Proteomic Insights into Phycobilisome Degradation, A Selective and Tightly Controlled Process in The Fast-Growing Cyanobacterium Synechococcus elongatus UTEX 2973

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strain and Growth Conditions
2.2. Nitrogen Starvation Conditions
2.3. Spectrophotometric Analysis
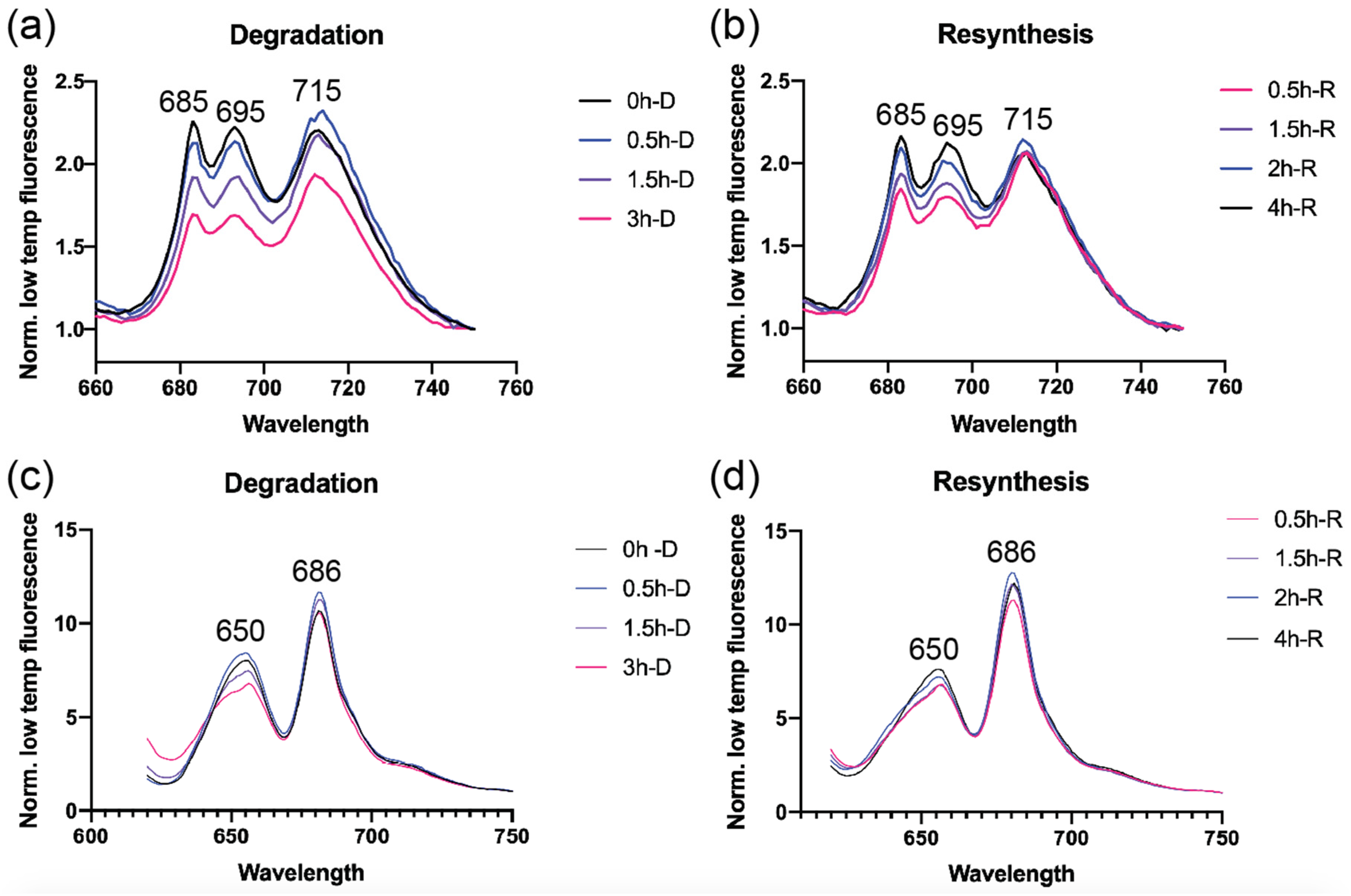
2.4. Low Temperature (77K) Fluorescence Spectroscopy
2.5. Preparation of PCB Proteins for Mass Spectrometry
2.6. Top-Down Mass Spectrometry of PCB Proteins
2.7. Selected Reaction Monitoring (SRM) Experimental Design and Analysis
3. Results
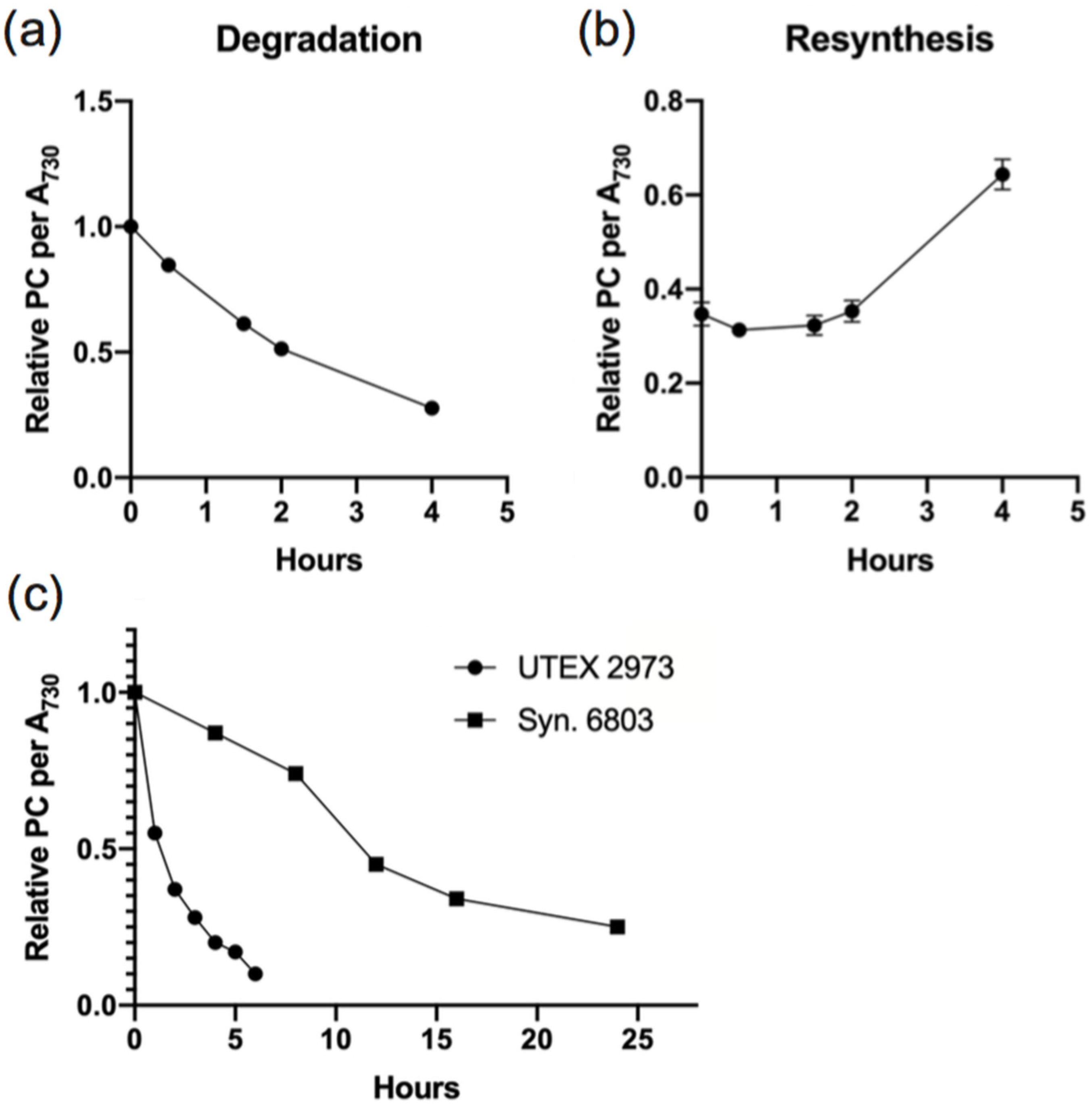
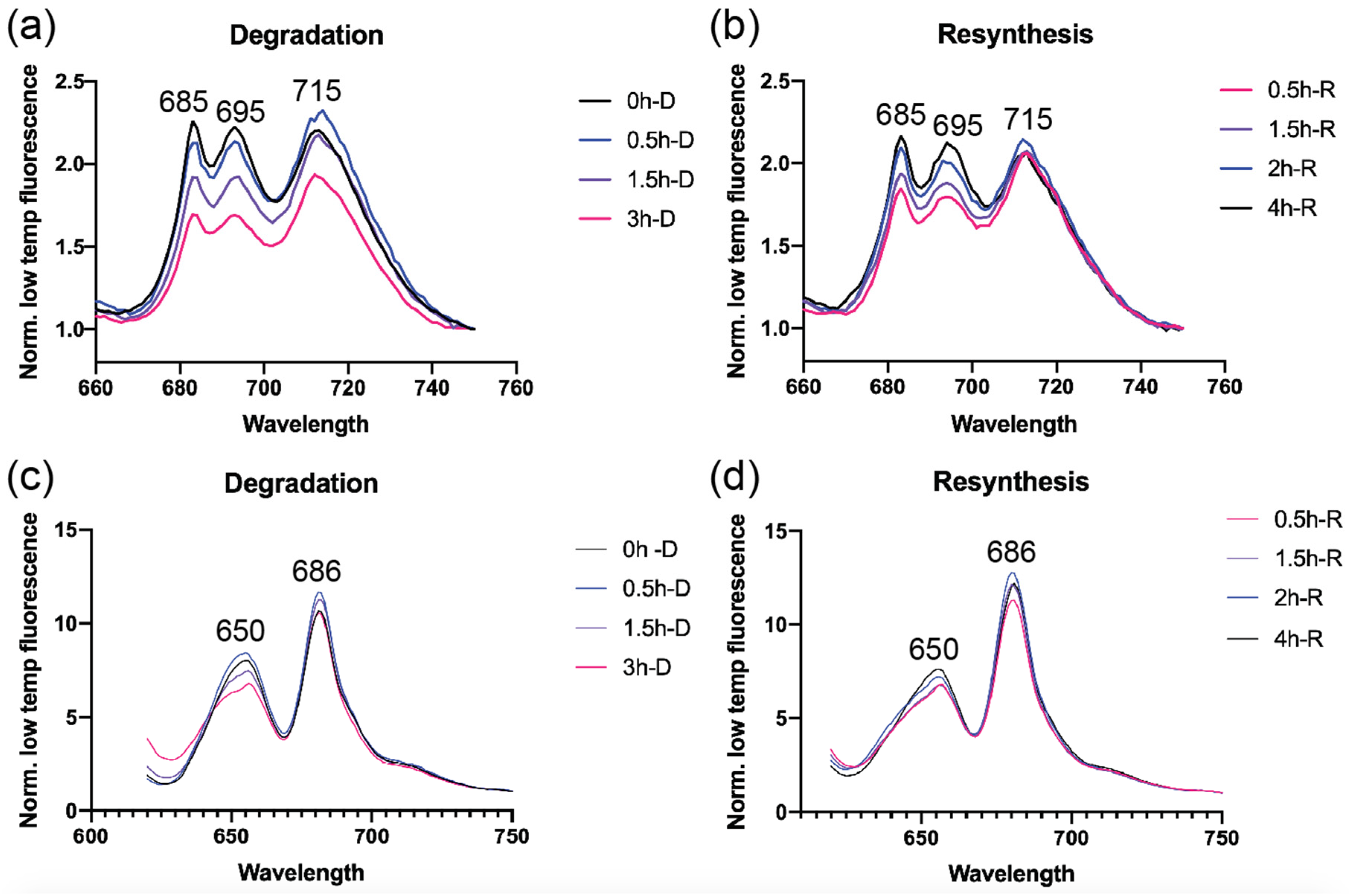
3.1. Rapid PBS Degradation and Re-Synthesis in UTEX 2973 during N Starvation
3.2. Photosystem Levels are Modulated as A Consequence of N Starvation and Re-Addition
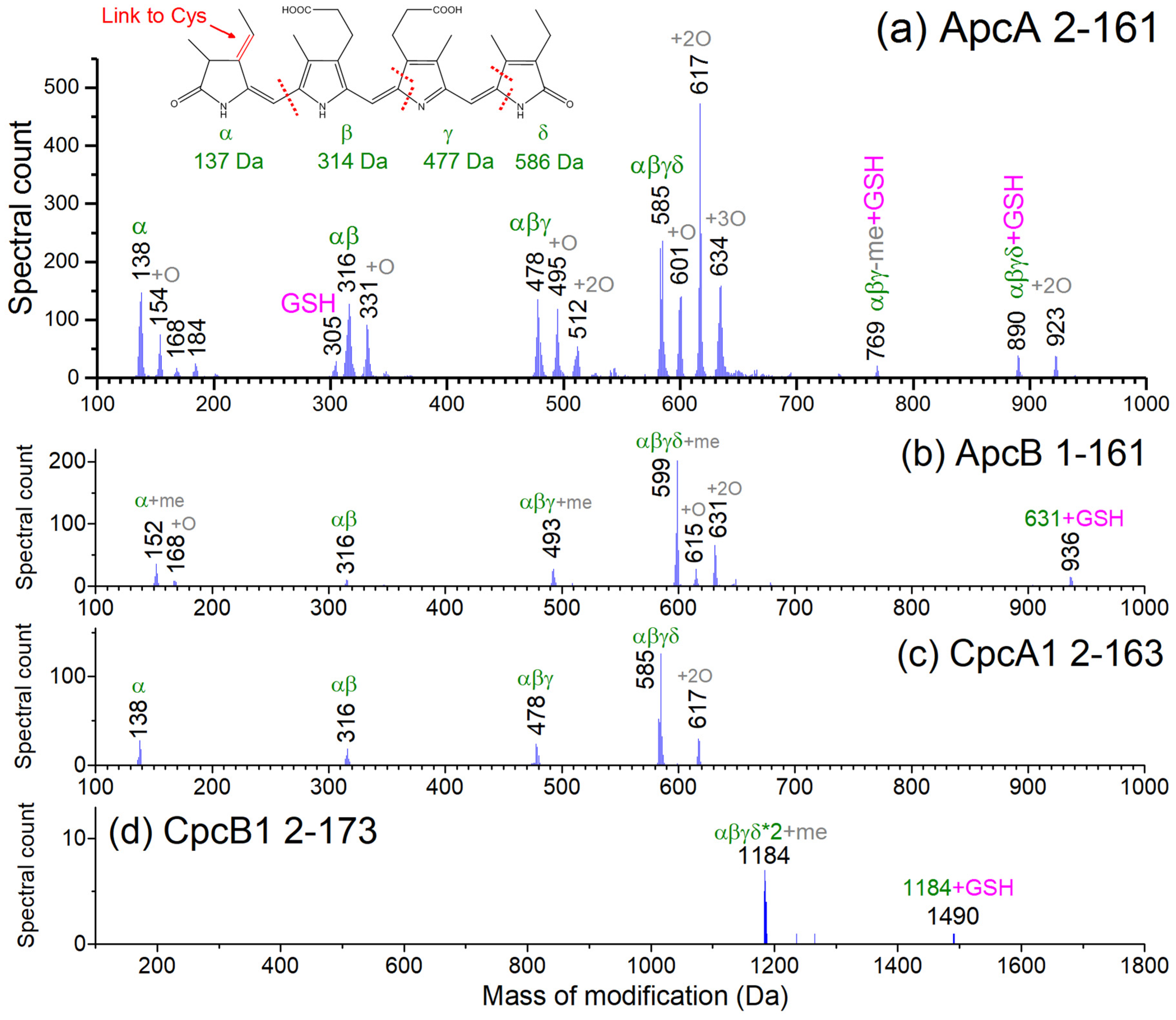
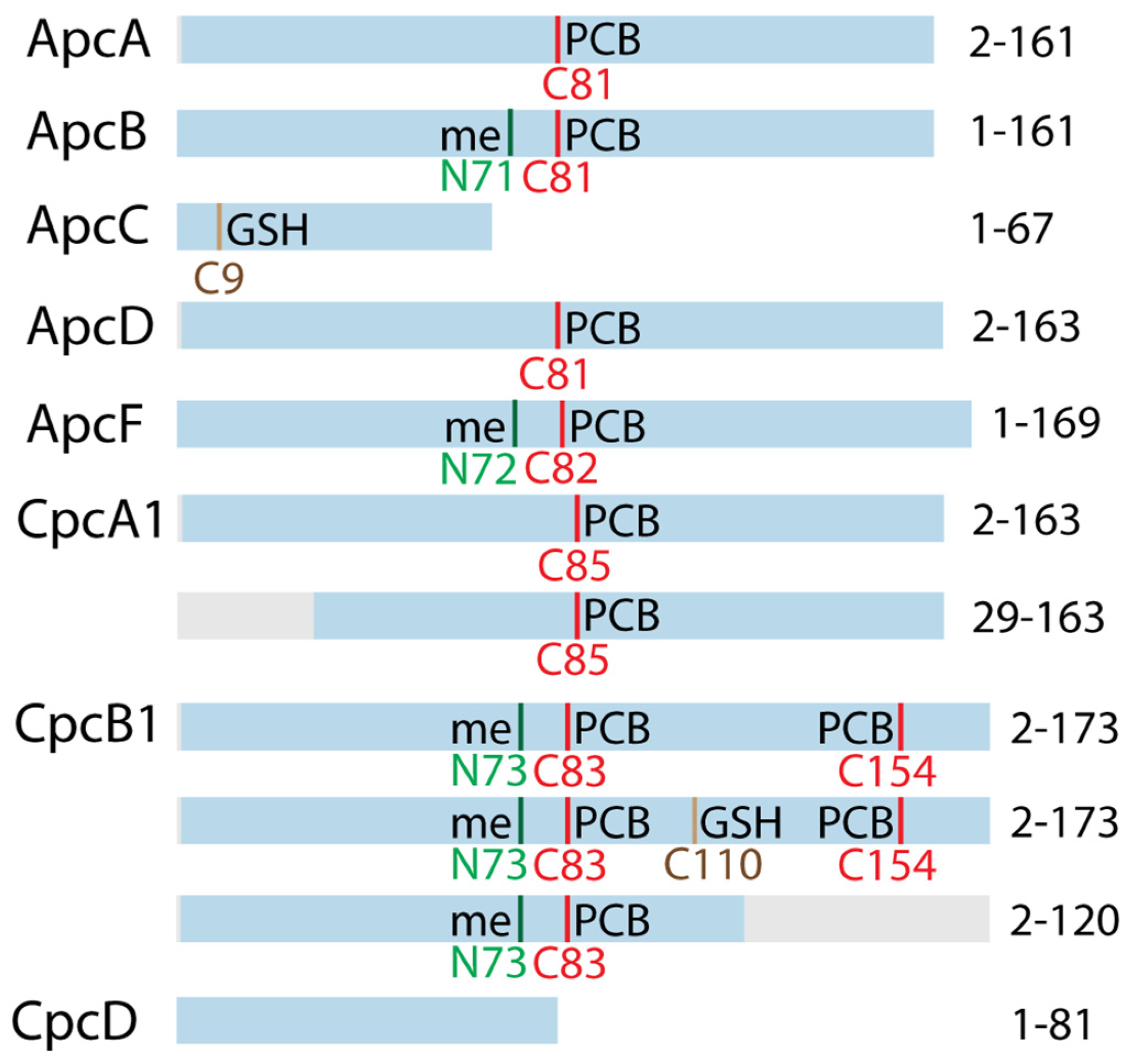
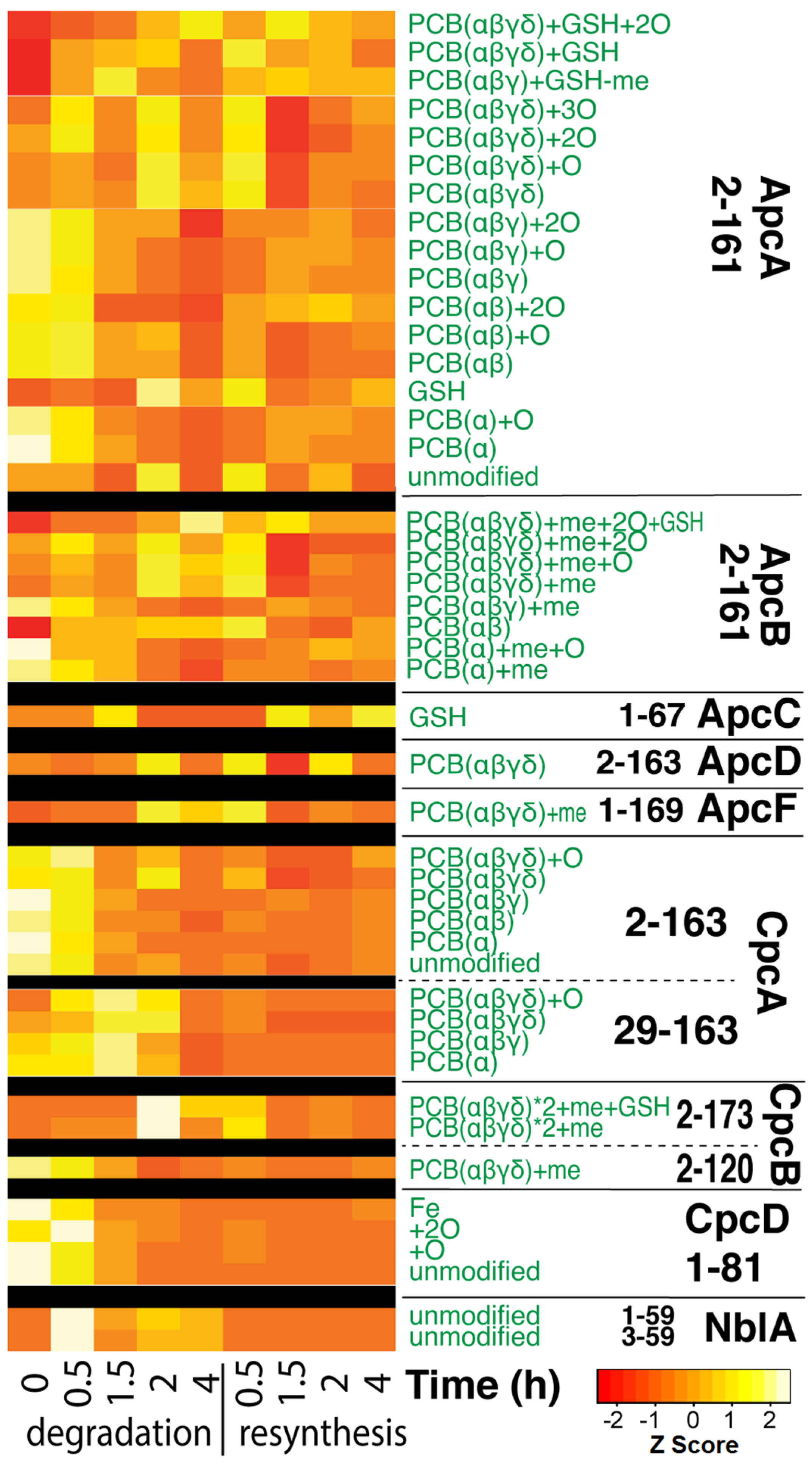
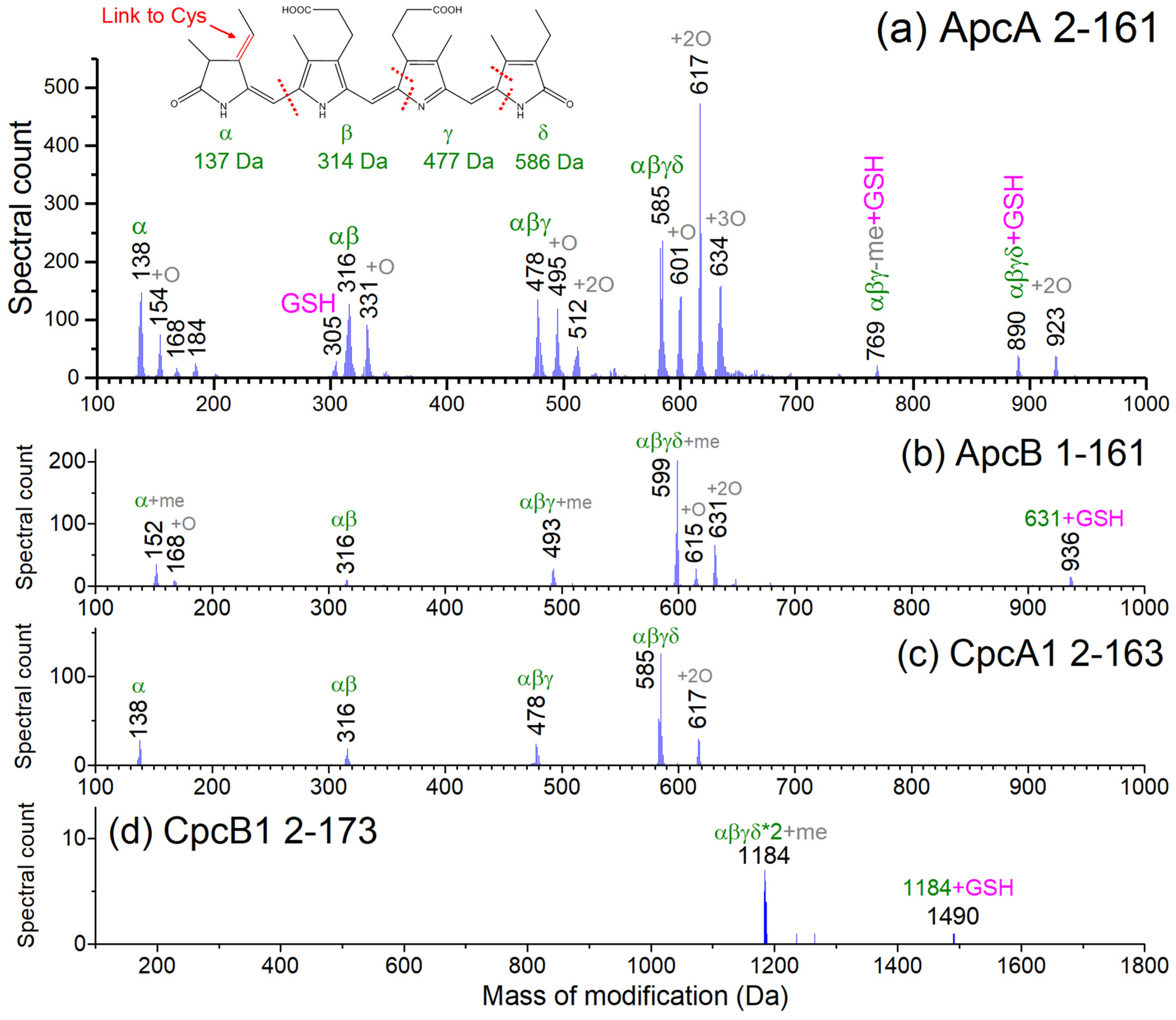
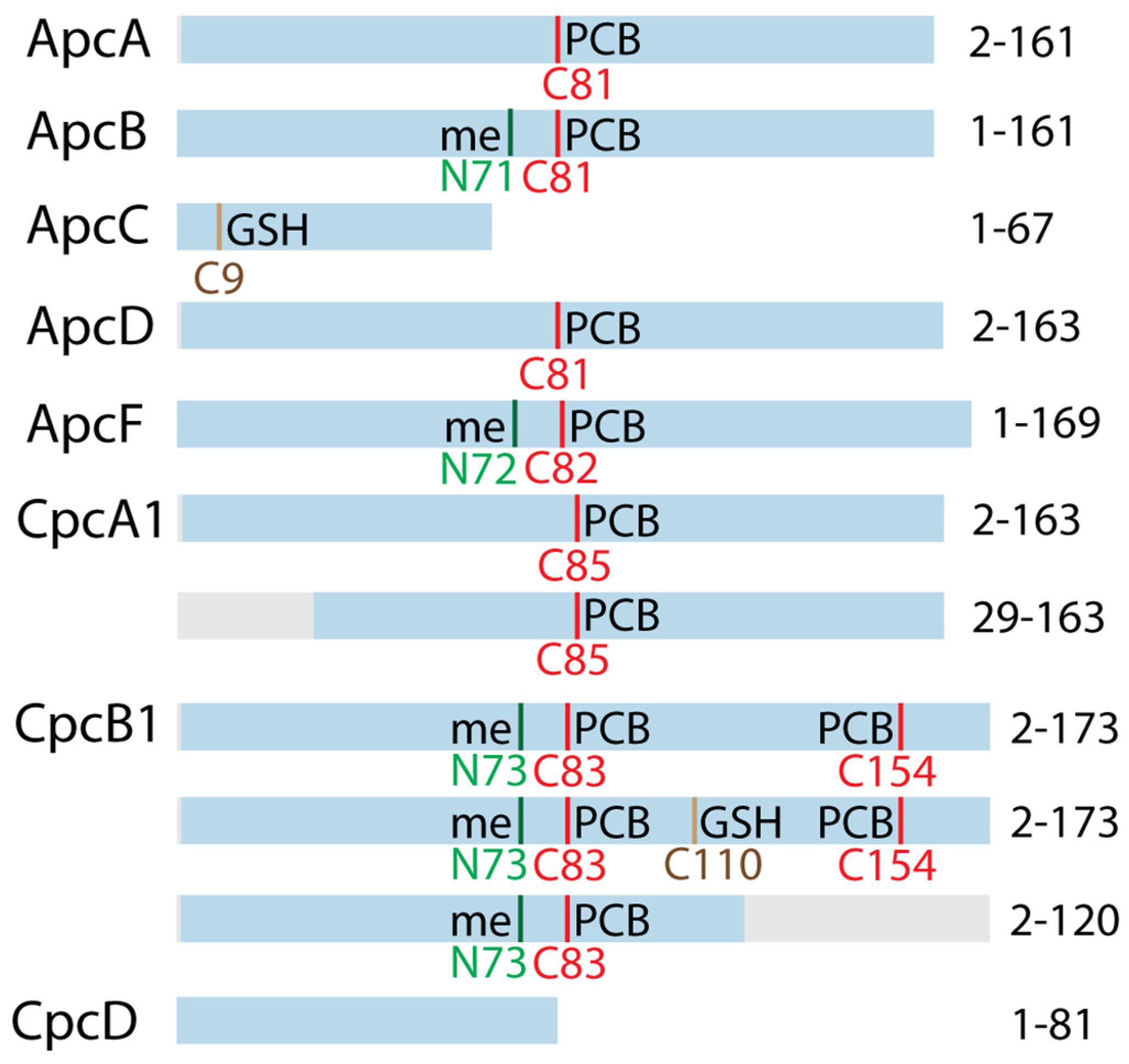
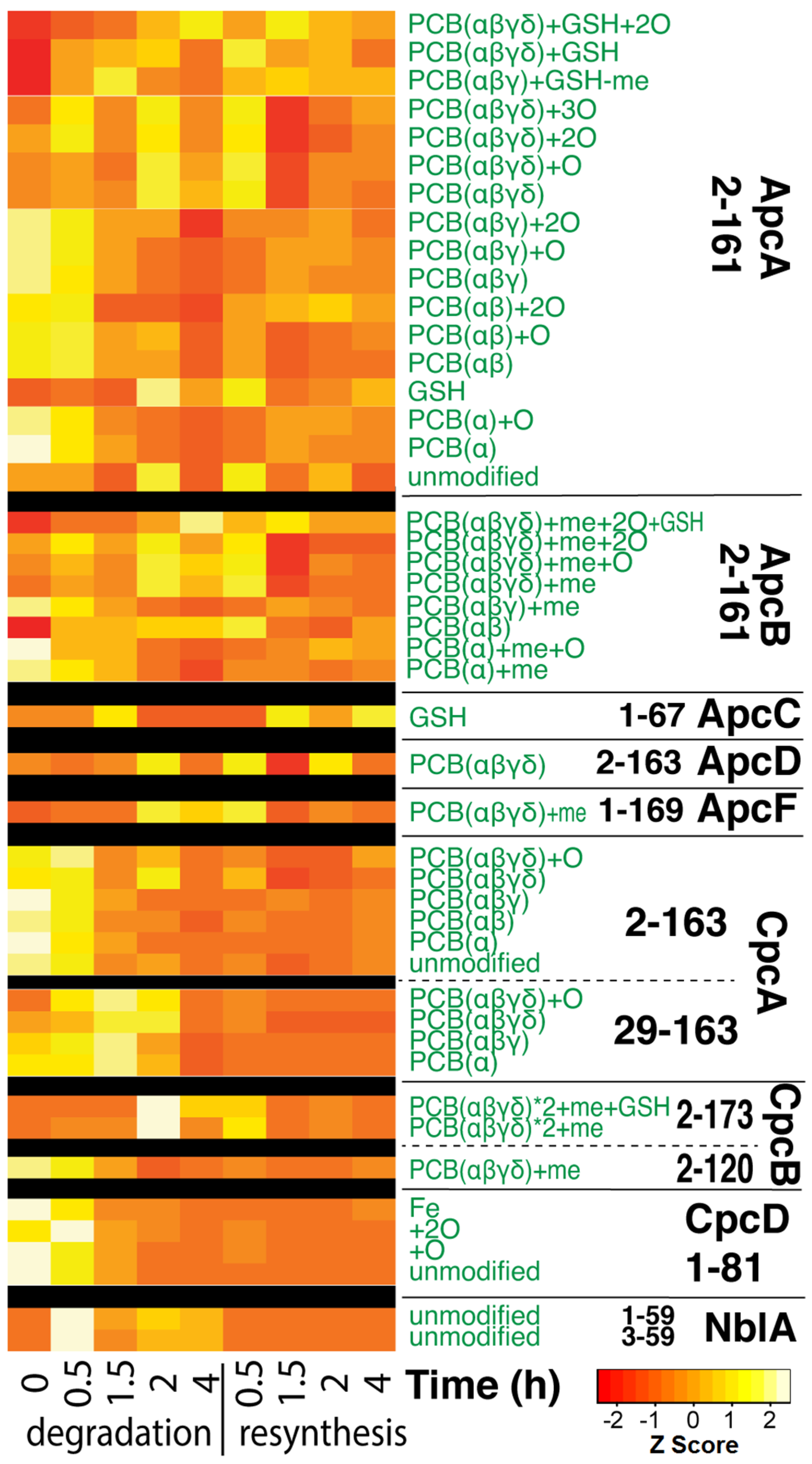
3.3. Top-Down Proteomics of PBP Proteoforms Linked to PCBs
3.4. Quantitative Changes of Proteins during PBS Degradation and Re-Synthesis
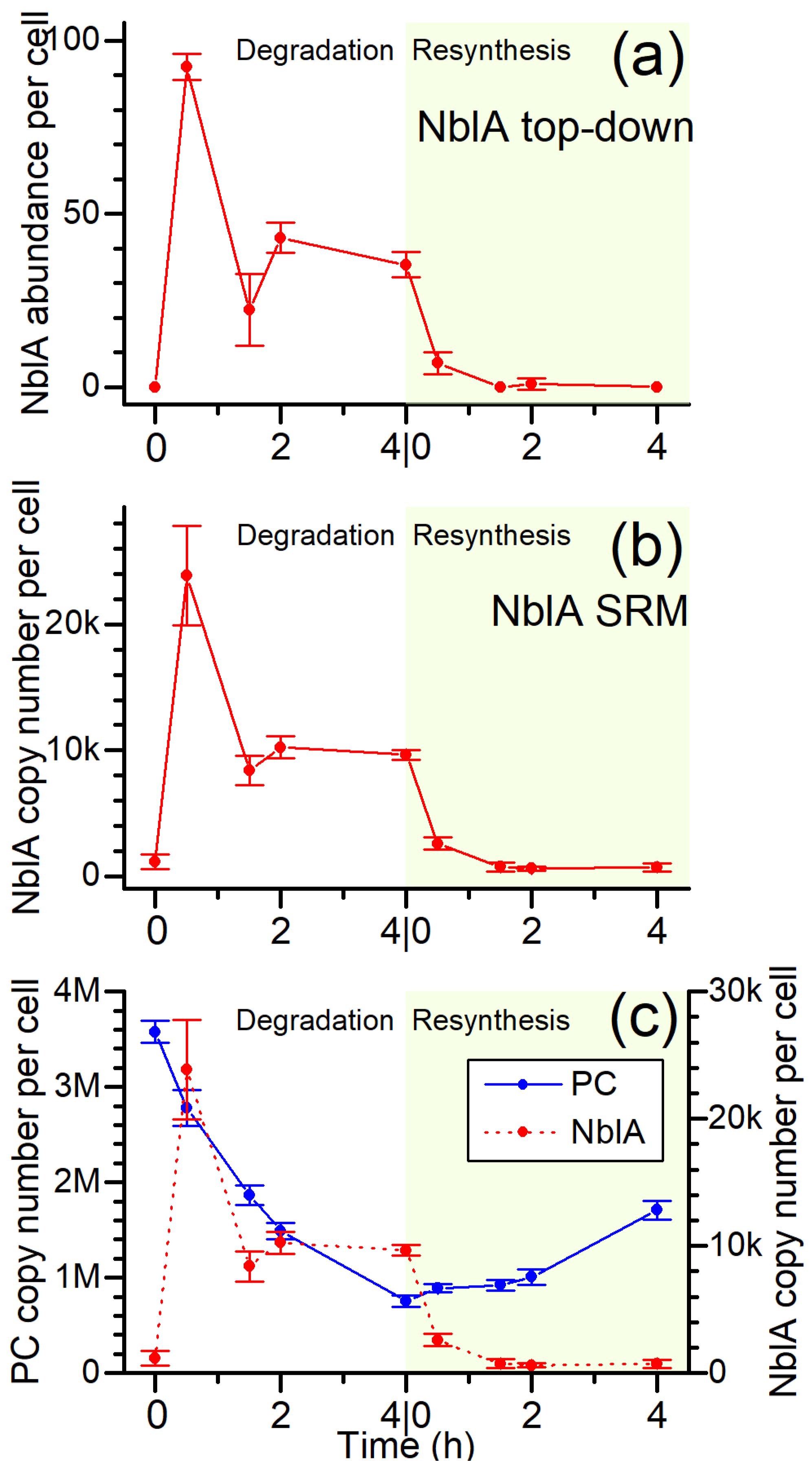
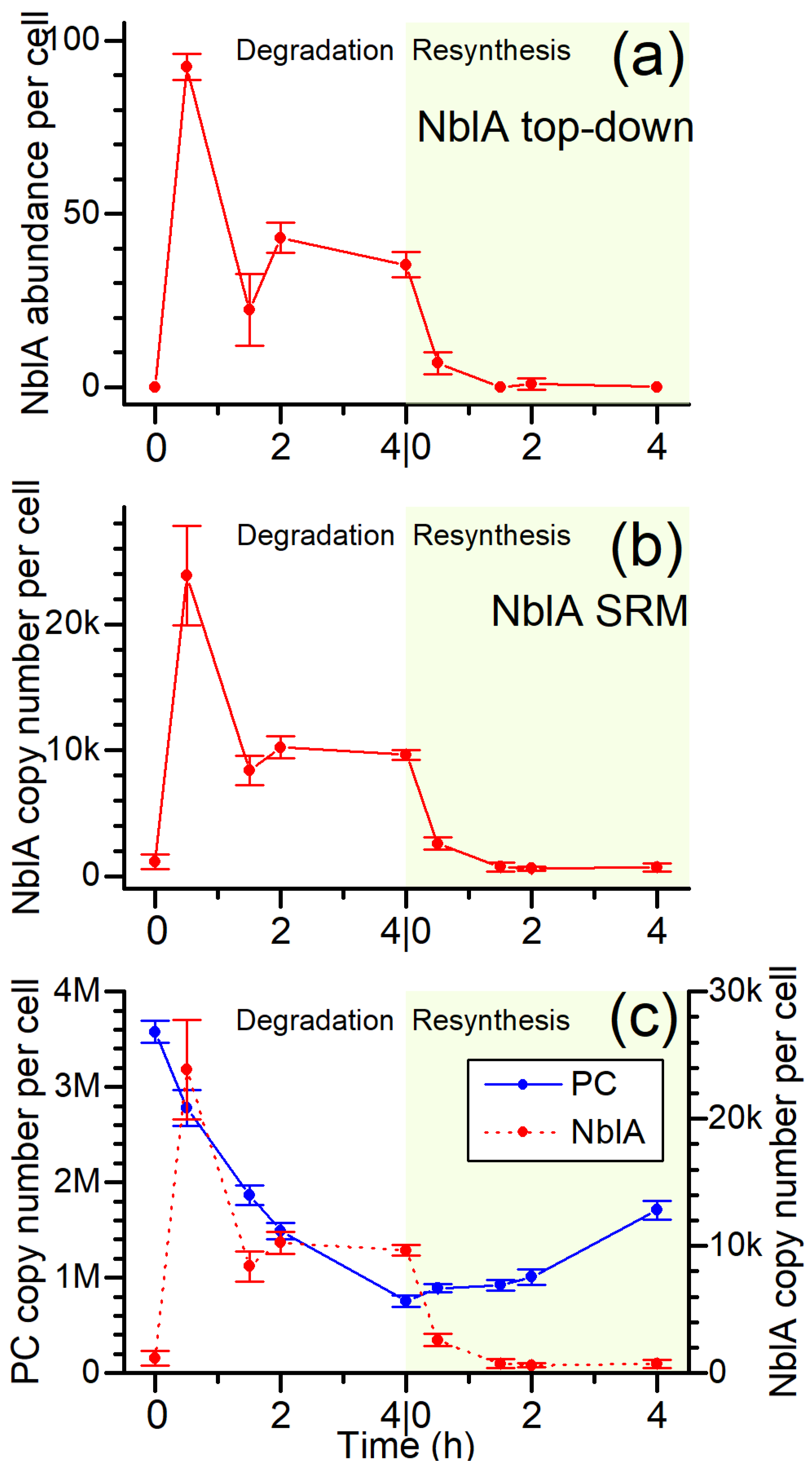
3.5. The NblA Protein Undergoes >50-Fold Induction under N Starvation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bogorad, L. Phycobiliproteins and complementary chromatic adaptation. Annu. Rev. Plant Physiol. 1975, 26, 369–401. [Google Scholar] [CrossRef]
- Gantt, E. Supramolecular membrane organization. In The Molecular Biology of Cyanobacteria; Bryant, D.A., Ed.; Springer: Dordrecht, The Netherlands, 1994; pp. 119–138. [Google Scholar]
- Yu, J.; Liberton, M.; Cliften, P.F.; Head, R.D.; Jacobs, J.M.; Smith, R.D.; Koppenaal, D.W.; Brand, J.J.; Pakrasi, H.B. Synechococcus elongatus UTEX 2973, a fast growing cyanobacterial chassis for biosynthesis using light and CO2. Sci. Rep. 2015, 5, 8132. [Google Scholar] [CrossRef] [PubMed]
