Cell Intrinsic and Extrinsic Mechanisms of Caveolin-1-Enhanced Metastasis

 ,
, 
Abstract
1. Introduction
2. Caveolins: Role in Physiological and Pathological Processes
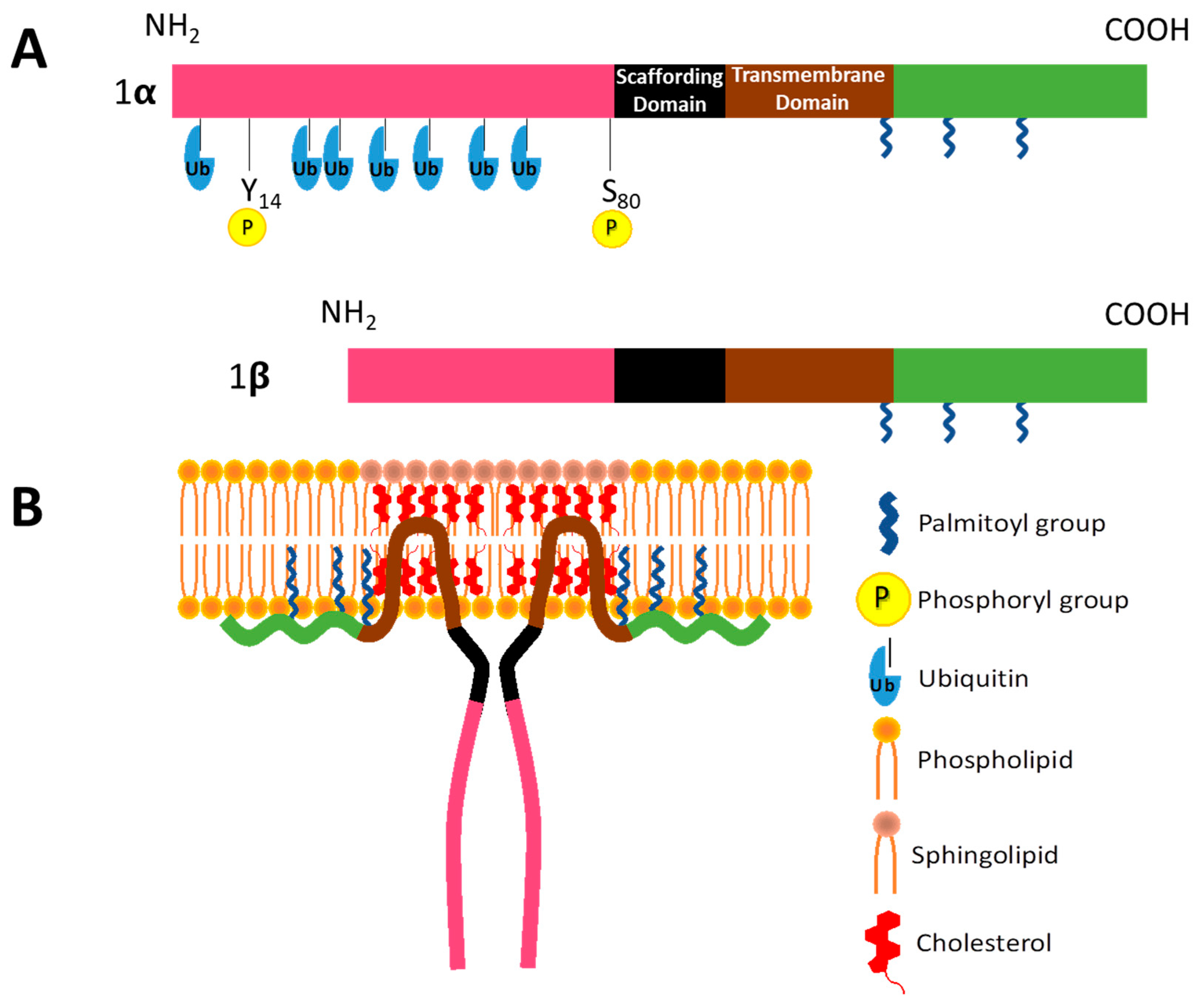
2.1. Caveolin-1 in Vesicular Transport
2.2. CAV1 in Cholesterol Homeostasis and Signal Transduction
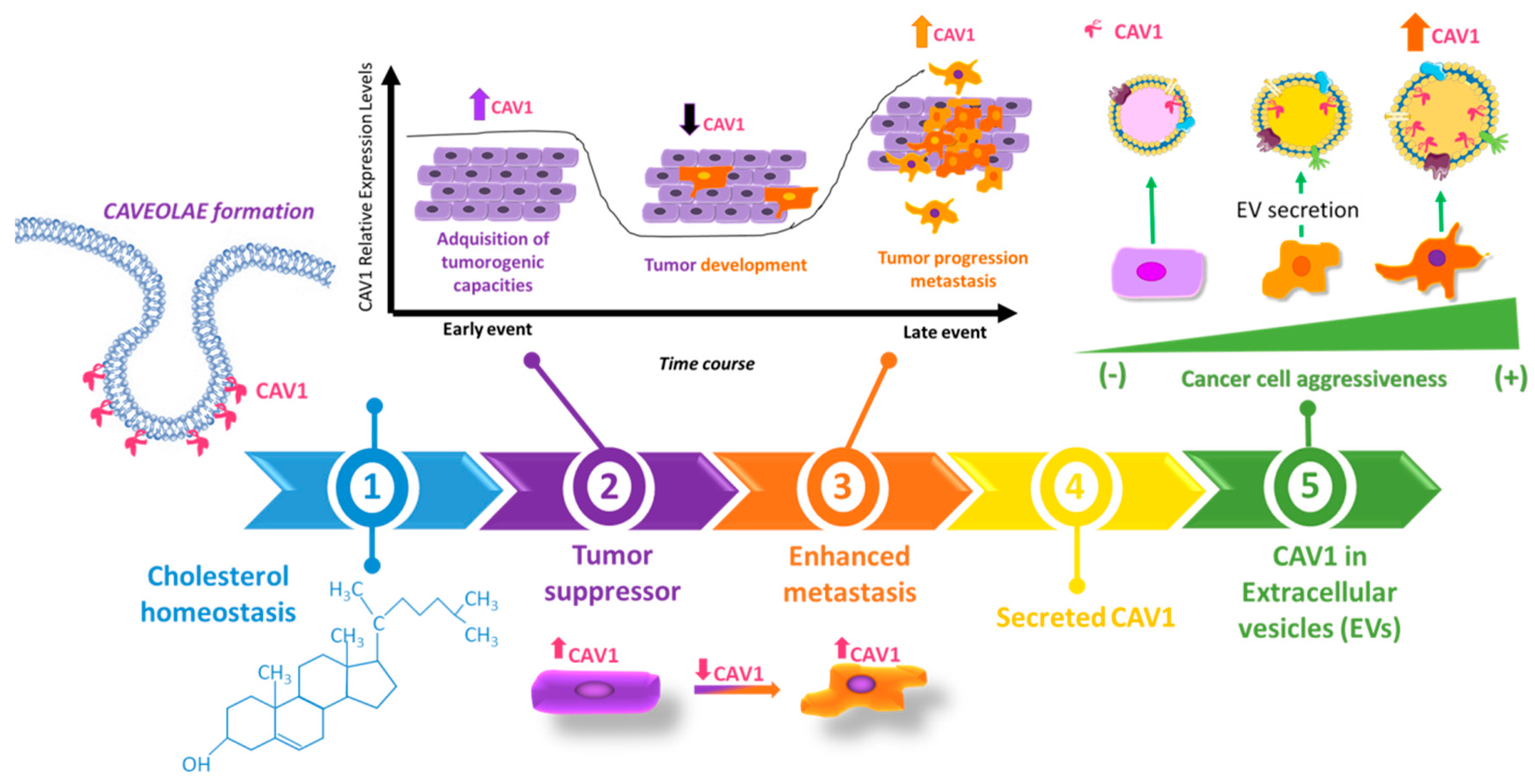
2.3. Dual Role of CAV1 in Cancer
2.4. Role of CAV1 in Metastatic Disease
2.5. Downstream Signaling of CAV1 in Advanced Cancer
2.6. CAV1 in Preclinical Studies
3. Caveolin-1 Outside of the Cell: CAV1 as a Secretable Protein
3.1. Secretable CAV1 Promotes the Acquisition of Malignant Traits in Recipient Cells
3.2. CAV1 Released in Extracellular Vesicles
3.3. CAV1-Containing EVs Promote Malignancy of Recipient Cells
4. Extracellular Vesicles in Cancer
4.1. Types of Extracellular Vesicles
4.2. Biogenesis of Exosomes
4.3. Exosomal Cargos
4.4. EV Secretion
4.5. EV Uptake
4.6. CAV1 Involved in EV Biogenesis and Protein Sorting
4.7. CAV1-Containing EVs Transport Proteins Which Promote Malignant Traits in Recipient Cells
5. Concluding Remarks
Funding
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Valastyan, S.; Weinberg, R.A. Tumor metastasis: Molecular insights and evolving paradigms. Cell 2011, 147, 275–292. [Google Scholar] [CrossRef] [PubMed]
- Syn, N.; Wang, L.; Sethi, G.; Thiery, J.P.; Goh, B.C. Exosome-Mediated Metastasis: From Epithelial-Mesenchymal Transition to Escape from Immunosurveillance. Trends Pharmacol. Sci. 2016, 37, 606–617. [Google Scholar] [CrossRef] [PubMed]
- Root, K.T.; Plucinsky, S.M.; Glover, K.J. Recent progress in the topology, structure, and oligomerization of caveolin: A building block of caveolae. Curr. Top. Membr. 2015, 75, 305–336. [Google Scholar] [PubMed]
- Rui, H.; Root, K.T.; Lee, J.; Glover, K.J.; Im, W. Probing the U-shaped conformation of caveolin-1 in a bilayer. Biophys. J. 2014, 106, 1371–1380. [Google Scholar] [CrossRef]
- Okamoto, T.; Schlegel, A.; Scherer, P.E.; Lisanti, M.P. Caveolins, a family of scaffolding proteins for organizing “preassembled signaling complexes” at the plasma membrane. J. Biol. Chem. 1998, 273, 5419–5422. [Google Scholar] [CrossRef]
- Krishna, A.; Sengupta, D. Interplay between Membrane Curvature and Cholesterol: Role of Palmitoylated Caveolin-1. Biophys. J. 2019, 116, 69–78. [Google Scholar] [CrossRef]
- Drab, M.; Verkade, P.; Elger, M.; Kasper, M.; Lohn, M.; Lauterbach, B.; Menne, J.; Lindschau, C.; Mende, F.; Luft, F.C.; et al. Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice. Science 2001, 293, 2449–2452. [Google Scholar] [CrossRef]
- Razani, B.; Engelman, J.A.; Wang, X.B.; Schubert, W.; Zhang, X.L.; Marks, C.B.; Macaluso, F.; Russell, R.G.; Li, M.; Pestell, R.G.; et al. Caveolin-1 null mice are viable but show evidence of hyperproliferative and vascular abnormalities. J. Biol. Chem. 2001, 276, 38121–38138. [Google Scholar]
- Han, B.; Copeland, C.A.; Tiwari, A.; Kenworthy, A.K. Assembly and Turnover of Caveolae: What Do We Really Know? Front. Cell Dev. Biol. 2016, 4, 68. [Google Scholar] [CrossRef]
- Fernandez, I.; Ying, Y.; Albanesi, J.; Anderson, R.G. Mechanism of caveolin filament assembly. Proc. Natl. Acad. Sci. USA 2002, 99, 11193–11198. [Google Scholar] [CrossRef] [PubMed]
- Hayer, A.; Stoeber, M.; Ritz, D.; Engel, S.; Meyer, H.H.; Helenius, A. Caveolin-1 is ubiquitinated and targeted to intralumenal vesicles in endolysosomes for degradation. J. Cell Biol. 2010, 191, 615–629. [Google Scholar] [CrossRef] [PubMed]
- Lamaze, C.; Tardif, N.; Dewulf, M.; Vassilopoulos, S.; Blouin, C.M. The caveolae dress code: Structure and signaling. Curr. Opin. Cell Biol. 2017, 47, 117–125. [Google Scholar] [CrossRef]
- Kirkham, M.; Fujita, A.; Chadda, R.; Nixon, S.J.; Kurzchalia, T.V.; Sharma, D.K.; Pagano, R.E.; Hancock, J.F.; Mayor, S.; Parton, R.G. Ultrastructural identification of uncoated caveolin-independent early endocytic vehicles. J. Cell Biol. 2005, 168, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Tagawa, A.; Mezzacasa, A.; Hayer, A.; Longatti, A.; Pelkmans, L.; Helenius, A. Assembly and trafficking of caveolar domains in the cell: Caveolae as stable, cargo-triggered, vesicular transporters. J. Cell Biol. 2005, 170, 769–779. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.W.; Hnasko, R.; Schubert, W.; Lisanti, M.P. Role of caveolae and caveolins in health and disease. Physiol. Rev. 2004, 84, 1341–1379. [Google Scholar] [CrossRef] [PubMed]
- Rothberg, K.G.; Heuser, J.E.; Donzell, W.C.; Ying, Y.S.; Glenney, J.R.; Anderson, R.G. Caveolin, a protein component of caveolae membrane coats. Cell 1992, 68, 673–682. [Google Scholar] [CrossRef]
- Bist, A.; Fielding, P.E.; Fielding, C.J. Two sterol regulatory element-like sequences mediate up-regulation of caveolin gene transcription in response to low density lipoprotein free cholesterol. Proc. Natl. Acad. Sci. USA 1997, 94, 10693–10698. [Google Scholar] [CrossRef] [PubMed]
- Hitkova, I.; Yuan, G.; Anderl, F.; Gerhard, M.; Kirchner, T.; Reu, S.; Röcken, C.; Schäfer, C.; Schmid, R.M.; Vogelmann, R.; et al. Caveolin-1 protects B6129 mice against Helicobacter pylori gastritis. PLoS Pathog. 2013, 9, e1003251. [Google Scholar] [CrossRef]
- Graf, G.A.; Connell, P.M.; van der Westhuyzen, D.R.; Smart, E.J. The class B, type I scavenger receptor promotes the selective uptake of high density lipoprotein cholesterol ethers into caveolae. J. Biol. Chem. 1999, 274, 12043–12048. [Google Scholar] [CrossRef]
- Bosch, M.; Marí, M.; Herms, A.; Fernández, A.; Fajardo, A.; Kassan, A.; Giralt, A.; Colell, A.; Balgoma, D.; Barbero, E.; et al. Caveolin-1 deficiency causes cholesterol-dependent mitochondrial dysfunction and apoptotic susceptibility. Curr. Biol. 2011, 21, 681–686. [Google Scholar] [CrossRef] [PubMed]
- Razani, B.; Woodman, S.E.; Lisanti, M.P. Caveolae: From cell biology to animal physiology. Pharmacol. Rev. 2002, 54, 431–467. [Google Scholar] [CrossRef] [PubMed]
- Ju, H.; Zou, R.; Venema, V.J.; Venema, R.C. Direct interaction of endothelial nitric-oxide synthase and caveolin-1 inhibits synthase activity. J. Biol. Chem. 1997, 272, 18522–18525. [Google Scholar] [CrossRef] [PubMed]
- Couet, J.; Sargiacomo, M.; Lisanti, M.P. Interaction of a receptor tyrosine kinase, EGF-R, with caveolins. Caveolin binding negatively regulates tyrosine and serine/threonine kinase activities. J. Biol. Chem. 1997, 272, 30429–30438. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Couet, J.; Lisanti, M.P. Src Tyrosine Kinases, Gα Subunits, and H-Ras Share a Common Membrane-Anchored Scaffolding Protein, Caveolin Caveolin Binding Negatively Regulates the Auto-Activation of Src Tyrosine Kinases. J. Biol. Chem. 1996, 271, 29182–29190. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Seitz, R.; Lisanti, M.P. Phosphorylation of Caveolin by Src Tyrosine Kinases the α-Isoform of Caveolin Is Selectively Phosphorylated by v-Src In Vivo. J. Biol. Chem. 1996, 271, 3863–3868. [Google Scholar] [CrossRef] [PubMed]
