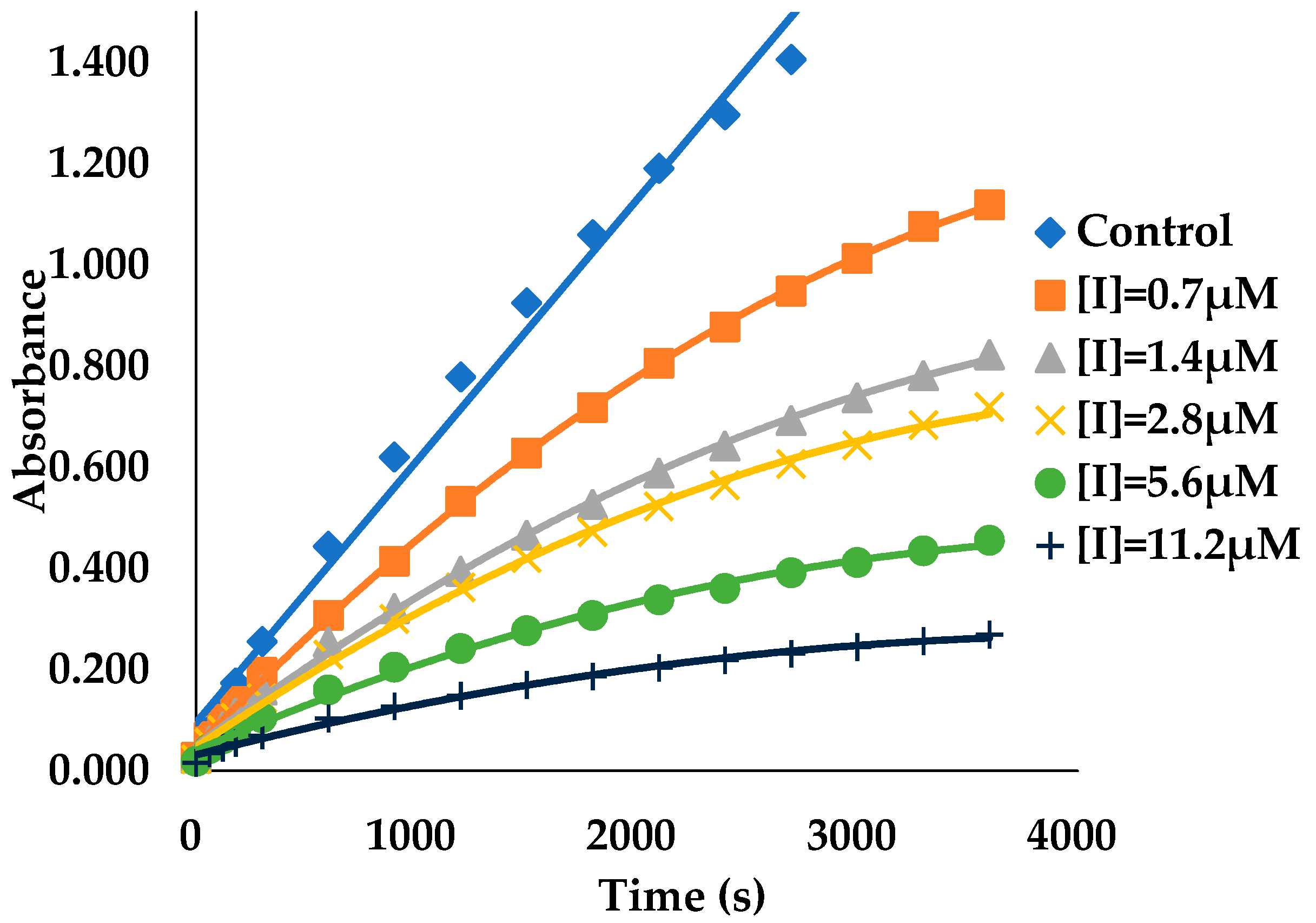
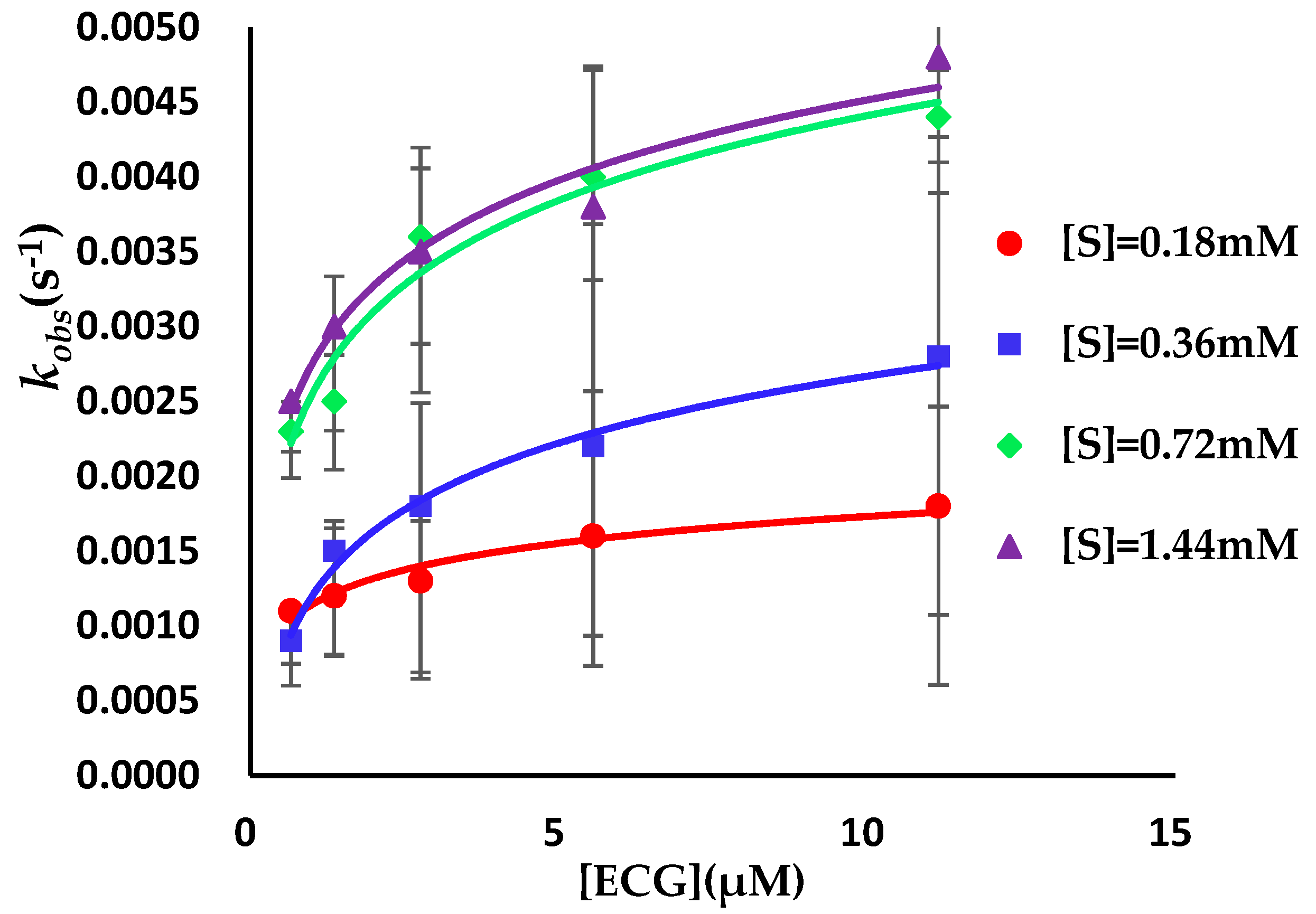
2.4. Binding Kinetics Survey of AGH
2.5. Micro-Pharmacokinetics Model of Free Compound in the Target Vicinity
2.5.1. The free Compound Concentration Versus Time Profile in the Intestine (Ci(t)) Using PBPK Model
2.5.2. The Free Compound Concentration Profile Over Time in the Unstirred Water Layer
2.6. The Development of BK-TPK Model
3. Results
3.1. Inhibition of Tea Polyphenols Against AGH
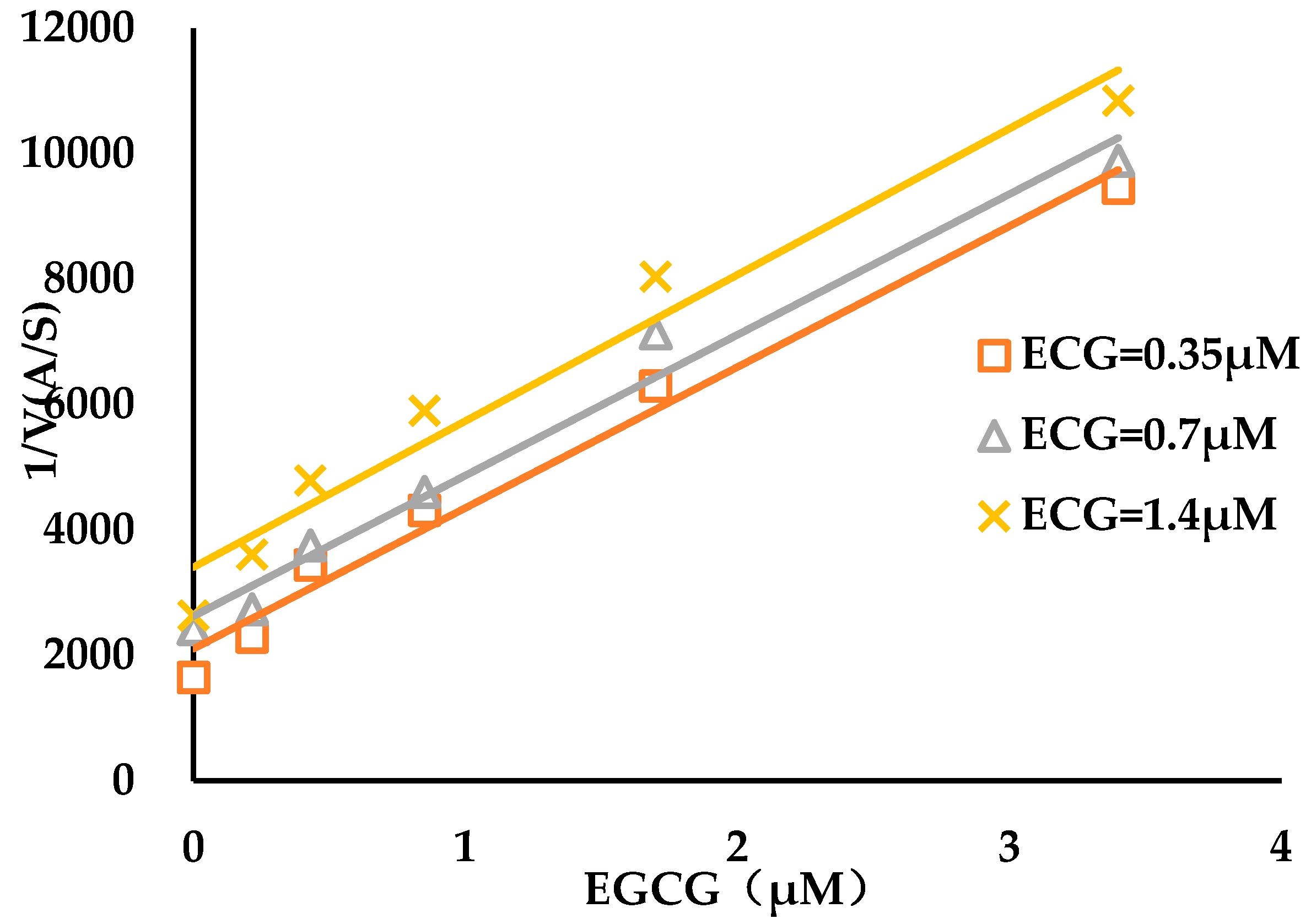
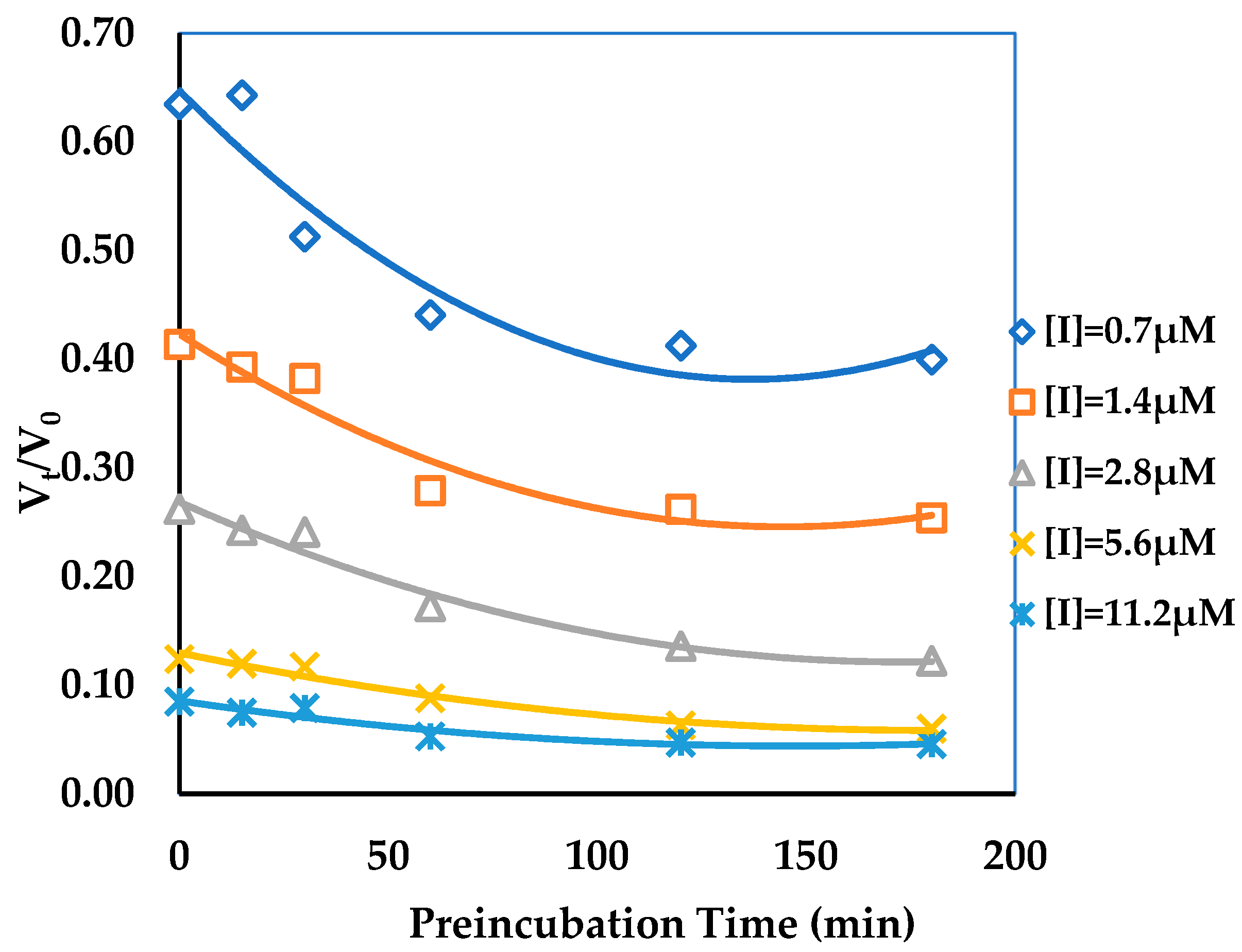
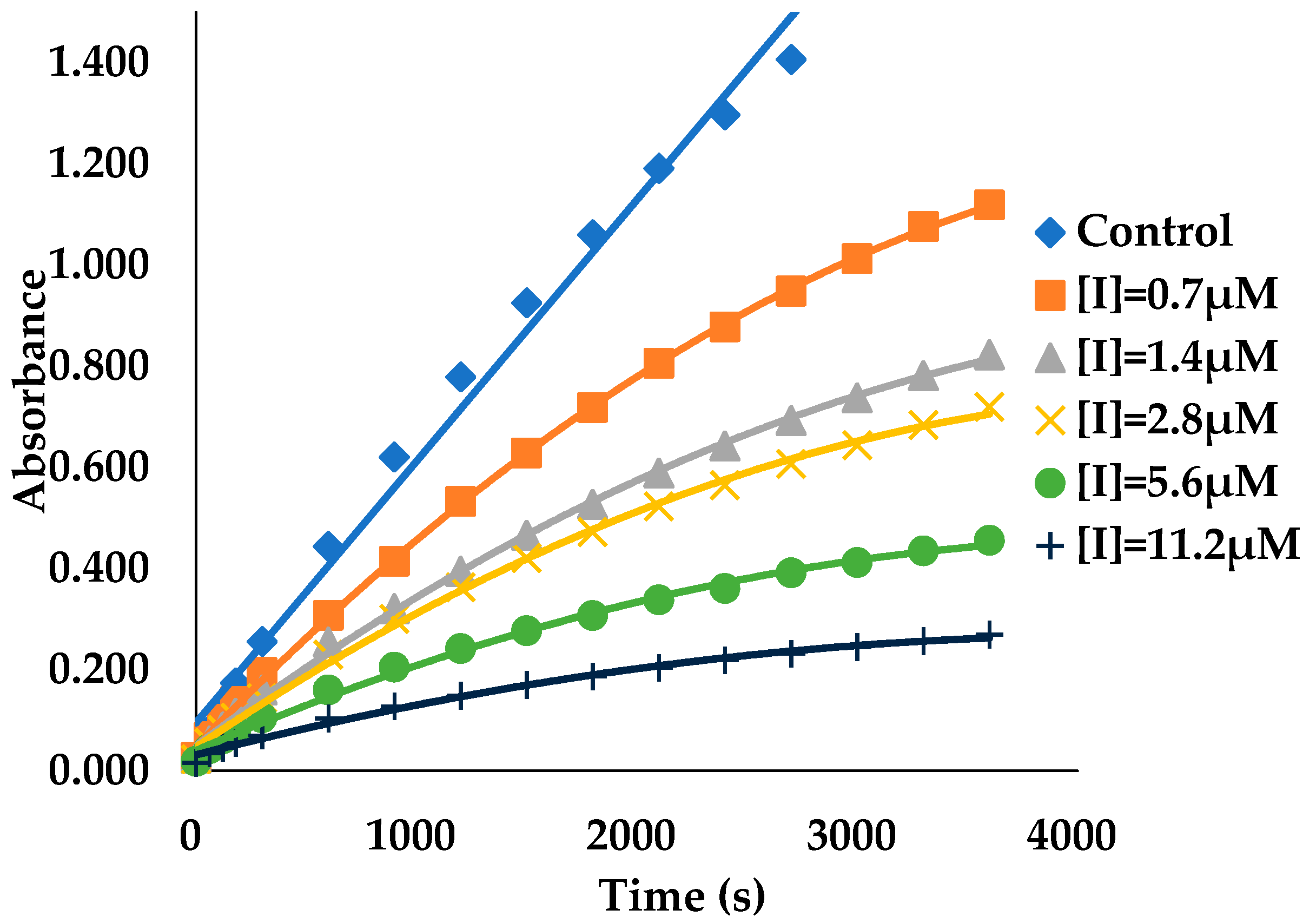
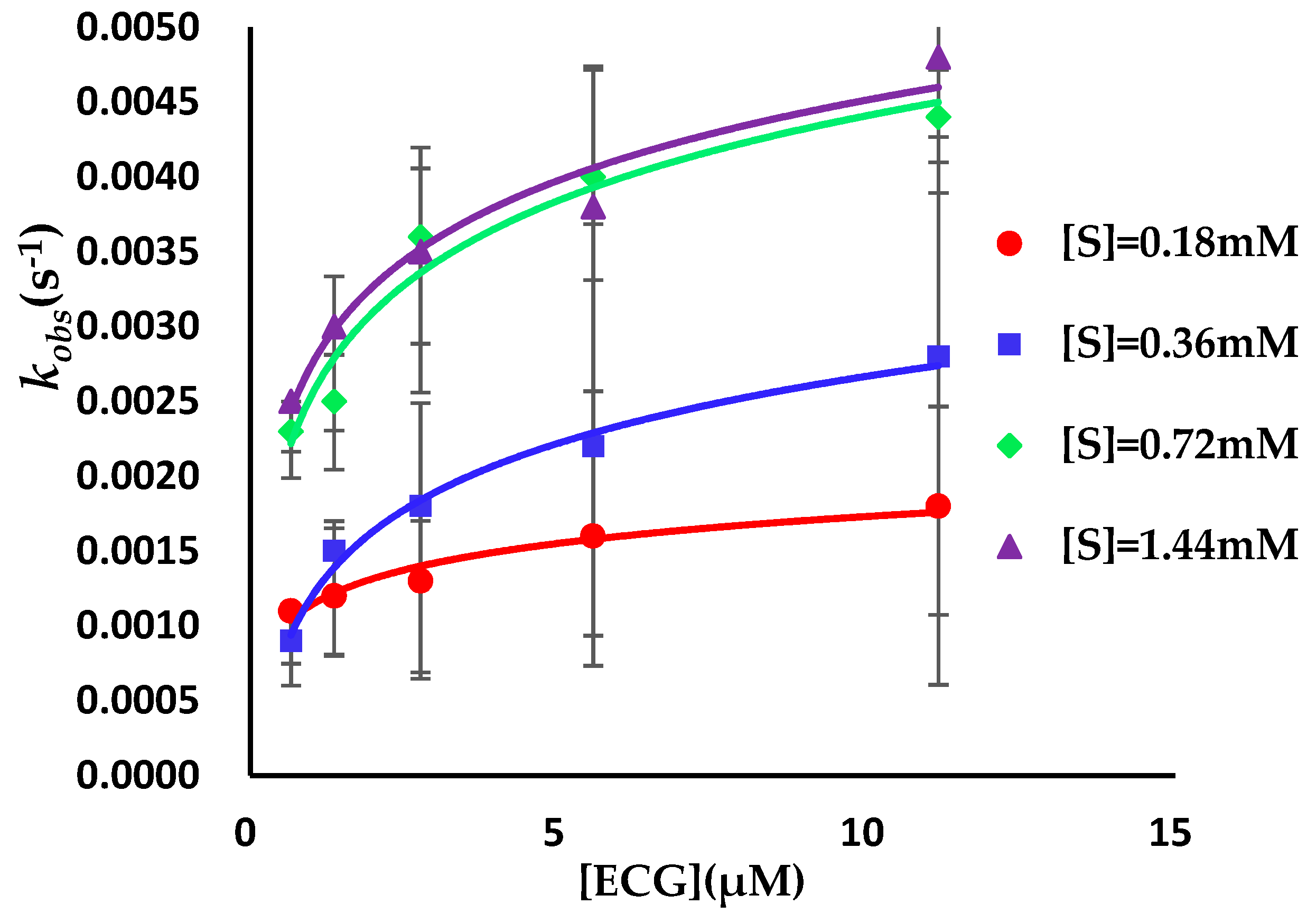
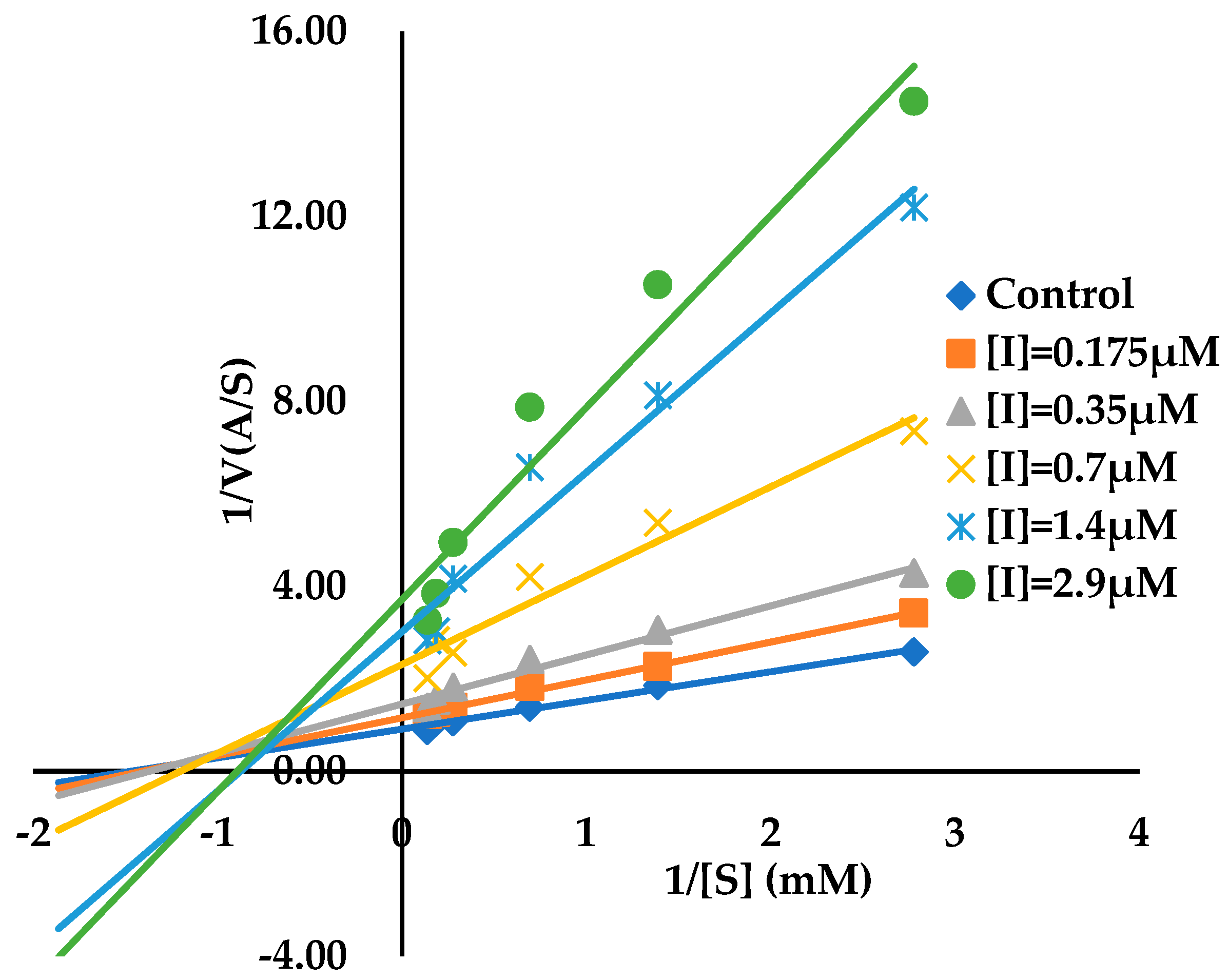
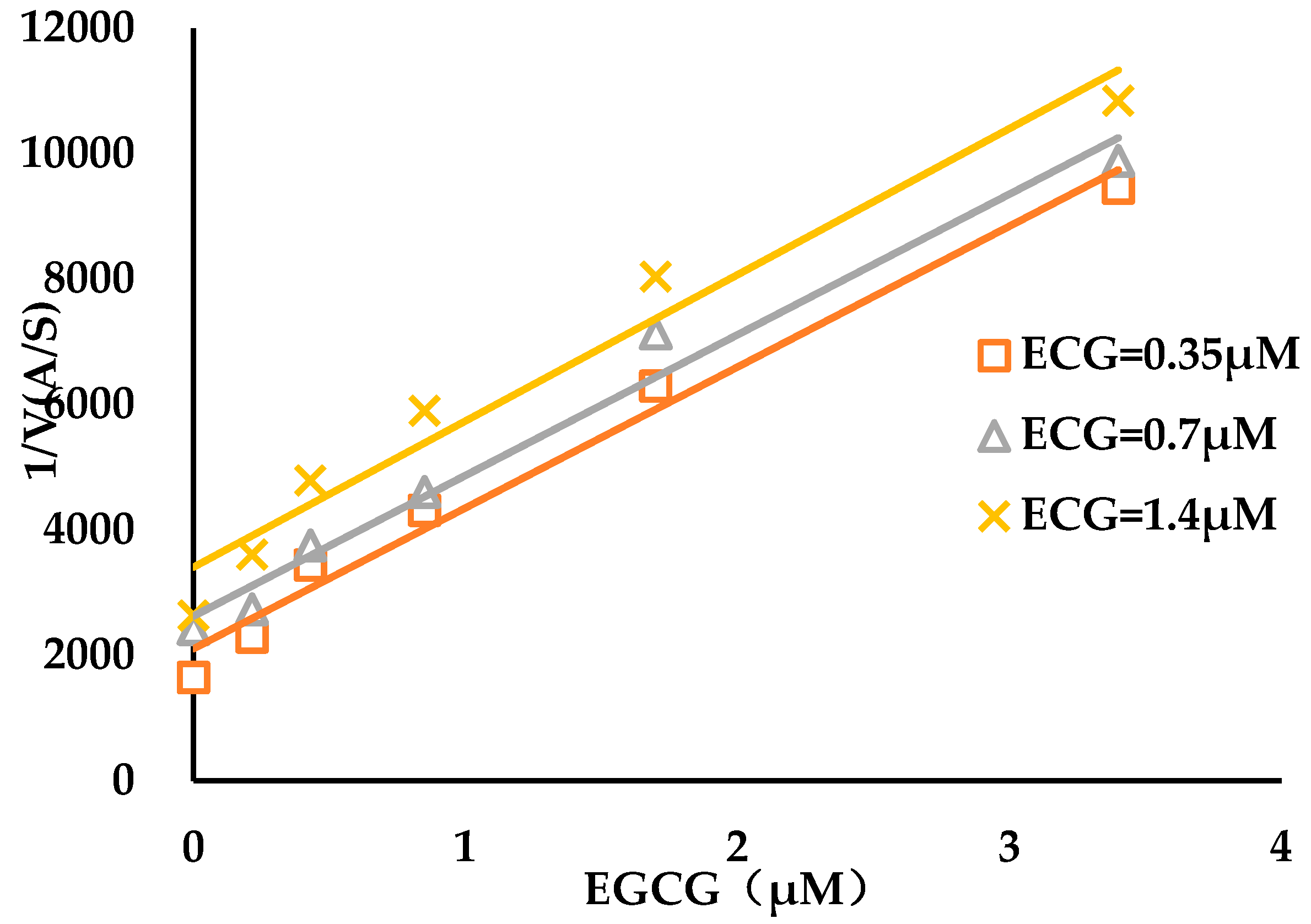
3.2. Binding Kinetics Survey of Tested Compounds Targeting the AGH by Progress Curve Analysis
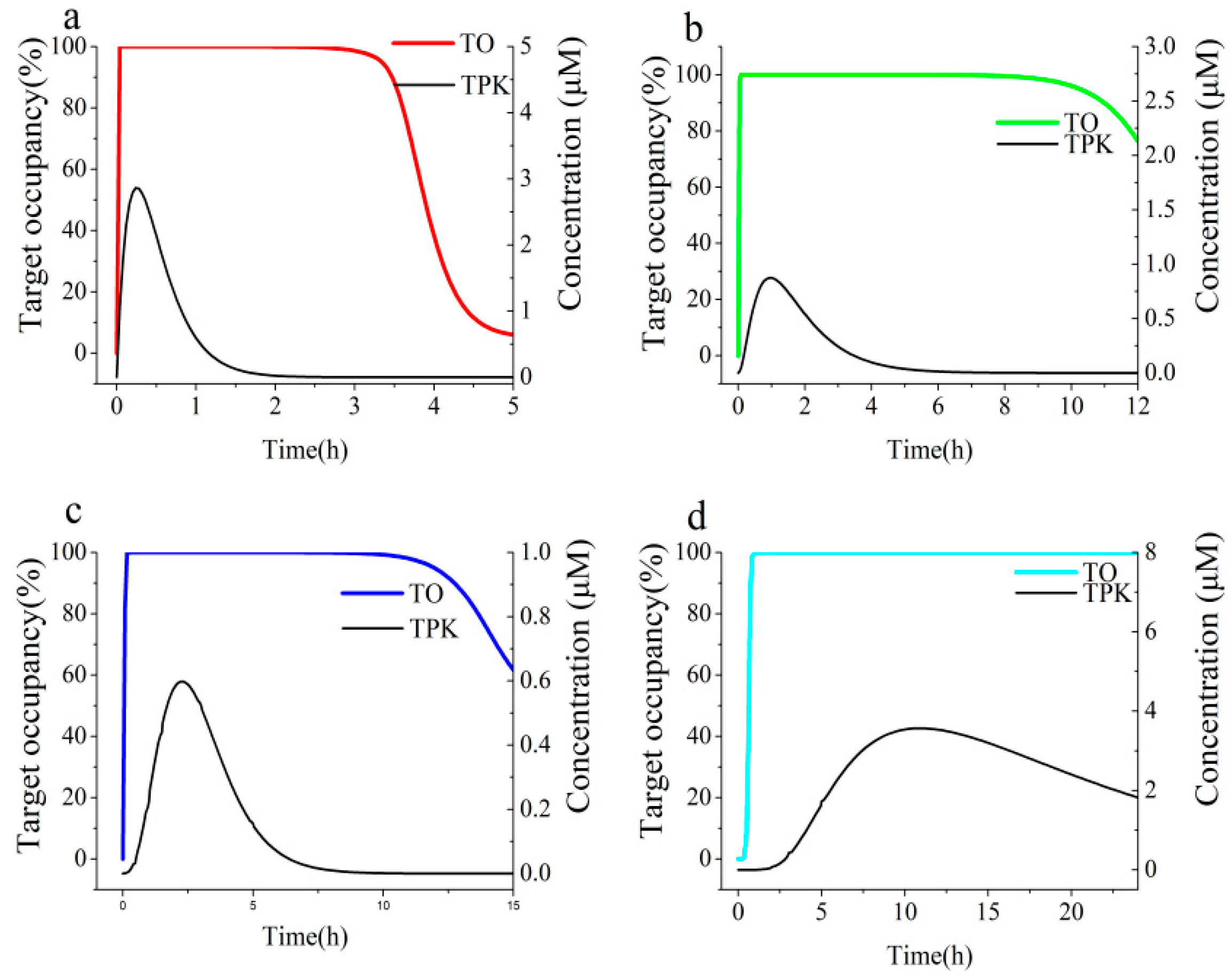
3.3. Micro-Pharmacokinetics Profiles Model of Free Compounds in the Target Vicinity
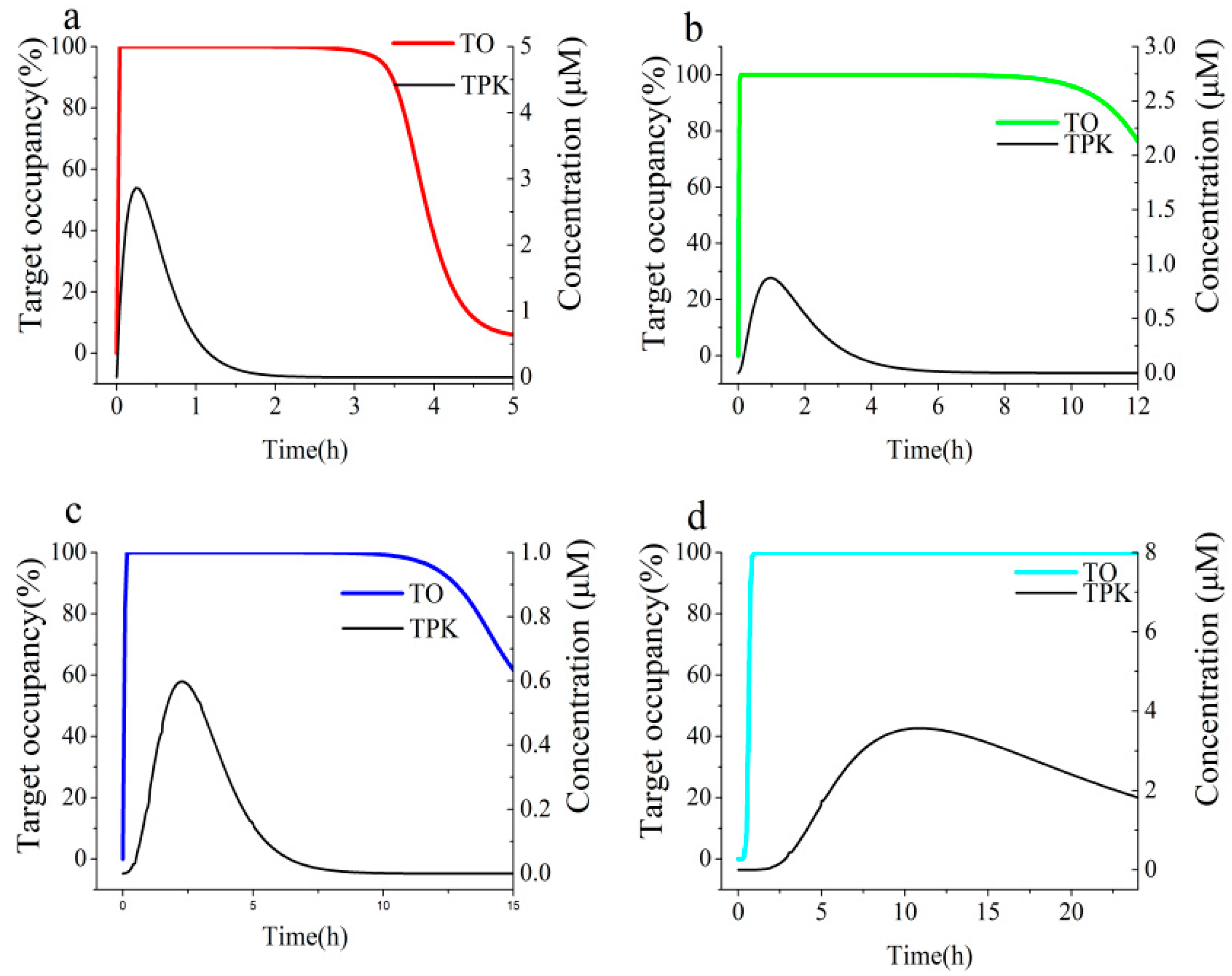
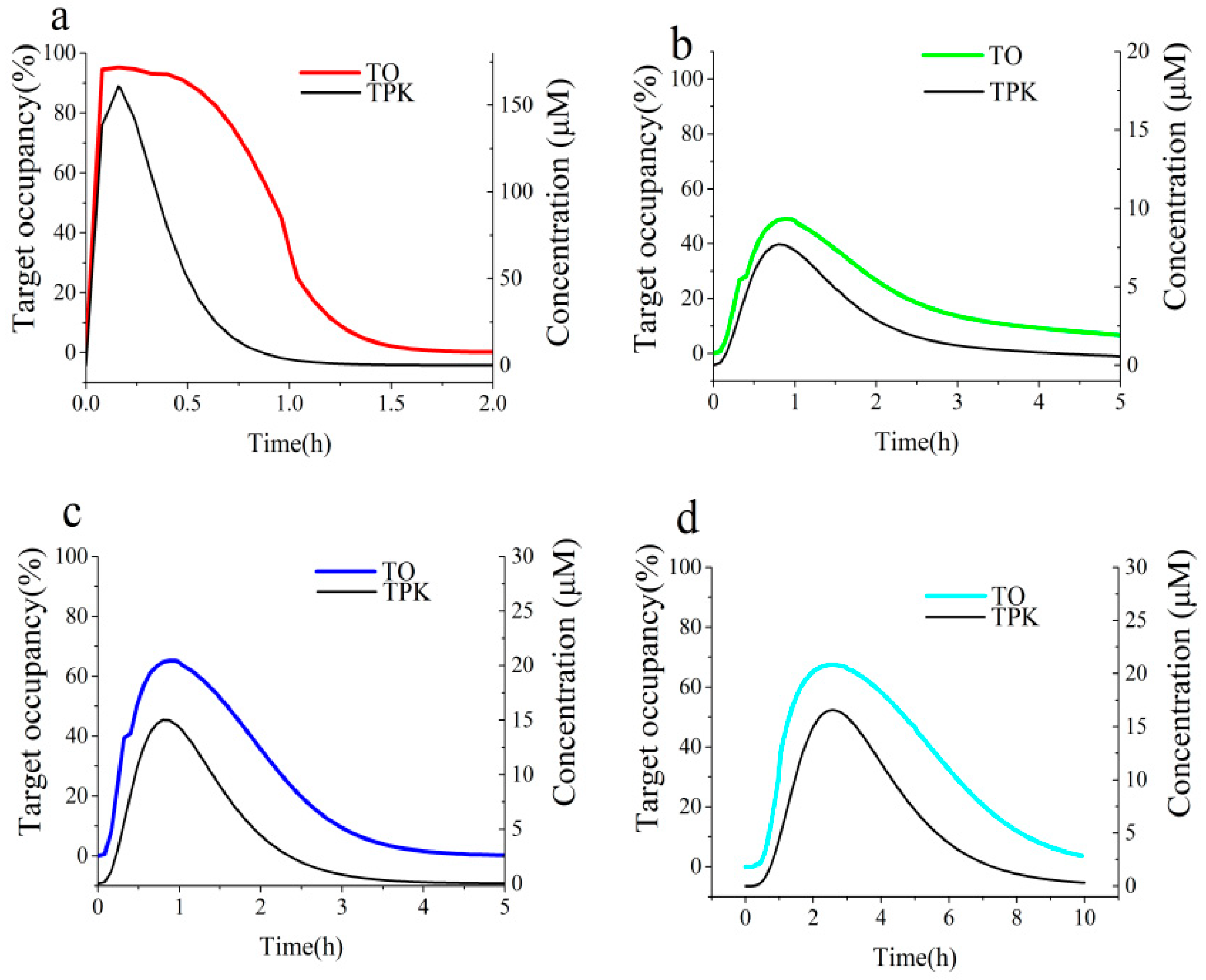
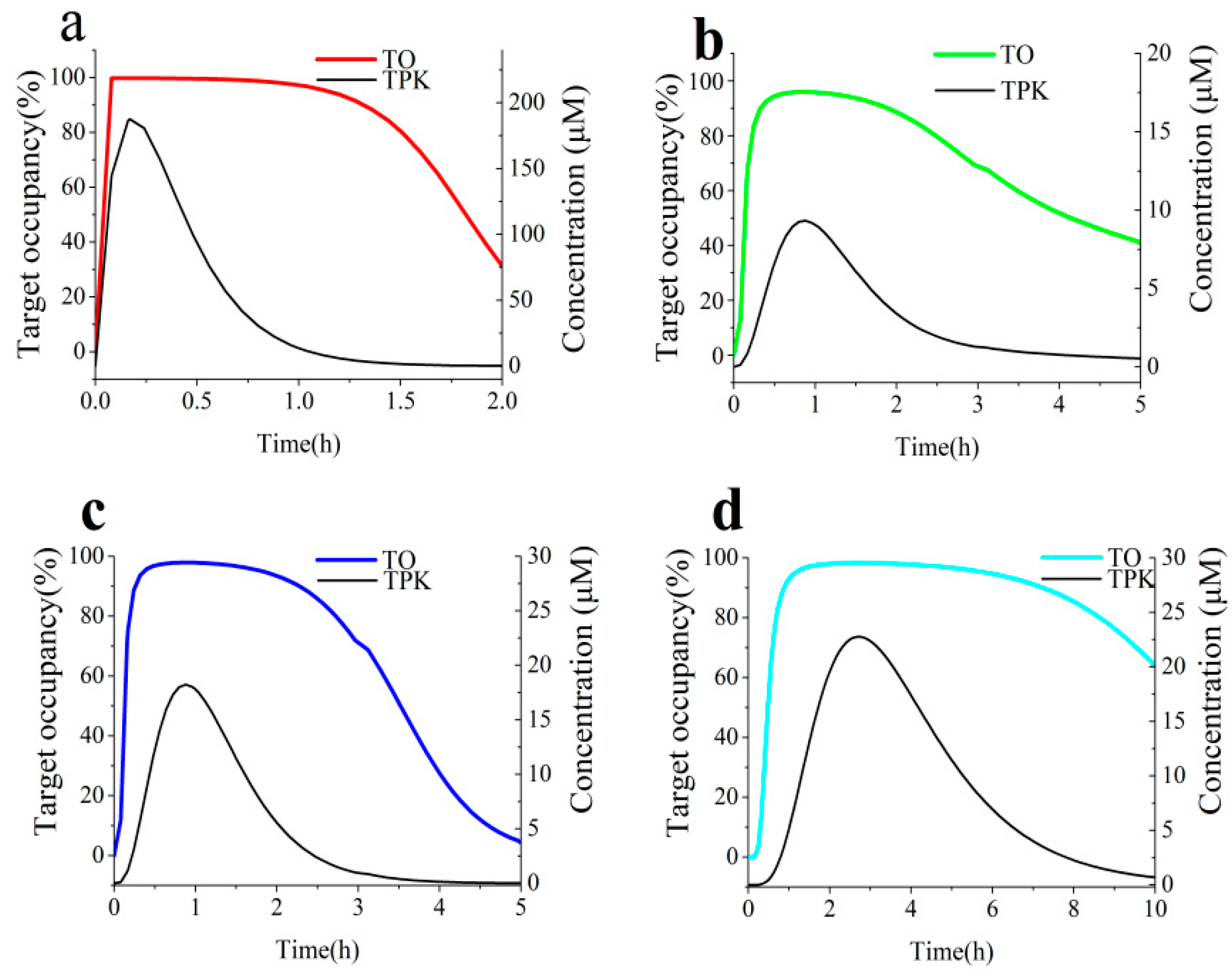
3.4. Link Binding Kinetics Rate Constants to in Vivo Target Occupancy with BK-TPK Model
4. Discussion
- The on-rate is >103 M−1s−1 for the fast association with AGH target.
- The off-rate is <10−4s−1 (corresponding to BK-t1/2 > 1.9 h) for the slow dissociation from AGH target.
- The molecule characterizations with a reasonable high aqueous solubility, a low drug permeability and high stability in intestinal tract are required to obtain high enough compound concentration in the UWLs.
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Matsui, T.; Ueda, T.; Oki, T.; Sugita, K.; Terahara, N.; Matsumoto, K. α-glucosidase inhibitory action of natural acylated anthocyanins. 1. Survey of natural pigments with potent inhibitory activity. J. Agric. Food Chem. 2001, 49, 1948–1951. [Google Scholar] [CrossRef] [PubMed]
- Zaharudin, N.; Staerk, D.; Dragsted, L.O. Inhibition of α-glucosidase activity by selected edible seaweeds and fucoxanthin. Food Chem. 2019, 270, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Dabhi, A.S.; Bhatt, N.R.; Shah, M.J. Voglibose: An alpha glucosidase inhibitor. J. Clin. Diagn. Res. 2013, 7, 3023–3027. [Google Scholar]