- Ungerer, J.; Lin, P.C.; Chen, H.Y.; Pakrasi, H.B. Adjustments to photosystem stoichiometry and electron transfer proteins are key to the remarkably fast growth of the cyanobacterium Synechococcus elongatus UTEX 2973. mBio 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Adir, N. Elucidation of the molecular structures of components of the phycobilisome: Reconstructing a giant. Photosynth. Res. 2005, 85, 15–32. [Google Scholar] [CrossRef] [PubMed]
- Adir, N.; Dines, M.; Klartag, M.; McGregor, A.; Melamed-Frank, M. Assembly and disassembly of phycobilisomes. In Complex Intracellular Structures in Prokaryotes; Springer: Berlin/Heidelberg, Germany, 2006; Volume 2, pp. 47–77. [Google Scholar]
- Grossman, A.R.; Schaefer, M.R.; Chiang, G.G.; Collier, J.L. The phycobilisome, a light-harvesting complex responsive to environmental conditions. Microbiol. Mol. Biol. Rev. 1993, 57, 725–749. [Google Scholar]
- Arnon, D.I.; McSwain, B.D.; Tsujimoto, Y.; Wada, K. Photochemical activity and components of membrane preparetion from blue-green algea.I. Coexistance of two photosystems in relation to chlorophyll a and removal of phycocyanin. Biochim. Biophys. Acta Bioenerg. 1974, 357, 231–245. [Google Scholar] [CrossRef]
- Myers, J.; Graham, J.R.; Wang, R.T. Light harvesting in Anacystis nidulans studied in pigment mutants. Plant Physiol. 1980, 66, 1144–1149. [Google Scholar] [CrossRef]
- Arteni, A.A.; Ajlani, G.; Boekema, E.J. Structural organisation of phycobilisomes from Synechocystis sp. strain PCC 6803 and their interaction with the membrane. BBA Bioenerg. 2009, 1787, 272–279. [Google Scholar] [CrossRef]
- Zhang, J.; Ma, J.; Liu, D.; Qin, S.; Sun, S.; Zhao, J.; Sui, S.F. Structure of phycobilisome from the red alga Griffithsia pacifica. Nature 2017, 551, 57–63. [Google Scholar] [CrossRef]
- Calzadilla, P.I.; Muzzopappa, F.; Setif, P.; Kirilovsky, D. Different roles for ApcD and ApcF in Synechococcus elongatus and Synechocystis sp. PCC 6803 phycobilisomes. Biochim. Biophys. Acta Bioenerg. 2019, 1860, 488–498. [Google Scholar] [CrossRef]