- Sanguinetti, A.R.; Mastick, C.C. c-Abl is required for oxidative stress-induced phosphorylation of caveolin-1 on tyrosine 14. Cell. Signal. 2003, 15, 289–298. [Google Scholar] [CrossRef]
- Sanguinetti, A.R.; Cao, H.; Corley Mastick, C. Fyn is required for oxidative- and hyperosmotic-stress-induced tyrosine phosphorylation of caveolin-1. Biochem. J. 2003, 376, 159–168. [Google Scholar] [CrossRef]
- Nunez-Wehinger, S.; Ortiz, R.J.; Diaz, N.; Diaz, J.; Lobos-Gonzalez, L.; Quest, A.F.G. Caveolin-1 in cell migration and metastasis. Curr. Mol. Med. 2014, 14, 255–274. [Google Scholar] [CrossRef]
- Schlegel, A.; Arvan, P.; Lisanti, M.P. Caveolin-1 binding to endoplasmic reticulum membranes and entry into the regulated secretory pathway are regulated by serine phosphorylation protein sorting at the level of the endoplasmic reticulum. J. Biol. Chem. 2001, 276, 4398–4408. [Google Scholar] [CrossRef]
- Fielding, P.E.; Chau, P.; Liu, D.; Spencer, T.A.; Fielding, C.J. Mechanism of platelet-derived growth factor-dependent caveolin-1 phosphorylation: Relationship to sterol binding and the role of serine-80. Biochemistry 2004, 43, 2578–2586. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Xu, H.; Li, Z.; Li, F. Interactions of caveolin-1 scaffolding and intramembrane regions containing a CRAC motif with cholesterol in lipid bilayers. Biochim. Biophys. Acta 2014, 1838, 2588–2599. [Google Scholar] [CrossRef] [PubMed]
- Parton, R.G.; Howes, M.T. Revisiting caveolin trafficking: The end of the caveosome. J. Cell Biol. 2010, 191, 439–441. [Google Scholar] [CrossRef] [PubMed]
- Bosch, M.; Marí, M.; Gross, S.P.; Fernández-Checa, J.C.; Pol, A. Mitochondrial cholesterol: A connection between caveolin, metabolism, and disease. Traffic 2011, 12, 1483–1489. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.R.; Jin, L.; Tian, M.X.; Jiang, X.F.; Yang, L.X.; Ding, Z.B.; Shen, Y.H.; Peng, Y.F.; Gao, D.M.; Zhou, J.; et al. Caveolin-1 promotes tumor growth and metastasis via autophagy inhibition in hepatocellular carcinoma. Clin. Res. Hepatol. Gastroenterol. 2016, 40, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Ha, T.K.; Her, N.G.; Lee, M.G.; Ryu, B.K.; Lee, J.H.; Han, J.; Jeong, S.I.; Kang, M.J.; Kim, N.H.; Kim, H.J.; et al. Caveolin-1 increases aerobic glycolysis in colorectal cancers by stimulating HMGA1-mediated GLUT3 transcription. Cancer Res. 2012, 72, 4097–4109. [Google Scholar] [CrossRef]
- Lu, Q.; Luo, X.; Mao, C.; Zheng, T.; Liu, B.; Dong, X.; Zhou, Y.; Xu, C.; Mou, X.; Wu, F.; et al. Caveolin-1 regulates autophagy activity in thyroid follicular cells and is involved in Hashimoto’s thyroiditis disease. Endocr. J. 2018. [Google Scholar] [CrossRef]
- Schwencke, C.; Braun-Dullaeus, R.C.; Wunderlich, C.; Strasser, R.H. Caveolae and caveolin in transmembrane signaling: Implications for human disease. Cardiovasc. Res. 2006, 70, 42–49. [Google Scholar] [CrossRef]
- Zou, H.; Stoppani, E.; Volonte, D.; Galbiati, F. Caveolin-1, cellular senescence and age-related diseases. Mech. Ageing Dev. 2011, 132, 533–542. [Google Scholar] [CrossRef]
- Quest, A.F.; Leyton, L.; Párraga, M. Caveolins, caveolae, and lipid rafts in cellular transport, signaling, and disease. Biochem. Cell Biol. 2004, 82, 129–144. [Google Scholar] [CrossRef]
- Quest, A.F.; Gutierrez-Pajares, J.L.; Torres, V.A. Caveolin-1: An ambiguous partner in cell signalling and cancer. J. Cell. Mol. Med. 2008, 12, 1130–1150. [Google Scholar] [CrossRef] [PubMed]
- Glenney, J.R.; Soppet, D. Sequence and expression of caveolin, a protein component of caveolae plasma membrane domains phosphorylated on tyrosine in Rous sarcoma virus-transformed fibroblasts. Proc. Natl. Acad. Sci. USA 1992, 89, 10517–10521. [Google Scholar] [CrossRef] [PubMed]
- Koleske, A.J.; Baltimore, D.; Lisanti, M.P. Reduction of caveolin and caveolae in oncogenically transformed cells. Proc. Natl. Acad. Sci. USA 1995, 92, 1381–1385. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Wykoff, C.C.; Yasuhara, S.; Song, K.S.; Okamoto, T.; Lisanti, M.P. Recombinant expression of caveolin-1 in oncogenically transformed cells abrogates anchorage-independent growth. J. Biol. Chem. 1997, 272, 16374–16381. [Google Scholar] [CrossRef] [PubMed]
- Racine, C.; Bélanger, M.; Hirabayashi, H.; Boucher, M.; Chakir, J.; Couet, J. Reduction of caveolin 1 gene expression in lung carcinoma cell lines. Biochem. Biophys. Res. Commun. 1999, 255, 580–586. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.W.; Reimer, C.L.; Oh, P.; Campbell, D.B.; Schnitzer, J.E. Tumor cell growth inhibition by caveolin re-expression in human breast cancer cells. Oncogene 1998, 16, 1391–1397. [Google Scholar] [CrossRef] [PubMed]
- Bender, F.C.; Reymond, M.A.; Bron, C.; Quest, A.F. Caveolin-1 levels are down-regulated in human colon tumors, and ectopic expression of caveolin-1 in colon carcinoma cell lines reduces cell tumorigenicity. Cancer Res. 2000, 60, 5870–5878. [Google Scholar] [PubMed]
- Torrejón, B.; Cristóbal, I.; Rojo, F.; García-Foncillas, J. Caveolin-1 Is Markedly Downregulated in Patients with Early-Stage Colorectal Cancer. World J. Surg. 2017, 41, 2625–2630. [Google Scholar] [CrossRef]
- Wiechen, K.; Diatchenko, L.; Agoulnik, A.; Scharff, K.M.; Schober, H.; Arlt, K.; Zhumabayeva, B.; Siebert, P.D.; Dietel, M.; Schäfer, R.; et al. Caveolin-1 is down-regulated in human ovarian carcinoma and acts as a candidate tumor suppressor gene. Am. J. Pathol. 2001, 159, 1635–1643. [Google Scholar] [CrossRef]
- Wiechen, K.; Sers, C.; Agoulnik, A.; Arlt, K.; Dietel, M.; Schlag, P.M.; Schneider, U. Down-regulation of caveolin-1, a candidate tumor suppressor gene, in sarcomas. Am. J. Pathol. 2001, 158, 833–839. [Google Scholar] [CrossRef]
- Manara, M.C.; Bernard, G.; Lollini, P.L.; Nanni, P.; Zuntini, M.; Landuzzi, L.; Benini, S.; Lattanzi, G.; Sciandra, M.; Serra, M.; et al. CD99 acts as an oncosuppressor in osteosarcoma. Mol. Biol. Cell 2006, 17, 1910–1921. [Google Scholar] [CrossRef] [PubMed]
- Quann, K.; Gonzales, D.M.; Mercier, I.; Wang, C.; Sotgia, F.; Pestell, R.G.; Lisanti, M.P.; Jasmin, J.F. Caveolin-1 is a negative regulator of tumor growth in glioblastoma and modulates chemosensitivity to temozolomide. Cell Cycle 2013, 12, 1510–1520. [Google Scholar] [CrossRef] [PubMed]