- Bischoff, H. Pharmacology of α-glucosidase inhibition. Eur. J. Clin. Investig. 2010, 24, 3–10. [Google Scholar]
- Li, D.Q.; Qian, Z.M.; Li, S.P. Inhibition of three selected beverage extracts on α-glucosidase and rapid identification of their active compounds using HPLC-DAD-MS/MS and biochemical detection. J. Agric. Food Chem. 2010, 58, 6608–6613. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Wang, Z.Y.; Li, H.; Chen, L.; Sun, Y.; Gobbo, S.; Balentine, D.A.; Yang, C.S. Analysis of plasma and urinary tea polyphenols in human subjects. Cancer Epidem. Biomar. 1995, 4, 393–399. [Google Scholar]
- Kale, A.; Gawande, S.; Kotwal, S.; Netke, S.; Roomi, W.; Ivanov, V.; Niedzwiecki, A.; Rath, M. Studies on the effects of oral administration of nutrient mixture, quercetin and red onions on the bioavailability of epigallocatechin gallate from green tea extract. Phytother. Res. 2010, 24, S48–S55. [Google Scholar] [CrossRef] [PubMed]
- Gan, R.Y.; Li, H.B.; Sui, Z.Q.; Corke, H. Absorption, metabolism, anti-cancer effect and molecular targets of epigallocatechin gallate (EGCG): An updated review. Crit. Rev. Food Sci. Nutr. 2018, 58, 924–941. [Google Scholar] [CrossRef] [PubMed]
- Gamberucci, A.; Konta, L.; Colucci, A.; Giunti, R.; Magyar, J.E.; Mandl, J.; Banhegyi, G.; Benedetti, A.; Csala, M. Green tea flavonols inhibit glucosidase II. Biochem. Pharmacol. 2006, 72, 640–646. [Google Scholar] [CrossRef]
- Gao, J.; Xu, P.; Wang, Y.; Wang, Y.; Hochstetter, D. Combined effects of green tea extracts, green tea polyphenols or epigallocatechin gallate with acarbose on inhibition against α-amylase and α-glucosidase in vitro. Molecules 2013, 18, 11614–11623. [Google Scholar] [CrossRef] [PubMed]
- Yilmazer-Musa, M.; Griffith, A.M.; Michels, A.J.; Schneider, E.; Frei, B. Grape seed and tea extracts and catechin 3-gallates are potent inhibitors of α-amylase and α-glucosidase activity. J. Agric. Food Chem. 2012, 60, 8924–8929. [Google Scholar] [CrossRef]