- Collier, J.L.; Herbert, S.K.; Fork, D.C.; Grossman, A.R. Changes in the cyanobacterial photosynthetic apparatus during acclimation to macronutrient deprivation. Photosynth. Res. 1994, 42, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Sauer, J.; Schreiber, U.; Schmid, R.; Volker, U.; Forchhammer, K. Nitrogen starvation-induced chlorosis in Synechococcus PCC 7942. Low-level photosynthesis as a mechanism of long-term survival. Plant Physiol. 2001, 126, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, R.; Forchhammer, K. Acclimation of unicellular cyanobacteria to macronutrient deficiency: Emergence of a complex network of cellular responses. Microbiology 2005, 151, 2503–2514. [Google Scholar] [CrossRef] [PubMed]
- Collier, J.L.; Grossman, A.R. Chlorosis induced by nutrient deprivation in Synechococcus sp. strain PCC 7942: Not all bleaching is the same. J. Bacteriol. 1992, 174, 4718–4726. [Google Scholar] [CrossRef] [PubMed]
- Duke, C.S.; Cezeaux, A.; Allen, M.M. Changes in polypeptide composition of Synechocystis sp. strain 6308 phycobilisomes induced by nitrogen starvation. J. Bacteriol. 1989, 171, 1960–1966. [Google Scholar] [CrossRef]
- Lau, R.H.; MacKenzie, M.M.; Doolittle, W.F. Phycocyanin synthesis and degradation in the blue-green bacterium Anacystis nidulans. J. Bacteriol. 1977, 132, 771–778. [Google Scholar] [PubMed]
- Li, H.; Sherman, L.A. Characterization of Synechocystis sp. strain PCC 6803 and ∆nblA mutants under nitrogen-deficient conditions. Arch. Microbiol. 2002, 178, 256–266. [Google Scholar] [CrossRef]
- Murton, J.; Nagarajan, A.; Nguyen, A.Y.; Liberton, M.; Hancock, H.A.; Pakrasi, H.B.; Timlin, J.A. Population-level coordination of pigment response in individual cyanobacterial cells under altered nitrogen levels. Photosynth. Res. 2017, 134, 165–174. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, R.; Grossman, A.R. A response regulator of cyanobacteria integrates diverse environmental signals and is critical for survival under extreme conditions. Proc. Natl. Acad. Sci. USA 1998, 95, 11008–11013. [Google Scholar] [CrossRef] [Green Version]
- Dolganov, N.; Grossman, A.R. A polypeptide with similarity to phycocyanin alpha-subunit phycocyanobilin lyase involved in degradation of phycobilisomes. J. Bacteriol. 1999, 181, 610–617. [Google Scholar]