- Quest, A.F.G.; Lobos-González, L.; Nuñez, S.; Sanhueza, C.; Fernández, J.-G.; Aguirre, A.; Rodríguez, D.; Leyton, L.; Torres, V. The caveolin-1 connection to cell death and survival. Mol. Med. 2013, 13, 266–281. [Google Scholar] [CrossRef]
- Yang, G.; Truong, L.D.; Timme, T.L.; Ren, C.; Wheeler, T.M.; Park, S.H.; Nasu, Y.; Bangma, C.H.; Kattan, M.W.; Scardino, P.T.; et al. Elevated expression of caveolin is associated with prostate and breast cancer. Clin. Cancer Res. 1998, 4, 1873–1880. [Google Scholar] [PubMed]
- Li, L.; Yang, G.; Ebara, S.; Satoh, T.; Nasu, Y.; Timme, T.L.; Ren, C.; Wang, J.; Tahir, S.A.; Thompson, T.C. Caveolin-1 mediates testosterone-stimulated survival/clonal growth and promotes metastatic activities in prostate cancer cells. Cancer Res. 2001, 61, 4386–4392. [Google Scholar]
- Yang, G.; Truong, L.D.; Wheeler, T.M.; Thompson, T.C. Caveolin-1 expression in clinically confined human prostate cancer: A novel prognostic marker. Cancer Res. 1999, 59, 5719–5723. [Google Scholar]
- Lobos-González, L.; Aguilar, L.; Diaz, J.; Diaz, N.; Urra, H.; Torres, V.A.; Silva, V.; Fitzpatrick, C.; Lladser, A.; Hoek, K.S.; et al. E-cadherin determines Caveolin-1 tumor suppression or metastasis enhancing function in melanoma cells. Pigment Cell Melanoma Res. 2013, 26, 555–570. [Google Scholar] [CrossRef]
- Lobos-Gonzalez, L.; Aguilar-Guzmán, L.; Fernandez, J.G.; Muñoz, N.; Hossain, M.; Bieneck, S.; Silva, V.; Burzio, V.; Sviderskaya, E.V.; Bennett, D.C.; et al. Caveolin-1 is a risk factor for postsurgery metastasis in preclinical melanoma models. Melanoma Res. 2014, 24, 108–119. [Google Scholar] [CrossRef]
- Logozzi, M.; De Milito, A.; Lugini, L.; Borghi, M.; Calabrò, L.; Spada, M.; Perdicchio, M.; Marino, M.L.; Federici, C.; Iessi, E.; et al. High levels of exosomes expressing CD63 and caveolin-1 in plasma of melanoma patients. PLoS ONE 2009, 4, e5219. [Google Scholar] [CrossRef]
- Janković, J.; Tatić, S.; Božić, V.; Živaljević, V.; Cvejić, D.; Paskaš, S. Inverse expression of caveolin-1 and EGFR in thyroid cancer patients. Hum. Pathol. 2017, 61, 164–172. [Google Scholar] [CrossRef]
- Torres, V.A.; Tapia, J.C.; Rodríguez, D.A.; Párraga, M.; Lisboa, P.; Montoya, M.; Leyton, L.; Quest, A.F. Caveolin-1 controls cell proliferation and cell death by suppressing expression of the inhibitor of apoptosis protein survivin. J. Cell Sci. 2006, 119, 1812–1823. [Google Scholar] [CrossRef] [PubMed]
- Torres, V.A.; Tapia, J.C.; Rodriguez, D.A.; Lladser, A.; Arredondo, C.; Leyton, L.; Quest, A.F. E-cadherin is required for caveolin-1-mediated down-regulation of the inhibitor of apoptosis protein survivin via reduced beta-catenin-Tcf/Lef-dependent transcription. Mol. Cell. Biol. 2007, 27, 7703–7717. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, D.A.; Tapia, J.C.; Fernandez, J.G.; Torres, V.A.; Muñoz, N.; Galleguillos, D.; Leyton, L.; Quest, A.F. Caveolin-1-mediated suppression of cyclooxygenase-2 via a beta-catenin-Tcf/Lef-dependent transcriptional mechanism reduced prostaglandin E2 production and survivin expression. Mol. Biol. Cell 2009, 20, 2297–2310. [Google Scholar] [CrossRef] [PubMed]
- Fernández, J.G.; Rodríguez, D.A.; Valenzuela, M.; Calderon, C.; Urzúa, U.; Munroe, D.; Rosas, C.; Lemus, D.; Díaz, N.; Wright, M.C.; et al. Survivin expression promotes VEGF-induced tumor angiogenesis via PI3K/Akt enhanced β-catenin/Tcf-Lef dependent transcription. Mol. Cancer 2014, 13, 209. [Google Scholar] [CrossRef] [PubMed]
- Senetta, R.; Stella, G.; Pozzi, E.; Sturli, N.; Massi, D.; Cassoni, P. Caveolin-1 as a promoter of tumour spreading: When, how, where and why. J. Cell. Mol. Med. 2013, 17, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Navarro, A.; Anand-Apte, B.; Parat, M.O. A role for caveolae in cell migration. FASEB J. 2004, 18, 1801–1811. [Google Scholar] [CrossRef] [PubMed]
- Urra, H.; Torres, V.A.; Ortiz, R.J.; Lobos, L.; Díaz, M.I.; Díaz, N.; Härtel, S.; Leyton, L.; Quest, A.F. Caveolin-1-enhanced motility and focal adhesion turnover require tyrosine-14 but not accumulation to the rear in metastatic cancer cells. PLoS ONE 2012, 7, e33085. [Google Scholar] [CrossRef] [PubMed]
- Joshi, B.; Strugnell, S.S.; Goetz, J.G.; Kojic, L.D.; Cox, M.E.; Griffith, O.L.; Chan, S.K.; Jones, S.J.; Leung, S.P.; Masoudi, H.; et al. Phosphorylated caveolin-1 regulates Rho/ROCK-dependent focal adhesion dynamics and tumor cell migration and invasion. Cancer Res. 2008, 68, 8210–8220. [Google Scholar] [CrossRef]
- Goetz, J.G.; Joshi, B.; Lajoie, P.; Strugnell, S.S.; Scudamore, T.; Kojic, L.D.; Nabi, I.R. Concerted regulation of focal adhesion dynamics by galectin-3 and tyrosine-phosphorylated caveolin-1. J. Cell Biol. 2008, 180, 1261–1275. [Google Scholar] [CrossRef]
- Shatz, M.; Lustig, G.; Reich, R.; Liscovitch, M. Caveolin-1 mutants P132L and Y14F are dominant negative regulators of invasion, migration and aggregation in H1299 lung cancer cells. Exp. Cell Res. 2010, 316, 1748–1762. [Google Scholar] [CrossRef]
- Ortiz, R.; Díaz, J.; Díaz, N.; Lobos-Gonzalez, L.; Cárdenas, A.; Contreras, P.; Díaz, M.I.; Otte, E.; Cooper-White, J.; Torres, V.; et al. Extracellular matrix-specific Caveolin-1 phosphorylation on tyrosine 14 is linked to augmented melanoma metastasis but not tumorigenesis. Oncotarget 2016, 7, 40571–40593. [Google Scholar] [CrossRef]
- Grande-García, A.; Echarri, A.; de Rooij, J.; Alderson, N.B.; Waterman-Storer, C.M.; Valdivielso, J.M.; del Pozo, M.A. Caveolin-1 regulates cell polarization and directional migration through Src kinase and Rho GTPases. J. Cell Biol. 2007, 177, 683–694. [Google Scholar] [CrossRef]
- Isshiki, M.; Ando, J.; Yamamoto, K.; Fujita, T.; Ying, Y.; Anderson, R.G. Sites of Ca(2+) wave initiation move with caveolae to the trailing edge of migrating cells. J. Cell Sci. 2002, 115, 475–484. [Google Scholar]
- Díaz, J.; Mendoza, P.; Ortiz, R.; Díaz, N.; Leyton, L.; Stupack, D.; Quest, A.F.; Torres, V.A. Rab5 is required in metastatic cancer cells for Caveolin-1-enhanced Rac1 activation, migration and invasion. J. Cell Sci. 2014, 127, 2401–2406. [Google Scholar] [CrossRef]
- Codenotti, S.; Faggi, F.; Ronca, R.; Chiodelli, P.; Grillo, E.; Guescini, M.; Megiorni, F.; Marampon, F.; Fanzani, A. Caveolin-1 enhances metastasis formation in a human model of embryonal rhabdomyosarcoma through Erk signaling cooperation. Cancer Lett. 2019, 449, 135–144. [Google Scholar] [CrossRef]
- Li, S.; Chen, Y.; Zhang, Y.; Jiang, X.; Jiang, Y.; Qin, X.; Yang, H.; Wu, C.; Liu, Y. Shear stress promotes anoikis resistance of cancer cells via caveolin-1-dependent extrinsic and intrinsic apoptotic pathways. J. Cell. Physiol. 2019, 234, 3730–3743. [Google Scholar] [CrossRef]