- Satoh, T.; Igarashi, M.; Yamada, S.; Takahashi, N.; Watanabe, K. Inhibitory effect of black tea and its combination with acarbose on small intestinal α-glucosidase activity. J. Ethnopharmacol. 2015, 161, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Li, W.; Chen, Z.; Guo, Q.; Wang, C.; Santhanam, R.K.; Chen, H. Inhibitory effect of epigallocatechin-3-O-gallate on α-glucosidase and its hypoglycemic effect via targeting PI3K/AKT signalingpathway in L6 skeletal muscle cells. Int. J. Biol. Macromol. 2019, 125, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.Q.; Li, M.; Zhu, F.; Liu, F.L.; Huang, J.B. Inhibitory potential of trilobatin from Lithocarpus polystachyus Rehd against α-glucosidase and α-amylase linked to type 2 diabetes. Food Chem. 2012, 130, 261–266. [Google Scholar] [CrossRef]
- Xu, D.; Wang, Q.; Zhang, W.; Hu, B.; Zhou, L.; Zeng, X.; Sun, Y. Inhibitory activities of caffeoylquinic acid derivatives from Ilex kudingcha C.J. Tseng on α-Glucosidase from saccharomyces cerevisiae. J. Agric. Food Chem. 2015, 63, 3694–3703. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Fang, C.; Zhu, R.; Peng, Q.; Li, D.; Wang, M. Inhibitory effect of phloretin on α-glucosidase: Kinetics, interaction mechanism and molecular docking. Int. J. Biol. Macromol. 2017, 95, 520–527. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.C.; Zhao, S.; Yang, B.Y.; Wang, Q.H.; Kuang, H.X. Anti-diabetic polysaccharides from natural sources: A review. Carbohydr. Polym. 2016, 148, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Lee, J.; Yue, L.M.; Lim, G.T.; Yang, J.M.; Ye, Z.M.; Park, Y.D. Inhibitory effect of pyrogallol on α-glucosidase: Integrating docking simulations with inhibition kinetics. Int. J. Biol. Macromol. 2018, 112, 686–693. [Google Scholar] [CrossRef]
- Li, Z.P.; Song, Y.H.; Uddin, Z.; Wang, Y.; Park, K.H. Inhibition of protein tyrosine phosphatase 1B (PTP1B) and α-glucosidase by xanthones from cratoxylum cochinchinense, and their kinetic characterization. Bioorg. Med. Chem. 2018, 26, 737–746. [Google Scholar] [CrossRef]