- Van Waasbergen, L.G.; Dolganov, N.; Grossman, A.R. nblS, a gene involved in controlling photosynthesis-related gene expression during high light and nutrient stress in Synechococcus elongatus PCC 7942. J. Bacteriol. 2002, 184, 2481–2490. [Google Scholar] [CrossRef] [PubMed]
- Forchhammer, K.; Schwarz, R. Nitrogen chlorosis in unicellular cyanobacteria—A developmental program for surviving nitrogen deprivation. Environ. Microbiol. 2019, 21, 1173–1184. [Google Scholar] [CrossRef] [PubMed]
- Collier, J.L.; Grossman, A.R. A small polypeptide triggers complete degradation of light-harvesting phycobiliproteins in nutrient-deprived cyanobacteria. EMBO J. 1994, 13, 1039–1047. [Google Scholar] [CrossRef] [PubMed]
- Baier, A.; Winkler, W.; Korte, T.; Lockau, W.; Karradt, A. Degradation of phycobilisomes in Synechocystis sp. PCC 6803: Evidence for essential formation of an NblA1/NblA2 heterodimer and its codegradation by a Clp protease complex. J. Biol. Chem. 2014, 289, 11755–11766. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Hein, M.Y.; Luber, C.A.; Paron, I.; Nagaraj, N.; Mann, M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteom. 2014, 13, 2513–2526. [Google Scholar] [CrossRef] [PubMed]
- Rauniyar, N.; Yates, J.R. Isobaric labeling-based relative quantification in shotgun proteomics. J. Proteome Res. 2014, 13, 5293–5309. [Google Scholar] [CrossRef] [PubMed]
- Schubert, O.T.; Röst, H.L.; Collins, B.C.; Rosenberger, G.; Aebersold, R. Quantitative proteomics: Challenges and opportunities in basic and applied research. Nat. Protoc. 2017, 12, 1289. [Google Scholar] [CrossRef] [PubMed]
- Schaffer, L.V.; Millikin, R.J.; Miller, R.M.; Anderson, L.C.; Fellers, R.T.; Ge, Y.; Kelleher, N.L.; LeDuc, R.D.; Liu, X.; Payne, S.H.; et al. Identification and quantification of proteoforms by mass spectrometry. Proteomics 2019, 19, 1800361. [Google Scholar] [CrossRef]
- Toby, T.K.; Fornelli, L.; Kelleher, N.L. Progress in top-down proteomics and the analysis of proteoforms. Ann. Rev. Anal. Chem. 2016, 9, 499–519. [Google Scholar] [CrossRef]