- Li, L.; Zhang, K.; Lu, C.; Sun, Q.; Zhao, S.; Jiao, L.; Han, R.; Lin, C.; Jiang, J.; Zhao, M.; et al. Caveolin-1-mediated STAT3 activation determines electrotaxis of human lung cancer cells. Oncotarget 2017, 8, 95741. [Google Scholar] [CrossRef]
- Mi, L.; Zhu, F.; Yang, X.; Lu, J.; Zheng, Y.; Zhao, Q.; Wen, X.; Lu, A.; Wang, M.; Zheng, M.; et al. The metastatic suppressor NDRG1 inhibits EMT, migration and invasion through interaction and promotion of caveolin-1 ubiquitylation in human colorectal cancer cells. Oncogene 2017, 36, 4323. [Google Scholar] [CrossRef]
- Ruan, H.; Li, X.; Yang, H.; Song, Z.; Tong, J.; Cao, Q.; Wang, K.; Xiao, W.; Xiao, H.; Chen, X.; et al. Enhanced expression of caveolin-1 possesses diagnostic and prognostic value and promotes cell migration, invasion and sunitinib resistance in the clear cell renal cell carcinoma. Exp. Cell Res. 2017, 358, 269–278. [Google Scholar] [CrossRef]
- Mao, X.; Wong, S.Y.S.; Tse, E.Y.T.; Ko, F.C.F.; Tey, S.K.; Yeung, Y.S.; Man, K.; Lo, R.C.L.; Ng, I.O.L.; Yam, J.W.P. Mechanisms through which hypoxia-induced caveolin-1 drives tumorigenesis and metastasis in hepatocellular carcinoma. Cancer Res. 2016, 76, 7242–7253. [Google Scholar] [CrossRef]
- Lagares-Tena, L.; García-Monclús, S.; López-Alemany, R.; Almacellas-Rabaiget, O.; Huertas-Martínez, J.; Sáinz-Jaspeado, M.; Mateo-Lozano, S.; Rodríguez-Galindo, C.; Rello-Varona, S.; Herrero-Martín, D.; et al. Caveolin-1 promotes Ewing sarcoma metastasis regulating MMP-9 expression through MAPK/ERK pathway. Oncotarget 2016, 7, 56889. [Google Scholar] [CrossRef]
- Luan, T.Y.; Zhu, T.N.; Cui, Y.J.; Zhang, G.; Song, X.J.; Gao, D.M.; Zhang, Y.M.; Zhao, Q.L.; Liu, S.; Su, T.Y.; et al. Correction: Mechanosensitive caveolin-1 activation-induced PI3K/Akt/mTOR signaling pathway promotes breast cancer motility, invadopodia formation and metastasis In Vivo. Oncotarget 2018, 9, 32730. [Google Scholar]
- Stenzel, M.; Tura, A.; Nassar, K.; Rohrbach, J.M.; Grisanti, S.; Lüke, M.; Lüke, J. Analysis of caveolin-1 and phosphoinositol-3 kinase expression in primary uveal melanomas. Clin. Exp. Ophthalmol. 2016, 44, 400–409. [Google Scholar] [CrossRef]
- Nassar, Z.D.; Hill, M.M.; Parton, R.G.; Francois, M.; Parat, M.O. Non-caveolar caveolin-1 expression in prostate cancer cells promotes lymphangiogenesis. Oncoscience 2015, 2, 635. [Google Scholar] [CrossRef][Green Version]
- Luan, T.Y.; Zhu, T.N.; Cui, Y.J.; Zhang, G.; Song, X.J.; Gao, D.M.; Zhang, Y.M.; Zhao, Q.L.; Liu, S.; Su, T.Y.; et al. Expression of caveolin-1 is correlated with lung adenocarcinoma proliferation, migration, and invasion. Med. Oncol. 2015, 32, 207. [Google Scholar] [CrossRef]
- Chatterjee, M.; Ben-Josef, E.; Thomas, D.G.; Morgan, M.A.; Zalupski, M.M.; Khan, G.; Robinson, C.A.; Griffith, K.A.; Chen, C.S.; Ludwig, T.; et al. Caveolin-1 is associated with tumor progression and confers a multi-modality resistance phenotype in pancreatic cancer. Sci. Rep. 2015, 5, 10867. [Google Scholar] [CrossRef]
- Joglekar, M.; Elbezanti, W.O.; Weitzman, M.D.; Lehman, H.L.; van Golen, K.L. Caveolin-1 mediates inflammatory breast cancer cell invasion via the Akt1 pathway and RhoC GTPase. J. Cell. Biochem. 2015, 116, 923–933. [Google Scholar] [CrossRef]
- Chanvorachote, P.; Pongrakhananon, V.; Halim, H. Caveolin-1 regulates metastatic behaviors of anoikis resistant lung cancer cells. Mol. Cell. Biochem. 2015, 399, 291–302. [Google Scholar] [CrossRef]
- Wang, R.; Li, Z.; Guo, H.; Shi, W.; Xin, Y.; Chang, W.; Huang, T. Caveolin 1 knockdown inhibits the proliferation, migration and invasion of human breast cancer BT474 cells. Mol. Med. Rep. 2014, 9, 1723–1728. [Google Scholar] [CrossRef][Green Version]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef]
- Bailey, K.M.; Liu, J. Caveolin-1 up-regulation during epithelial to mesenchymal transition is mediated by focal adhesion kinase. J. Biol. Chem. 2008, 283, 13714–13724. [Google Scholar] [CrossRef]
- Diaz, J.E.; Díaz, N.; Leyton, L.; Torres, V.A.; Quest, A.F. Molecular insights to a novel Caveolin-1-Rab5-Rac-1 signaling pathway important for metastatic cancer cell migration and invasion. Cancer Cell Microenviron. 2014, 1. [Google Scholar] [CrossRef]
- Wang, K.; Zhu, X.; Chen, Y.; Yin, Y.; Ma, T. Tubeimoside V sensitizes human triple negative breast cancer MDA-MB-231 cells to anoikis via regulating caveolin-1-related signaling pathways. Arch. Biochem. Biophys. 2018, 646, 10–15. [Google Scholar] [CrossRef]
- Lv, W.; Chen, N.; Lin, Y.; Ma, H.; Ruan, Y.; Li, Z.; Li, X.; Pan, X.; Tian, X. Macrophage migration inhibitory factor promotes breast cancer metastasis via activation of HMGB1/TLR4/NF kappa B axis. Cancer Lett. 2016, 375, 245–255. [Google Scholar] [CrossRef]
- Gai, X.; Lu, Z.; Tu, K.; Liang, Z.; Zheng, X. Caveolin-1 is up-regulated by GLI1 and contributes to GLI1-driven EMT in hepatocellular carcinoma. PLoS ONE 2014, 9, e84551. [Google Scholar] [CrossRef]
- Felicetti, F.; Parolini, I.; Bottero, L.; Fecchi, K.; Errico, M.C.; Raggi, C.; Biffoni, M.; Spadaro, F.; Lisanti, M.P.; Sargiacomo, M.; et al. Caveolin-1 tumor-promoting role in human melanoma. Int. J. Cancer 2009, 125, 1514–1522. [Google Scholar] [CrossRef]
- Ho, C.C.; Huang, P.H.; Huang, H.Y.; Chen, Y.H.; Yang, P.C.; Hsu, S.M. Up-regulated caveolin-1 accentuates the metastasis capability of lung adenocarcinoma by inducing filopodia formation. Am. J. Pathol. 2002, 161, 1647–1656. [Google Scholar] [CrossRef]
- Jaffe, A.B.; Hall, A. Rho GTPases: Biochemistry and biology. Annu. Rev. Cell Dev. Biol. 2005, 21, 247–269. [Google Scholar] [CrossRef]
- Davis, M.J.; Ha, B.H.; Holman, E.C.; Halaban, R.; Schlessinger, J.; Boggon, T.J. RAC1P29S is a spontaneously activating cancer-associated GTPase. Proc. Natl. Acad. Sci. USA 2013, 110, 912–917. [Google Scholar] [CrossRef]
- Beardsley, A.; Fang, K.; Mertz, H.; Castranova, V.; Friend, S.; Liu, J. Loss of caveolin-1 polarity impedes endothelial cell polarization and directional movement. J. Biol. Chem. 2005, 280, 3541–3547. [Google Scholar] [CrossRef]
- Wei, Y.; Yang, X.; Liu, Q.; Wilkins, J.A.; Chapman, H.A. A role for caveolin and the urokinase receptor in integrin-mediated adhesion and signaling. J. Cell Biol. 1999, 144, 1285–1294. [Google Scholar] [CrossRef]
- Schmidt-Glenewinkel, H.; Reinz, E.; Bulashevska, S.; Beaudouin, J.; Legewie, S.; Alonso, A.; Eils, R. Multiparametric image analysis reveals role of Caveolin1 in endosomal progression rather than internalization of EGFR. FEBS Lett. 2012, 586, 1179–1189. [Google Scholar] [CrossRef]