- Sekar, V.; Chakraborty, S.; Mani, S.; Sali, V.K.; Vasanthi, H.R. Mangiferin from mangifera indica fruits reduces post-prandial glucose level by inhibiting α-glucosidase and α-amylase activity. South Afr. J. Bot. 2018, 120, 129–134. [Google Scholar] [CrossRef]
- Wang, M.; Jiang, J.; Tian, J.; Chen, S.; Ye, X.; Hu, Y.; Chen, J. Inhibitory mechanism of novel allosteric inhibitor, Chinese bayberry (Myrica rubra Sieb. et Zucc.) leaves proanthocyanidins against α-glucosidase. J. Funct. Foods 2019, 56, 286–294. [Google Scholar] [CrossRef]
- Copeland, R.A.; Pompliano, D.L.; Meek, T.D. Drug–target residence time and its implications for lead optimization. Nat. Rev. Drug Discov. 2006, 5, 730–739. [Google Scholar] [CrossRef] [PubMed]
- Copeland, R.A. The drug–target residence time model: A 10-year retrospective. Nat. Rev. Drug Discov. 2015, 15, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Wittmann, H.J.; Strasser, A. Competitive association binding kinetic assays: A new tool to detect two different binding orientations of a ligand to its target protein under distinct conditions? Naunyn-Schmiedeberg’s Arch. Pharmacol. 2017, 390, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Copeland, R.A. Slow Binding Inhibitors. In Evaluation of Enzyme Inhibitors in Drug Discovery: A Guide for Medicinal Chemists and Pharmacologists, 1st ed.; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2005; Volume 6, pp. 141–177. [Google Scholar]
- Hanozet, G.; Pircher, H.P.; Vanni, P.; Oesch, B.; Semenza, G. An example of enzyme hysteresis. The slow and tight interaction of some fully competitive inhibitors with small intestinal sucrase. J. Biol. Chem. 1981, 256, 3703–3711. [Google Scholar] [PubMed]
- Lu, H.; Tonge, P.J. Drug–target residence time: Critical information for lead optimization. Curr. Opin. Chem. Biol. 2010, 14, 467–474. [Google Scholar] [CrossRef]