- Skinner, O.S.; Haverland, N.A.; Fornelli, L.; Melani, R.D.; Do Vale, L.H.F.; Seckler, H.S.; Doubleday, P.F.; Schachner, L.F.; Srzentić, K.; Kelleher, N.L.; et al. Top-down characterization of endogenous protein complexes with native proteomics. Nat. Chem. Biol. 2017, 14, 36. [Google Scholar] [CrossRef]
- Huang, H.; Lin, S.; Garcia, B.A.; Zhao, Y. Quantitative proteomic analysis of histone modifications. Chem. Rev. 2015, 115, 2376–2418. [Google Scholar] [CrossRef] [PubMed]
- Ntai, I.; Kim, K.; Fellers, R.T.; Skinner, O.S.; Smith, A.D.T.; Early, B.P.; Savaryn, J.P.; LeDuc, R.D.; Thomas, P.M.; Kelleher, N.L. Applying label-free quantitation to top down proteomics. Anal. Chem. 2014, 86, 4961–4968. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Guy, M.J.; Norman, H.S.; Chen, Y.C.; Xu, Q.; Dong, X.; Guner, H.; Wang, S.; Kohmoto, T.; Young, K.H.; et al. Top-down quantitative proteomics identified phosphorylation of cardiac troponin I as a candidate biomarker for chronic heart failure. J. Proteome Res. 2011, 10, 4054–4065. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.M. Simple conditions for growth of unicellular blue-green algae on plates. J. Phycol. 1968, 4, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Kou, Q.; Xun, L.; Liu, X. TopPIC: A software tool for top-down mass spectrometry-based proteoform identification and characterization. Bioinformatics 2016, 32, 3495–3497. [Google Scholar] [CrossRef]
- Liu, X.; Sirotkin, Y.; Shen, Y.; Anderson, G.; Tsai, Y.S.; Ting, Y.S.; Goodlett, D.R.; Smith, R.D.; Bafna, V.; Pevzner, P.A. Protein identification using top-down spectra. Mol. Cell. Proteom. 2012, 11, M111.008524. [Google Scholar] [CrossRef]
- Park, J.; Piehowski, P.D.; Wilkins, C.; Zhou, M.; Mendoza, J.; Fujimoto, G.M.; Gibbons, B.C.; Shaw, J.B.; Shen, Y.; Shukla, A.K.; et al. Informed-Proteomics: Open-source software package for top-down proteomics. Nat. Methods 2017, 14, 909. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef]
- Fusaro, V.A.; Mani, D.R.; Mesirov, J.P.; Carr, S.A. Prediction of high-responding peptides for targeted protein assays by mass spectrometry. Nat. Biotechnol. 2009, 27, 190. [Google Scholar] [CrossRef]