- Shi, F.; Sottile, J. Caveolin-1-dependent beta1 integrin endocytosis is a critical regulator of fibronectin turnover. J. Cell Sci. 2008, 121, 2360–2371. [Google Scholar] [CrossRef]
- Orlichenko, L.; Weller, S.G.; Cao, H.; Krueger, E.W.; Awoniyi, M.; Beznoussenko, G.; Buccione, R.; McNiven, M.A. Caveolae mediate growth factor-induced disassembly of adherens junctions to support tumor cell dissociation. Mol. Biol. Cell 2009, 20, 4140–4152. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef]
- Yang, B.; Wang, N.; Wang, S.; Li, X.; Zheng, Y.; Li, M.; Song, J.; Zhang, F.; Mei, W.; Lin, Y.; et al. Network-pharmacology-based identification of caveolin-1 as a key target of Oldenlandia diffusa to suppress breast cancer metastasis. Biomed. Pharmacother. 2019, 112, 108607. [Google Scholar] [CrossRef]
- Tahir, S.A.; Yang, G.; Ebara, S.; Timme, T.L.; Satoh, T.; Li, L.; Goltsov, A.; Ittmann, M.; Morrisett, J.D.; Thompson, T.C. Secreted caveolin-1 stimulates cell survival/clonal growth and contributes to metastasis in androgen-insensitive prostate cancer. Cancer Res. 2001, 61, 3882–3885. [Google Scholar]
- Huang, C.; Qiu, Z.; Wang, L.; Peng, Z.; Jia, Z.; Logsdon, C.D.; Le, X.; Wei, D.; Huang, S.; Xie, K. A novel FoxM1-caveolin signaling pathway promotes pancreatic cancer invasion and metastasis. Cancer Res. 2012, 72, 655–665. [Google Scholar] [CrossRef]
- Thomas, S.; Overdevest, J.B.; Nitz, M.D.; Williams, P.D.; Owens, C.R.; Sanchez-Carbayo, M.; Frierson, H.F.; Schwartz, M.A.; Theodorescu, D. Src and caveolin-1 reciprocally regulate metastasis via a common downstream signaling pathway in bladder cancer. Cancer Res. 2011, 71, 832–841. [Google Scholar] [CrossRef]
- Liu, P.; Li, W.P.; Machleidt, T.; Anderson, R.G. Identification of caveolin-1 in lipoprotein particles secreted by exocrine cells. Nat. Cell Biol. 1999, 1, 369–375. [Google Scholar] [CrossRef]
- Li, W.P.; Liu, P.; Pilcher, B.K.; Anderson, R.G. Cell-specific targeting of caveolin-1 to caveolae, secretory vesicles, cytoplasm or mitochondria. J. Cell Sci. 2001, 114, 1397–1408. [Google Scholar]
- Bartz, R.; Zhou, J.; Hsieh, J.T.; Ying, Y.; Li, W.; Liu, P. Caveolin-1 secreting LNCaP cells induce tumor growth of caveolin-1 negative LNCaP cells In Vivo. Int. J. Cancer 2008, 122, 520–525. [Google Scholar] [CrossRef]
- Sengupta, A.; Mateo-Lozano, S.; Tirado, O.M.; Notario, V. Auto-stimulatory action of secreted caveolin-1 on the proliferation of Ewing’s sarcoma cells. Int. J. Oncol. 2011, 38, 1259–1265. [Google Scholar]
- Lugini, L.; Matarrese, P.; Tinari, A.; Lozupone, F.; Federici, C.; Iessi, E.; Gentile, M.; Luciani, F.; Parmiani, G.; Rivoltini, L.; et al. Cannibalism of live lymphocytes by human metastatic but not primary melanoma cells. Cancer Res. 2006, 66, 3629–3638. [Google Scholar] [CrossRef]
- Llorente, A.; de Marco, M.C.; Alonso, M.A. Caveolin-1 and MAL are located on prostasomes secreted by the prostate cancer PC-3 cell line. J. Cell Sci. 2004, 117, 5343–5351. [Google Scholar] [CrossRef]
- Sawada, N.; Taketani, Y.; Amizuka, N.; Ichikawa, M.; Ogawa, C.; Nomoto, K.; Nashiki, K.; Sato, T.; Arai, H.; Isshiki, M.; et al. Caveolin-1 in extracellular matrix vesicles secreted from osteoblasts. Bone 2007, 41, 52–58. [Google Scholar] [CrossRef]
- Parolini, I.; Federici, C.; Raggi, C.; Lugini, L.; Palleschi, S.; De Milito, A.; Coscia, C.; Iessi, E.; Logozzi, M.; Molinari, A.; et al. Microenvironmental pH is a key factor for exosome traffic in tumor cells. J. Biol. Chem. 2009, 284, 34211–34222. [Google Scholar] [CrossRef]
- Di Vizio, D.; Morello, M.; Dudley, A.C.; Schow, P.W.; Adam, R.M.; Morley, S.; Mulholland, D.; Rotinen, M.; Hager, M.H.; Insabato, L.; et al. Large oncosomes in human prostate cancer tissues and in the circulation of mice with metastatic disease. Am. J. Pathol. 2012, 181, 1573–1584. [Google Scholar] [CrossRef]
- Crewe, C.; Joffin, N.; Rutkowski, J.M.; Kim, M.; Zhang, F.; Towler, D.A.; Gordillo, R.; Scherer, P.E. An endothelial-to-adipocyte extracellular vesicle axis governed by metabolic state. Cell 2018, 175, 695–708. [Google Scholar] [CrossRef]
- Kucharzewska, P.; Christianson, H.C.; Welch, J.E.; Svensson, K.J.; Fredlund, E.; Ringnér, M.; Mörgelin, M.; Bourseau-Guilmain, E.; Bengzon, J.; Belting, M. Exosomes reflect the hypoxic status of glioma cells and mediate hypoxia-dependent activation of vascular cells during tumor development. Proc. Natl. Acad. Sci. USA 2013, 110, 7312–7317. [Google Scholar] [CrossRef]
- He, M.; Qin, H.; Poon, T.C.W.; Sze, S.C.; Ding, X.; Co, N.N.; Ngai, S.M.; Chan, T.F.; Wong, N. Hepatocellular carcinoma-derived exosomes promote motility of immortalized hepatocyte through transfer of oncogenic proteins and RNAs. Carcinogenesis 2015, 36, 1008–1018. [Google Scholar] [CrossRef]
- Campos, A.; Salomon, C.; Bustos, R.; Díaz, J.; Martínez, S.; Silva, V.; Reyes, C.; Díaz-Valdivia, N.; Varas-Godoy, M.; Lobos-González, L.; et al. Caveolin-1-containing extracellular vesicles transport adhesion proteins and promote malignancy in breast cancer cell lines. Nanomedicine 2018, 13, 2597–2609. [Google Scholar] [CrossRef]
- Shao, H.; Im, H.; Castro, C.M.; Breakefield, X.; Weissleder, R.; Lee, H. New Technologies for Analysis of Extracellular Vesicles. Chem. Rev. 2018, 118, 1917–1950. [Google Scholar] [CrossRef]
- Johnstone, R.M.; Adam, M.; Hammond, J.R.; Orr, L.; Turbide, C. Vesicle formation during reticulocyte maturation. Association of plasma membrane activities with released vesicles (exosomes). J. Biol. Chem. 1987, 262, 9412–9420. [Google Scholar]
- Sagredo, A.I.; Sepulveda, S.A.; Roa, J.C.; Oróstica, L. Exosomes in bile as potential pancreatobiliary tumor biomarkers. Transl. Cancer Res. 2017, 6, S1371–S1383. [Google Scholar] [CrossRef]
- Czernek, L.; Duchler, M. Functions of Cancer-Derived Extracellular Vesicles in Immunosuppression. Arch. Immunol. Ther. Exp. 2017, 65, 311–323. [Google Scholar] [CrossRef]
- Liu, M.X.; Liao, J.; Xie, M.; Gao, Z.K.; Wang, X.H.; Zhang, Y.; Shang, M.H.; Yin, L.H.; Pu, Y.P.; Liu, R. miR-93-5p Transferred by Exosomes Promotes the Proliferation of Esophageal Cancer Cells via Intercellular Communication by Targeting PTEN. Biomed. Environ. Sci. 2018, 31, 171–185. [Google Scholar]
- La Shu, S.; Yang, Y.; Allen, C.L.; Maguire, O.; Minderman, H.; Sen, A.; Ciesielski, M.J.; Collins, K.A.; Bush, P.J.; Singh, P.; et al. Metabolic reprogramming of stromal fibroblasts by melanoma exosome microRNA favours a pre-metastatic microenvironment. Sci. Rep. 2018, 8, 12905. [Google Scholar] [CrossRef]