- Copeland, R.A. The dynamics of drug-target interactions: Drug-target residence time and its impact on efficacy and safety. Expert Opin. Drug Discov. 2010, 5, 305–310. [Google Scholar] [CrossRef]
- Guo, D.; Hillger, J.M.; Ijzerman, A.P.; Heitman, L.H. Drug-target residence time—A case for g protein-coupled receptors. Med. Res. Rev. 2014, 34, 856–892. [Google Scholar] [CrossRef]
- Vauquelin, G. Effects of target binding kinetics on in vivo drug efficacy: koff, kon and rebinding. Br. J. Pharmacol. 2016, 173, 2319–2334. [Google Scholar] [CrossRef]
- Vauquelin, G. Distinct in vivo target occupancy by bivalent and induced-fit-like binding drugs. Br. J. Pharmacol. 2017, 174, 4233–4246. [Google Scholar] [CrossRef]
- Georgi, V.; Schiele, F.; Berger, B.T.; Steffen, A.; Marin Zapata, P.A.; Briem, H.; Menz, S.; Preusse, C.; Vasta, J.D.; Robers, M.B.; et al. Binding kinetics survey of the drugged kinome. J. Am. Chem. Soc. 2018, 140, 15774–15782. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.W.; Morrison, J.F. The kinetics of reversible tight-binding inhibition. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 1979; Volume 63, pp. 437–467. [Google Scholar]
- Copeland, R.A. Experimental measures of enzyme activity. In Enzyme: A practical Introduction to Structure, Mechanism, and Data Analysis, 2nd ed.; John Wiley & Sons, Inc.: New York, NY, USA, 2000; Volume 7, pp. 188–265. [Google Scholar]
- Kim, Y.B.; Kopcho, L.M.; Kirby, M.S.; Hamann, L.G.; Weigelt, C.A.; Metzler, W.J.; Marcinkeviciene, J. Mechanism of Gly-Pro-pNA cleavage catalyzed by dipeptidyl peptidase-IV and its inhib-ition bysaxagliptin (BMS-477118). Arch. Biochem. Biophys. 2006, 445, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.Y.; Nancy, Y.W. Enzyme kinetics for clinically relevant CYP inhibition. Curr. Drug Metab. 2005, 6, 241–357. [Google Scholar] [CrossRef] [PubMed]
- Theorell, H.; Yonetani, T. Studies on liver alcohol dehydrogenase. Arch. Biochem. Biophys. 1964, 106, 243–251. [Google Scholar] [CrossRef]