- Eyers, C.E.; Lawless, C.; Wedge, D.C.; Lau, K.W.; Gaskell, S.J.; Hubbard, S.J. CONSeQuence: Prediction of reference peptides for absolute quantitative proteomics using consensus machine learning approaches. Mol. Cell. Proteom. 2011, 10, M110.003384. [Google Scholar] [CrossRef]
- Shi, T.; Su, D.; Liu, T.; Tang, K.; Camp, D.G.; Qian, W.J.; Smith, R.D. Advancing the sensitivity of selected reaction monitoring-based targeted quantitative proteomics. Proteomics 2012, 12, 1074–1092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, T.; Sun, X.; Gao, Y.; Fillmore, T.L.; Schepmoes, A.A.; Zhao, R.; He, J.; Moore, R.J.; Kagan, J.; Rodland, K.D.; et al. Targeted quantification of low ng/mL level proteins in human serum without immunoaffinity depletion. J. Proteome Res. 2013, 12, 3353–3361. [Google Scholar] [CrossRef] [PubMed]
- MacLean, B.; Tomazela, D.M.; Shulman, N.; Chambers, M.; Finney, G.L.; Frewen, B.; Kern, R.; Tabb, D.L.; Liebler, D.C.; MacCoss, M.J. Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010, 26, 966–968. [Google Scholar] [CrossRef] [PubMed]
- Andrizhiyevskaya, E.G.; Chojnicka, A.; Bautista, J.A.; Diner, B.A.; van Grondelle, R.; Dekker, J.P. Origin of the F685 and F695 fluorescence in photosystem II. Photosynth. Res. 2005, 84, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Spät, P.; Klotz, A.; Rexroth, S.; Maček, B.; Forchhammer, K. Chlorosis as a developmental program in cyanobacteria: The proteomic fundament for survival and awakening. Mol. Cell. Proteom. 2018, 17, 1650–1669. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Wu, Q.; Mao, H.; Zhao, N.; Vermaas, W.F. Effects of chlorophyll availability on phycobilisomes in Synechocystis sp. PCC 6803. IUBMB Life 1999, 48, 625–630. [Google Scholar] [CrossRef] [PubMed]
- Shen, G.; Schluchter, W.M.; Bryant, D.A. Biogenesis of Phycobiliproteins: I. cpcS-I and cpcU mutants of the cyanobacterium Synechococcus sp. PCC 7002 define a heterodimeric phyococyanobilin lyase specific for β-phycocyanin and allophycocyanin subunits. J. Biol. Chem. 2008, 283, 7503–7512. [Google Scholar] [CrossRef]
- Klotz, A.V.; Glazer, A.N. gamma-N-methylasparagine in phycobiliproteins. Occurrence, location, and biosynthesis. J. Biol. Chem. 1987, 262, 17350–17355. [Google Scholar]