- Pfeiler, S.; Thakur, M.; Grünauer, P.; Megens, R.T.A.; Joshi, U.; Coletti, R.; Samara, V.; Müller-Stoy, G.; Ishikawa-Ankerhold, H.; Stark, K.; et al. CD36-triggered cell invasion and persistent tissue colonization by tumor microvesicles during metastasis. FASEB J. 2018, 33, 1860–1872. [Google Scholar] [CrossRef]
- Kahlert, C.; Kalluri, R. Exosomes in tumor microenvironment influence cancer progression and metastasis. J. Mol. Med. 2013, 91, 431–437. [Google Scholar] [CrossRef]
- van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- Zijlstra, A.; Di Vizio, D. Size matters in nanoscale communication. Nat. Cell Biol. 2018, 20, 228–230. [Google Scholar] [CrossRef]
- Tricarico, C.; Clancy, J.; D’Souza-Schorey, C. Biology and biogenesis of shed microvesicles. Small GTPases 2017, 8, 220–232. [Google Scholar] [CrossRef]
- Raposo, G.; Nijman, H.W.; Stoorvogel, W.; Liejendekker, R.; Harding, C.V.; Melief, C.J.; Geuze, H.J. B lymphocytes secrete antigen-presenting vesicles. J. Exp. Med. 1996, 183, 1161–1172. [Google Scholar] [CrossRef]
- Raiborg, C.; Rusten, T.E.; Stenmark, H. Protein sorting into multivesicular endosomes. Curr. Opin. Cell Biol. 2003, 15, 446–455. [Google Scholar] [CrossRef]
- Colombo, M.; Moita, C.; van Niel, G.; Kowal, J.; Vigneron, J.; Benaroch, P.; Manel, N.; Moita, L.F.; Thery, C.; Raposo, G. Analysis of ESCRT functions in exosome biogenesis, composition and secretion highlights the heterogeneity of extracellular vesicles. J. Cell Sci. 2013, 126, 5553–5565. [Google Scholar] [CrossRef]
- Trajkovic, K.; Hsu, C.; Chiantia, S.; Rajendran, L.; Wenzel, D.; Wieland, F.; Schwille, P.; Brugger, B.; Simons, M. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 2008, 319, 1244–1247. [Google Scholar] [CrossRef]
- Chairoungdua, A.; Smith, D.L.; Pochard, P.; Hull, M.; Caplan, M.J. Exosome release of beta-catenin: A novel mechanism that antagonizes Wnt signaling. J. Cell Biol. 2010, 190, 1079–1091. [Google Scholar] [CrossRef]
- Kajimoto, T.; Okada, T.; Miya, S.; Zhang, L.; Nakamura, S. Ongoing activation of sphingosine 1-phosphate receptors mediates maturation of exosomal multivesicular endosomes. Nat. Commun. 2013, 4, 2712. [Google Scholar] [CrossRef]
- Phuyal, S.; Hessvik, N.P.; Skotland, T.; Sandvig, K.; Llorente, A. Regulation of exosome release by glycosphingolipids and flotillins. FEBS J. 2014, 281, 2214–2227. [Google Scholar] [CrossRef]
- Rappa, G.; Mercapide, J.; Anzanello, F.; Pope, R.M.; Lorico, A. Biochemical and biological characterization of exosomes containing prominin-1/CD133. Mol. Cancer 2013, 12, 62. [Google Scholar] [CrossRef]
- Buschow, S.I.; Nolte-‘t Hoen, E.N.M.; Van Niel, G.; Pols, M.S.; Ten Broeke, T.; Lauwen, M.; Ossendorp, F.; Melief, C.J.M.; Raposo, G.; Wubbolts, R.; et al. MHC II in dendritic cells is targeted to lysosomes or T cell-induced exosomes via distinct multivesicular body pathways. Traffic 2009, 10, 1528–1542. [Google Scholar] [CrossRef]
- Lee, T.H.; Chennakrishnaiah, S.; Meehan, B.; Montermini, L.; Garnier, D.; D’Asti, E.; Hou, W.; Magnus, N.; Gayden, T.; Jabado, N.; et al. Barriers to horizontal cell transformation by extracellular vesicles containing oncogenic H-ras. Oncotarget 2016, 7, 51991. [Google Scholar] [CrossRef]
- Jenjaroenpun, P.; Kremenska, Y.; Nair, V.M.; Kremenskoy, M.; Joseph, B.; Kurochkin, I.V. Characterization of RNA in exosomes secreted by human breast cancer cell lines using next-generation sequencing. PeerJ 2013, 1, e201. [Google Scholar] [CrossRef]
- Villarroya-Beltri, C.; Gutierrez-Vazquez, C.; Sanchez-Cabo, F.; Perez-Hernandez, D.; Vazquez, J.; Martin-Cofreces, N.; Martinez-Herrera, D.J.; Pascual-Montano, A.; Mittelbrunn, M.; Sanchez-Madrid, F. Sumoylated hnRNPA2B1 controls the sorting of miRNAs into exosomes through binding to specific motifs. Nat. Commun. 2013, 4, 2980. [Google Scholar] [CrossRef]
- Statello, L.; Maugeri, M.; Garre, E.; Nawaz, M.; Wahlgren, J.; Papadimitriou, A.; Lundqvist, C.; Lindfors, L.; Collén, A.; Sunnerhagen, P.; et al. Identification of RNA-binding proteins in exosomes capable of interacting with different types of RNA: RBP-facilitated transport of RNAs into exosomes. PLoS ONE 2018, 13, e0195969. [Google Scholar] [CrossRef]
- Melo, S.A.; Sugimoto, H.; O’Connell, J.T.; Kato, N.; Villanueva, A.; Vidal, A.; Qiu, L.; Vitkin, E.; Perelman, L.T.; Melo, C.A.; et al. Cancer exosomes perform cell-independent microRNA biogenesis and promote tumorigenesis. Cancer Cell 2014, 26, 707–721. [Google Scholar] [CrossRef]
- Deschamps, T.; Kalamvoki, M. Extracellular Vesicles Released by Herpes Simplex Virus 1-Infected Cells Block Virus Replication in Recipient Cells in a STING-Dependent Manner. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Ginestra, A.; Monea, S.; Seghezzi, G.; Dolo, V.; Nagase, H.; Mignatti, P.; Vittorelli, M.L. Urokinase plasminogen activator and gelatinases are associated with membrane vesicles shed by human HT1080 fibrosarcoma cells. J. Biol. Chem. 1997, 272, 17216–17222. [Google Scholar] [CrossRef]
- Colombo, M.; Raposo, G.; Théry, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef]
- Ostrowski, M.; Carmo, N.B.; Krumeich, S.; Fanget, I.; Raposo, G.; Savina, A.; Moita, C.F.; Schauer, K.; Hume, A.N.; Freitas, R.P.; et al. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat. Cell Biol. 2010, 12, 19. [Google Scholar] [CrossRef]
- Hsu, C.; Morohashi, Y.; Yoshimura, S.I.; Manrique-Hoyos, N.; Jung, S.; Lauterbach, M.A.; Bakhti, M.; Grønborg, M.; Möbius, W.; Rhee, J.; et al. Regulation of exosome secretion by Rab35 and its GTPase-activating proteins TBC1D10A–C. J. Cell Biol. 2010, 189, 223–232. [Google Scholar] [CrossRef]
- Savina, A.; Fader, C.M.; Damiani, M.T.; Colombo, M.I. Rab11 promotes docking and fusion of multivesicular bodies in a calcium-dependent manner. Traffic 2005, 6, 131–143. [Google Scholar] [CrossRef]
- Jahn, R.; Scheller, R.H. SNAREs—Engines for membrane fusion. Nat. Rev. Mol. Cell Biol. 2006, 7, 631–643. [Google Scholar] [CrossRef]
- Verweij, F.J.; Bebelman, M.P.; Jimenez, C.R.; Garcia-Vallejo, J.J.; Janssen, H.; Neefjes, J.; Knol, J.C.; de Goeij-de Haas, R.; Piersma, S.R.; Baglio, S.R.; et al. Quantifying exosome secretion from single cells reveals a modulatory role for GPCR signaling. J. Cell Biol. 2018, 217, 1129–1142. [Google Scholar] [CrossRef]
- Barres, C.; Blanc, L.; Bette-Bobillo, P.; Andre, S.; Mamoun, R.; Gabius, H.J.; Vidal, M. Galectin-5 is bound onto the surface of rat reticulocyte exosomes and modulates vesicle uptake by macrophages. Blood 2010, 115, 696–705. [Google Scholar] [CrossRef]
- Tian, T.; Wang, Y.; Wang, H.; Zhu, Z.; Xiao, Z. Visualizing of the cellular uptake and intracellular trafficking of exosomes by live-cell microscopy. J. Cell. Biochem. 2010, 111, 488–496. [Google Scholar] [CrossRef]