- Inoue, M.B.; Inoue, M.; Fernando, Q.; Valcic, S.; Timmermann, B.N. Potentiometric and 1H NMR studies of complexation of Al3+ with (−)-epigallocatechin gallate, a major active constituent of green tea. J. Inorg. Biochem. 2002, 88, 7–13. [Google Scholar] [CrossRef]
- Vaidyanathan, J.B.; Walle, T. Cellular uptake and efflux of the tea flavonoid (−) epicatechin-3-gallate in the human intestinal cell line caco-2. J. Pharmacol. Exp. Ther. 2003, 307, 745–752. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Chen, Y.; Li, R.C. Pharmacokinetics and system linearity of tea catechins in rat. Xenobiotica 2001, 31, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.S. The Influence of Gastrointestinal Physiology Factors on Main Ingredients of Tea Polyphenols and the Study on Absorption Process of EGCG. Master’s Thesis, Guangzhou University of Chinese Medicine, Guangzhou, China, 2016. [Google Scholar]
- Okumu, A.; DiMaso, M.; Lobenberg, R. Dynamic dissolution testing to establish in vitro/in vivo correlations for montelukast sodium, a poorly soluble drug. Pharm. Res. 2008, 25, 2778–2785. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Kou, L.; Ma, P.; Gao, L.; Li, B.; Li, R.; Luo, C.; Shentu, J.; He, Z.; Sun, J. Interspecies prediction of oral pharmacokinetics of different lacidipine formulations from dogs to human: Physiologically based pharmacokinetic modelling combined with biorelevant dissolution. RSC Adv. 2015, 5, 19844–19852. [Google Scholar] [CrossRef]
- Radwan, A.; Zaid, A.N.; Jaradat, N.; Odeh, Y. Food effect: The combined effect of media pH and viscosity on the gastrointestinal absorption of ciprofloxacin tablet. Eur. J. Pharm. Sci. 2017, 101, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Loftsson, T. Drug permeation through biomembranes: Cyclodextrins and the unstirred water layer. Pharmazie 2012, 67, 363–370. [Google Scholar] [PubMed]
- Loftsson, T.; Brewster, M.E. Pharmaceutical applications of cyclodextrins: Effects on drug permeation through biological membranes. J. Pharm. Pharmacol. 2011, 63, 1119–1135. [Google Scholar] [CrossRef] [PubMed]