- Swanson, R.V.; Glazer, A.N. Phycobiliprotein methylation: Effect of the γ-N-methylasparagine residue on energy transfer in phycocyanin and the phycobilisome. J. Mol. Biol. 1990, 214, 787–796. [Google Scholar] [CrossRef]
- De Lorimier, R.; Bryant, D.A.; Stevens, S.E., Jr. Genetic analysis of a 9 kDa phycocyanin-associated linker polypeptide. Biochim. Biophys. Acta 1990, 1019, 29–41. [Google Scholar] [CrossRef]
- Paul, K.; Nieto, V.; Carlquist, W.C.; Blair, D.F.; Harshey, R.M. The c-di-GMP binding protein YcgR controls flagellar motor direction and speed to affect chemotaxis by a “backstop brake” mechanism. Mol. Cell 2010, 38, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Komenda, J.; Sobotka, R. Cyanobacterial high-light-inducible proteins—Protectors of chlorophyll–protein synthesis and assembly. Biochim. Biophys. Acta Bioenerg. 2016, 1857, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Richaud, C.; Zabulon, G.; Joder, A.; Thomas, J.C. Nitrogen or sulfur starvation differentially affects phycobilisome degradation and expression of the nblA gene in Synechocystis strain PCC 6803. J. Bacteriol. 2001, 183, 2989–2994. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.Y.; Bricker, W.P.; Zhang, H.; Weisz, D.A.; Gross, M.L.; Pakrasi, H.B. The proteolysis adaptor, NblA, binds to the N-terminus of beta-phycocyanin: Implications for the mechanism of phycobilisome degradation. Photosynth. Res. 2017, 132, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Bienert, R.; Baier, K.; Volkmer, R.; Lockau, W.; Heinemann, U. Crystal structure of NblA from Anabaena sp. PCC 7120, a small protein playing a key role in phycobilisome degradation. J. Biol. Chem. 2006, 281, 5216–5223. [Google Scholar] [CrossRef] [PubMed]
- Spät, P.; Maček, B.; Forchhammer, K. Phosphoproteome of the cyanobacterium Synechocystis sp. PCC 6803 and its dynamics during nitrogen starvation. Front. Microbiol. 2015, 6, 248. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Shortreed, M.R.; Scalf, M.; Frey, B.L.; Cesnik, A.J.; Solntsev, S.; Schaffer, L.V.; Smith, L.M. Elucidating Escherichia coli proteoform families using intact-mass proteomics and a global PTM discovery database. J. Proteome Res. 2017, 16, 4156–4165. [Google Scholar] [CrossRef]