- Costa Verdera, H.; Gitz-Francois, J.J.; Schiffelers, R.M.; Vader, P. Cellular uptake of extracellular vesicles is mediated by clathrin-independent endocytosis and macropinocytosis. J. Control. Release 2017, 266, 100–108. [Google Scholar] [CrossRef]
- Horibe, S.; Tanahashi, T.; Kawauchi, S.; Murakami, Y.; Rikitake, Y. Mechanism of recipient cell-dependent differences in exosome uptake. BMC Cancer 2018, 18, 47. [Google Scholar] [CrossRef]
- Mulcahy, L.A.; Pink, R.C.; Carter, D.R. Routes and mechanisms of extracellular vesicle uptake. J. Extracell. Vesicles 2014, 3, 24641. [Google Scholar] [CrossRef]
- Nanbo, A.; Kawanishi, E.; Yoshida, R.; Yoshiyama, H. Exosomes derived from Epstein-Barr virus-infected cells are internalized via caveola-dependent endocytosis and promote phenotypic modulation in target cells. J. Virol. 2013, 87, 10334–10347. [Google Scholar] [CrossRef]
- Svensson, K.J.; Christianson, H.C.; Wittrup, A.; Bourseau-Guilmain, E.; Lindqvist, E.; Svensson, L.M.; Mörgelin, M.; Belting, M. Exosome uptake depends on ERK1/2-heat shock protein 27 signaling and lipid Raft-mediated endocytosis negatively regulated by caveolin-1. J. Biol. Chem. 2013, 288, 17713–17724. [Google Scholar] [CrossRef]
- Montecalvo, A.; Larregina, A.T.; Shufesky, W.J.; Stolz, D.B.; Sullivan, M.L.; Karlsson, J.M.; Baty, C.J.; Gibson, G.A.; Erdos, G.; Wang, Z.; et al. Mechanism of transfer of functional microRNAs between mouse dendritic cells via exosomes. Blood 2012, 119, 756–766. [Google Scholar] [CrossRef]
- Heusermann, W.; Hean, J.; Trojer, D.; Steib, E.; Von Bueren, S.; Graff-Meyer, A.; Genoud, C.; Martin, K.; Pizzato, N.; Voshol, J.; et al. Exosomes surf on filopodia to enter cells at endocytic hot spots, traffic within endosomes, and are targeted to the ER. J. Cell Biol. 2016, 213, 173–184. [Google Scholar] [CrossRef]
- Hill, M.M.; Bastiani, M.; Luetterforst, R.; Kirkham, M.; Kirkham, A.; Nixon, S.J.; Hancock, J.F. PTRF-Cavin, a conserved cytoplasmic protein required for caveola formation and function. Cell 2008, 132, 113–124. [Google Scholar] [CrossRef]
- del Pozo, M.A.; Balasubramanian, N.; Alderson, N.B.; Kiosses, W.B.; Grande-García, A.; Anderson, R.G.; Schwartz, M.A. Phospho-caveolin-1 mediates integrin-regulated membrane domain internalization. Nat. Cell Biol. 2005, 7, 901. [Google Scholar] [CrossRef]
- Grande-García, A.; del Pozo, M.A. Caveolin-1 in cell polarization and directional migration. Eur. J. Cell Biol. 2008, 87, 641–647. [Google Scholar] [CrossRef]
- Kirchner, P.; Bug, M.; Meyer, H. Ubiquitination of the N-terminal region of caveolin-1 regulates endosomal sorting by the VCP/p97 AAA-ATPase. J. Biol. Chem. 2013, 288, 7363–7372. [Google Scholar] [CrossRef]
- Burana, D.; Yoshihara, H.; Tanno, H.; Yamamoto, A.; Saeki, Y.; Tanaka, K.; Komada, M. The Ankrd13 family of ubiquitin-interacting motif-bearing proteins regulates valosin-containing protein/p97 protein-mediated lysosomal trafficking of caveolin 1. J. Biol. Chem. 2016, 291, 6218–6231. [Google Scholar] [CrossRef]
- Ritz, D.; Vuk, M.; Kirchner, P.; Bug, M.; Schütz, S.; Hayer, A.; Bremer, S.; Lusk, C.; Baloh, R.H.; Lee, H.; et al. Endolysosomal sorting of ubiquitylated caveolin-1 is regulated by VCP and UBXD1 and impaired by VCP disease mutations. Nat. Cell Biol. 2011, 13, 1116. [Google Scholar] [CrossRef]
- Mundy, D.I.; Li, W.P.; Luby-Phelps, K.; Anderson, R.G. Caveolin targeting to late endosome/lysosomal membranes is induced by perturbations of lysosomal pH and cholesterol content. Mol. Biol. Cell 2012, 23, 864–880. [Google Scholar] [CrossRef]
- Parat, M.O.; Anand-Apte, B.; Fox, P.L. Differential caveolin-1 polarization in endothelial cells during migration in two and three dimensions. Mol. Biol. Cell 2003, 14, 3156–3168. [Google Scholar] [CrossRef]
- Hessvik, N.P.; Øverbye, A.; Brech, A.; Torgersen, M.L.; Jakobsen, I.S.; Sandvig, K.; Llorente, A. PIKfyve inhibition increases exosome release and induces secretory autophagy. Cell. Mol. Life Sci. 2016, 73, 4717–4737. [Google Scholar] [CrossRef]
- Lu, Q.; Zhang, J.; Allison, R.; Gay, H.; Yang, W.X.; Bhowmick, N.A.; Frelix, G.; Shappell, S.; Chen, Y.H. Identification of extracellular δ-catenin accumulation for prostate cancer detection. Prostate 2009, 69, 411–418. [Google Scholar]
- Kosaka, N.; Kogure, A.; Yamamoto, T.; Urabe, F.; Usuba, W.; Prieto-Vila, M.; Ochiya, T. Exploiting the message from cancer: The diagnostic value of extracellular vesicles for clinical applications. Exp. Mol. Med. 2019, 51, 31. [Google Scholar] [CrossRef]
- Harris, D.A.; Patel, S.H.; Gucek, M.; Hendrix, A.; Westbroek, W.; Taraska, J.W. Exosomes released from breast cancer carcinomas stimulate cell movement. PLoS ONE 2015, 10, e0117495. [Google Scholar] [CrossRef]
- Holbourn, K.P.; Acharya, K.R.; Perbal, B. The CCN family of proteins: Structure-function relationships. Trends Biochem. Sci. 2008, 33, 461–473. [Google Scholar] [CrossRef]
- Cheng, T.Y.; Wu, M.S.; Hua, K.T.; Kuo, M.L.; Lin, M.T. Cyr61/CTGF/Nov family proteins in gastric carcinogenesis. World J. Gastroenterol. 2014, 20, 1694–1700. [Google Scholar] [CrossRef]
- Tsai, M.S.; Bogart, D.F.; Castaneda, J.M.; Li, P.; Lupu, R. Cyr61 promotes breast tumorigenesis and cancer progression. Oncogene 2002, 21, 8178–8185. [Google Scholar] [CrossRef]
- Huang, Y.T.; Lan, Q.; Lorusso, G.; Duffey, N.; Ruegg, C. The matricellular protein CYR61 promotes breast cancer lung metastasis by facilitating tumor cell extravasation and suppressing anoikis. Oncotarget 2017, 8, 9200–9215. [Google Scholar] [CrossRef]
- Lin, M.T.; Chang, C.C.; Lin, B.R.; Yang, H.Y.; Chu, C.Y.; Wu, M.H.; Kuo, M.L. Elevated expression of Cyr61 enhances peritoneal dissemination of gastric cancer cells through integrin α2β1. J. Biol. Chem. 2007, 282, 34594–34604. [Google Scholar] [CrossRef]
- Holloway, S.E.; Beck, A.W.; Girard, L.; Jaber, M.R.; Barnett, C.C., Jr.; Brekken, R.A.; Fleming, J.B. Increased expression of Cyr61 (CCN1) identified in peritoneal metastases from human pancreatic cancer. J. Am. Coll. Surg. 2005, 200, 371–377. [Google Scholar] [CrossRef]
- Jin, Y.; Kim, H.P.; Cao, J.; Zhang, M.; Ifedigbo, E.; Choi, A.M. Caveolin-1 regulates the secretion and cytoprotection of Cyr61 in hyperoxic cell death. FASEB J. 2009, 23, 341–350. [Google Scholar] [CrossRef]
- Bergenfelz, C.; Gaber, A.; Allaoui, R.; Mehmeti, M.; Jirström, K.; Leanderson, T.; Leandersson, K. S100A9 expressed in ER−PgR− breast cancers induces inflammatory cytokines and is associated with an impaired overall survival. Br. J. Cancer 2015, 113, 1234. [Google Scholar] [CrossRef]