- Winne, D. Dependence of intestinal absorption in vivo on the unstirred layer. Naunyn-Schmiedeberg’s Arch. Pharmacol. 1978, 304, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Copeland, R.A. Assay considerations for compound library screening. In Evaluation of Enzyme Inhibitors in Drug Discovery: A Guide for Medicinal Chemists and Pharmacologists, 2nd ed.; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2013; Volume 4, pp. 123–168. [Google Scholar]
- Brandt, I.; Joossens, J.; Chen, X.; Maes, M.B.; Scharpe, S.; De Meester, I.; Lambeir, A.M. Inhibition of dipeptidyl-peptidase IV catalyzed peptide truncation by Vildagliptin ((2S)-{[(3-hydroxyadamantan-1-yl)amino]acetyl}-pyrrolidine-2-carbonitrile). Biochem. Pharmacol. 2005, 70, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.; Herman, D.; Gottesfeld, J. Pimelic diphenylamide 106 is a slow, tight-binding inhibitor of class I histone deacetylases. J. Biol. Chem. 2008, 283, 35402–35409. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; England, K.; Am Ende, C.; Truglio, J.J.; Luckner, S.; Reddy, B.G.; Marlenee, N.L.; Knudson, S.E.; Knudson, D.L.; Bowen, R.A.; et al. Slow-onset inhibition of the fabi enoyl reductase from francisella tularensis: Residence time and in vivo activity. ACS Chem. Biol. 2009, 4, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Kong, F. Evaluation of the in vitro α-glucosidase inhibitory activity of green tea polyphenols and different tea types. J. Sci. Food Agric. 2016, 96, 777–782. [Google Scholar] [CrossRef]
- Lai, Z.; Yang, T.; Kim, Y.B.; Sielecki, T.M.; Diamond, M.A.; Strack, P.; Rolfe, M.; Caligiuri, M.; Benfield, P.A.; Auger, K.R.; et al. Differentiation of Hdm2-mediated p53 ubiquitination and Hdm2 autoubiquitination activity by small molecular weight inhibitors. Proc. Natl. Acad. Sci. USA 2002, 99, 14734–14739. [Google Scholar] [CrossRef] [Green Version]
- Zhu, M.; Chen, Y.; Li, R.C. Oral absorption and bioavailability of tea catechins. Planta Med. 2000, 66, 444–447. [Google Scholar] [CrossRef]
- Package Insert. In Precose (Acarbose); Bayer HealthCare Pharmaceuticals Inc.: Whippany, NJ, USA, 2014.