- Brown, K.A.; Chen, B.; Guardado-Alvarez, T.M.; Lin, Z.; Hwang, L.; Ayaz-Guner, S.; Jin, S.; Ge, Y. A photocleavable surfactant for top-down proteomics. Nat. Methods 2019, 16, 417–420. [Google Scholar] [CrossRef] [PubMed]
- Ungerer, J.; Wendt, K.E.; Hendry, J.I.; Maranas, C.D.; Pakrasi, H.B. Comparative genomics reveals the molecular determinants of rapid growth of the cyanobacterium Synechococcus elongatus UTEX 2973. Proc. Natl. Acad. Sci. USA 2018, 115, E11761–E11770. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagarajan, A.; Zhou, M.; Nguyen, A.Y.; Liberton, M.; Kedia, K.; Shi, T.; Piehowski, P.; Shukla, A.; Fillmore, T.L.; Nicora, C.; et al. Proteomic Insights into Phycobilisome Degradation, A Selective and Tightly Controlled Process in The Fast-Growing Cyanobacterium Synechococcus elongatus UTEX 2973. Biomolecules 2019, 9, 374. https://doi.org/10.3390/biom9080374
Nagarajan A, Zhou M, Nguyen AY, Liberton M, Kedia K, Shi T, Piehowski P, Shukla A, Fillmore TL, Nicora C, et al. Proteomic Insights into Phycobilisome Degradation, A Selective and Tightly Controlled Process in The Fast-Growing Cyanobacterium Synechococcus elongatus UTEX 2973. Biomolecules. 2019; 9(8):374. https://doi.org/10.3390/biom9080374
Chicago/Turabian StyleNagarajan, Aparna, Mowei Zhou, Amelia Y. Nguyen, Michelle Liberton, Komal Kedia, Tujin Shi, Paul Piehowski, Anil Shukla, Thomas L. Fillmore, Carrie Nicora, and et al. 2019. "Proteomic Insights into Phycobilisome Degradation, A Selective and Tightly Controlled Process in The Fast-Growing Cyanobacterium Synechococcus elongatus UTEX 2973" Biomolecules 9, no. 8: 374. https://doi.org/10.3390/biom9080374
APA StyleNagarajan, A., Zhou, M., Nguyen, A. Y., Liberton, M., Kedia, K., Shi, T., Piehowski, P., Shukla, A., Fillmore, T. L., Nicora, C., Smith, R. D., Koppenaal, D. W., Jacobs, J. M., & Pakrasi, H. B. (2019). Proteomic Insights into Phycobilisome Degradation, A Selective and Tightly Controlled Process in The Fast-Growing Cyanobacterium Synechococcus elongatus UTEX 2973. Biomolecules, 9(8), 374. https://doi.org/10.3390/biom9080374