- Miller, P.; Kidwell, K.M.; Thomas, D.; Sabel, M.; Rae, J.M.; Hayes, D.F.; Hudson, B.I.; El-Ashry, D.; Lippman, M.E. Elevated S100A8 protein expression in breast cancer cells and breast tumor stroma is prognostic of poor disease outcome. Breast Cancer Res. Treat. 2017, 166, 85–94. [Google Scholar] [CrossRef]
- Hiratsuka, S.; Watanabe, A.; Aburatani, H.; Maru, Y. Tumour-mediated upregulation of chemoattractants and recruitment of myeloid cells predetermines lung metastasis. Nat. Cell Biol. 2006, 8, 1369–1375. [Google Scholar] [CrossRef]
- Onion, D.; Isherwood, M.; Shridhar, N.; Xenophontos, M.; Craze, M.L.; Day, L.J.; García-Márquez, M.A.; Pineda, R.G.; Reece-Smith, A.M.; Saunders, J.H.; et al. Multicomponent analysis of the tumour microenvironment reveals low CD8 T cell number, low stromal caveolin-1 and high tenascin-C and their combination as significant prognostic markers in non-small cell lung cancer. Oncotarget 2018, 9, 1760. [Google Scholar] [CrossRef]
- Yoshida, T.; Ishihara, A.; Hirokawa, Y.; Kusakabe, M.; Sakakura, T. Tenascin in breast cancer development–Is epithelial tenascin a marker for poor prognosis? Cancer Lett. 1995, 90, 65–73. [Google Scholar] [CrossRef]
- Adams, M.; Jones, J.; Walker, R.A.; Pringle, J.H.; Bell, S.C. Changes in tenascin-C isoform expression in invasive and preinvasive breast disease. Cancer Res. 2002, 62, 3289–3297. [Google Scholar]
- Oskarsson, T.; Acharyya, S.; Zhang, X.H.F.; Vanharanta, S.; Tavazoie, S.F.; Morris, P.G.; Downey, R.J.; Manova-Todorova, K.; Brogi, E.; Massagué, J. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat. Med. 2011, 17, 867. [Google Scholar]
- Calvo, A.; Catena, R.; Noble, M.S.; Carbott, D.; Gil-Bazo, I.; Gonzalez-Moreno, O.; Merlino, G. Identification of VEGF-regulated genes associated with increased lung metastatic potential: Functional involvement of tenascin-C in tumor growth and lung metastasis. Oncogene 2008, 27, 5373. [Google Scholar] [CrossRef]
- Tanaka, K.; Hiraiwa, N.; Hashimoto, H.; Yamazaki, Y.; Kusakabe, M. Tenascin-C regulates angiogenesis in tumor through the regulation of vascular endothelial growth factor expression. Int. J. Cancer 2004, 108, 31–40. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Model | Study Type | Major Finding | Reference |
|---|---|---|---|
| Embryonal rhabdomyosarcoma | In vivo/in vitro | Cav-1 overexpression enhances tumor formation and metastasis to the lung | [75] |
| Human breast carcinoma cells MDA-MB-231 | In vitro | CAV1 was overexpressed in low shear stressed cells and prevented tumor cells from anoikis, while depletion of CAV1 restored sensitivity to anoikis. | [76] |
| Lung cancer | In vivo/in vitro | CAV1 and STAT3 are involved in electrotaxis playing a role in cell migration guidance | [77] |
| Human colorectal cancer | In vivo/in vitro | CAV1 ubiquitylation and subsequent degradation is promoted by NDRG1, which inhibits Epithelial-Mesenchymal transition (EMT), migration and invasion | [78] |
| Clear cell renal cell carcinoma | In vitro/clinical | CAV1 is overexpressed in renal cell carcinoma and has diagnostic and prognostic value. In vitro, it promotes cell migration and invasion | [79] |
| Hepatocellular carcinoma | In vitro/in vivo/clinical | Hypoxia upregulates CAV1 expression, which acts on calcium-binding protein S100P and promotes metastasis | [80] |
| Melanoma | In vitro/in vivo | CAV1 is phosphorylated on tyrosine-14 in an extracellular matrix-specific manner, and this is required to promote melanoma | [71] |
| Ewing sarcoma | In vitro/in vivo | CAV1 regulates MMP-9 expression through MAPK/ERK pathway, in this way regulating Ewing’s sarcoma metastasis | [81] |
| Breast carcinoma MDA-MB-231 cells | In vitro/in vivo | CAV1 is mechanosensitive to low shear-stress exposure, and its activation induces PI3K/Akt/mTOR signaling, which promotes motility, invadopodia formation and metastasis | [82] |
| Uveal melanomas | Clinical | High expression of CAV1 is associated with metastatic disease, larger tumor size, lymph node metastasis and invasion of the optic nerve head | [83] |
| Prostate cancer | In vitro | Non-caveolar CAV1 enhances lymphatic endothelial cell proliferation, migration and differentiation, thus promoting lymphagiogenesis | [84] |
| Hepatocellular carcinoma | In vitro | CAV1 inhibits autophagy, thus promoting tumor growth and metastasis | [35] |
| Lung adenocarcinoma | In vitro/in vivo/clinical | Overexpression of CAV-1 increased proliferation, migration and invasion. CAV1-expressing cell tumors were larger in an in vivo xenograft model. In patients, CAV1 expression correlated positively with lymph node metastasis and cancer stage | [85] |
| Pancreatic cancer | In vitro/in vivo/clinical | CAV1 is overexpressed in human pancreatic cancer cell lines, mouse models, and patients tumors, and is associated with worse tumor grade | [86] |
| Inflammatory Breast Cancer Cell | In vitro | CAV1 down regulation reduces cell invasion. Activation of Akt1 is also decreased leading to reduced phosphorylation of RhoC GTPase | [87] |
| Lung cancer | In vitro | CAV1 levels positively correlate with anoikis resistance, anchorage-independent growth, migration, and invasion in a variety of lung carcinoma cells | [88] |
| Human breast cancer BT474 cells | In vitro | CAV1 knockdown decreases cell proliferation, migration, and invasion. In addition, activity of the extracellular signal-regulated kinase 1/2 pathway was reduced. Likewise, expression of the cell cycle-associated proteins (cyclin D1, c-Fos and β-catenin), and metalloproteinases (MMP-1, -2, -9), were also decreased, while E-cadherin increased | [89] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Campos, A.; Burgos-Ravanal, R.; González, M.F.; Huilcaman, R.; Lobos González, L.; Quest, A.F.G. Cell Intrinsic and Extrinsic Mechanisms of Caveolin-1-Enhanced Metastasis. Biomolecules 2019, 9, 314. https://doi.org/10.3390/biom9080314
Campos A, Burgos-Ravanal R, González MF, Huilcaman R, Lobos González L, Quest AFG. Cell Intrinsic and Extrinsic Mechanisms of Caveolin-1-Enhanced Metastasis. Biomolecules. 2019; 9(8):314. https://doi.org/10.3390/biom9080314
Chicago/Turabian StyleCampos, America, Renato Burgos-Ravanal, María Fernanda González, Ricardo Huilcaman, Lorena Lobos González, and Andrew Frederick Geoffery Quest. 2019. "Cell Intrinsic and Extrinsic Mechanisms of Caveolin-1-Enhanced Metastasis" Biomolecules 9, no. 8: 314. https://doi.org/10.3390/biom9080314
APA StyleCampos, A., Burgos-Ravanal, R., González, M. F., Huilcaman, R., Lobos González, L., & Quest, A. F. G. (2019). Cell Intrinsic and Extrinsic Mechanisms of Caveolin-1-Enhanced Metastasis. Biomolecules, 9(8), 314. https://doi.org/10.3390/biom9080314