- Acarbose, PMDA Review Report. Pharmaceuticals and Medical Devices Agency, PMDA Review Report. Pharmaceuticals and Medical Devices Agency. Available online: http://www.info.pmda.go.jp/go/interview/3/830001_3969003F1077_3_004_1F.pdf (accessed on 9 August 2019).
- Dash, R.P.; Babu, R.J.; Srinivas, N.R. Reappraisal and perspectives of clinical drug–drug interaction potential of α-glucosidase inhibitors such as acarbose, voglibose and miglitol in the treatment of type 2 diabetes mellitus. Xenobiotica 2018, 48, 89–108. [Google Scholar] [CrossRef]
- Acarbose, PMDA Review Report. Pharmaceuticals and Medical Devices Agency, PMDA Review Report. Pharmaceuticals and Medical Devices Agency. Available online: http://www.info.pmda.go.jp/go/interview/1/630004_3969003F3037_1_001_1F.pdf (accessed on 9 August 2019).
- Copeland, R.A. Quantitative biochemistry in the pharmacological evaluation of drugs. In Evaluation of Enzyme Inhibitors in Drug Discovery: A Guide for Medicinal Chemists and Pharmacologists, 2nd ed.; John Wiley & Sons: Hoboken, NJ, USA, 2013; Volume 10, pp. 383–469. [Google Scholar]
- Grimwood, S.; Hartig, P.R. Target site occupancy: Emerging generalizations from clinical and preclinical studies. Pharmacol. Ther. 2009, 122, 281–301. [Google Scholar] [CrossRef] [PubMed]
- Merget, B.; Sotriffer, C.A. Slow-onset inhibition of mycobacterium tuberculosis InhA: Revealing molecular determinants of residence time by MD simulations. PLoS ONE 2015, 10, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Miyahisa, I.; Suzuki, H.; Mizukami, A.; Tanaka, Y.; Ono, M.; Hixon, M.S.; Matsui, J. T-3364366 targets the desaturase domain of delta-5 desaturase with nanomolar potency and a multihour residence time. ACS Med. Chem. Lett. 2016, 7, 868–872. [Google Scholar] [CrossRef] [PubMed]
- Uitdehaag, J.C.M.; De Man, J.; Willemsen-Seegers, N.; Prinsen, M.B.W.; Libouban, M.A.A.; Sterrenburg, J.G.; De Wit, J.J.P.; De Vetter, J.R.F.; De Roos, J.A.D.M.; Buijsman, R.C.; et al. Target residence time-guided optimization on TTK kinase results in inhibitors with potent anti-proliferative activity. J. Mol. Biol. 2017, 429, 2211–2230. [Google Scholar] [CrossRef] [PubMed]
- Reggie, B.; Jelle, V.D.B.; Vischer, H.F.; Luis, L.; Rob, L. The long duration of action of the second generation antihistamine bilastine coincides with its long residence time at the histamine H1 receptor. Eur. J. Pharmacol. 2018, 838, 107–111. [Google Scholar]
- Hao, L.; Iuliano, J.N.; Tonge, P.J. Structure–kinetic relationships that control the residence time of drug–target complexes: Insights from molecular structure and dynamics. Curr. Opin. Chem. Biol. 2018, 44, 101–109. [Google Scholar]
- Vauquelin, G.; Charlton, S.J. Long-lasting target binding and rebinding as mechanisms to prolong in vivo drug action. Br. J. Pharmacol. 2010, 161, 488–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | kon (103 M−1s−1) | koff (10−3 s−1) | Ki* (μM) |
|---|---|---|---|
| ECG | 0.2 ± 0.05 | 1.6 ± 0.3 | 1.0 ± 0.3 |
| EGCG | 2.4 ± 0.3 | 1.9 ± 0.2 | 0.16 ± 0.03 |
| Parameters | Cmax (μM) | AUC0-inf (103 μM·h) a | Terminal UWL Half-Lifes (h) a | Dissociation Half-Life (h) b | Ratio c | |
|---|---|---|---|---|---|---|
| ECG | Duodenum | 160.9 | 63 | 0.13 | 0.12 | 0.92 |
| Jejunum | 7.7 | 14 | 1.1 | 0.11 | ||
| Ileum | 15.0 | 21 | 0.28 | 0.43 | ||
| Colon | 16.6 | 63 | 1.7 | 0.07 | ||
| EGCG | Duodenum | 187.5 | 94 | 0.17 | 0.10 | 0.59 |
| Jejunum | 9.3 | 17 | 1.5 | 0.07 | ||
| Ileum | 18.2 | 25 | 0.17 | 0.59 | ||
| Colon | 22.8 | 92 | 1.1 | 0.09 | ||
| Acarbose | Duodenum | 2.86 | 1.97 | 0.81 | 10.1 | 12.5 |
| Jejunum | 0.88 | 1.92 | 0.82 | 12.3 | ||
| Ileum | 0.60 | 1.90 | 0.87 | 11.6 | ||
| Colon | 3.57 | 82 | 9.6 | 1.1 | ||
| Parameters | TOmax | The Duration of >70% TO (h) | AUC0-inf a | |
|---|---|---|---|---|
| ECG | Duodenum | 95.3% | 0.64 | 84 |
| Jejunum | 48.9% | 0 | 118 | |
| Ileum | 65.2% | 0 | 121 | |
| Colon | 67.5% | 0 | 346 | |
| EGCG | Duodenum | 99.8% | 1.5 | 180 |
| Jejunum | 96.0% | 2.6 | 449 | |
| Ileum | 97.9% | 2.9 | 330 | |
| Colon | 98.3% | 8.9 | 1164 | |
| Acarbose | Duodenum | 100.0% | 3.6 | 402 |
| Jejunum | 100.0% | >12 | 1500 | |
| Ileum | 100.0% | 14.2 | 1885 | |
| Colon | 100.0% | >24 | >2330 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, G.; Ji, Y.; Li, X.; Wang, Q.; Gong, H.; Wang, B.; Liu, Y.; Pan, Y. Utilizing the Combination of Binding Kinetics and Micro-Pharmacokinetics Link in Vitro α-Glucosidase Inhibition to in Vivo Target Occupancy. Biomolecules 2019, 9, 493. https://doi.org/10.3390/biom9090493
Wang G, Ji Y, Li X, Wang Q, Gong H, Wang B, Liu Y, Pan Y. Utilizing the Combination of Binding Kinetics and Micro-Pharmacokinetics Link in Vitro α-Glucosidase Inhibition to in Vivo Target Occupancy. Biomolecules. 2019; 9(9):493. https://doi.org/10.3390/biom9090493
Chicago/Turabian StyleWang, Guopeng, Yanhua Ji, Xueyan Li, Qian Wang, Hang Gong, Baoshun Wang, Yang Liu, and Yanli Pan. 2019. "Utilizing the Combination of Binding Kinetics and Micro-Pharmacokinetics Link in Vitro α-Glucosidase Inhibition to in Vivo Target Occupancy" Biomolecules 9, no. 9: 493. https://doi.org/10.3390/biom9090493
APA StyleWang, G., Ji, Y., Li, X., Wang, Q., Gong, H., Wang, B., Liu, Y., & Pan, Y. (2019). Utilizing the Combination of Binding Kinetics and Micro-Pharmacokinetics Link in Vitro α-Glucosidase Inhibition to in Vivo Target Occupancy. Biomolecules, 9(9), 493. https://doi.org/10.3390/biom9090493