Inhibiting Extracellular Cathepsin D Reduces Hepatic Steatosis in Sprague–Dawley Rats †


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Design and Development of CTD-002
2.2. Isolation and Culturing of Bone Marrow-Derived Macrophages
2.3. Rats, Diet and Intervention
2.4. Histological Analyses
2.5. Plasma Measurements
2.6. Liver Tumor Necrosis Factor-Alpha Levels
2.7. Statistical Analyses
3. Results
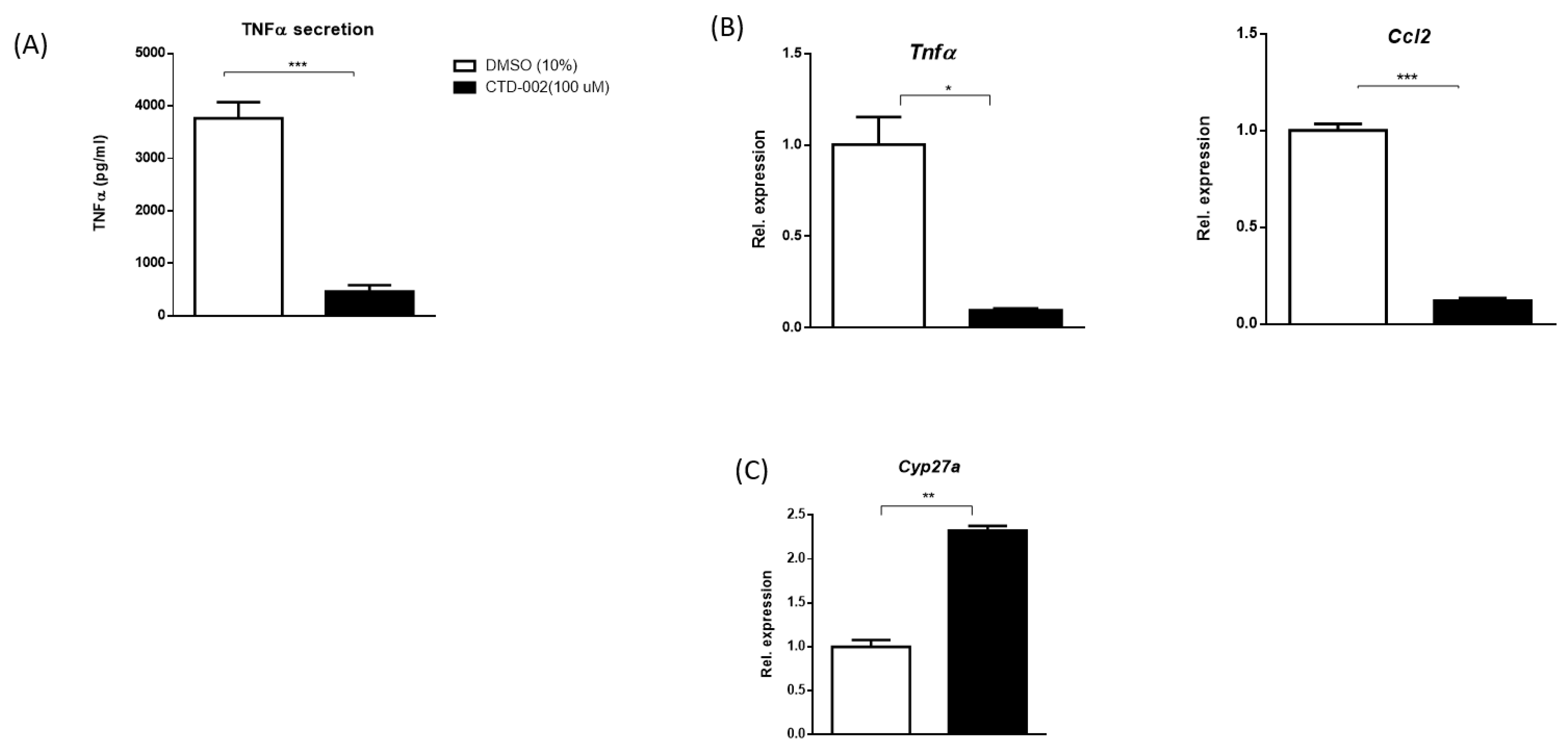
3.1. Decreased Inflammation after Inhibition of Extracellular CTSD in oxLDL-Loaded Primary Mouse Macrophages
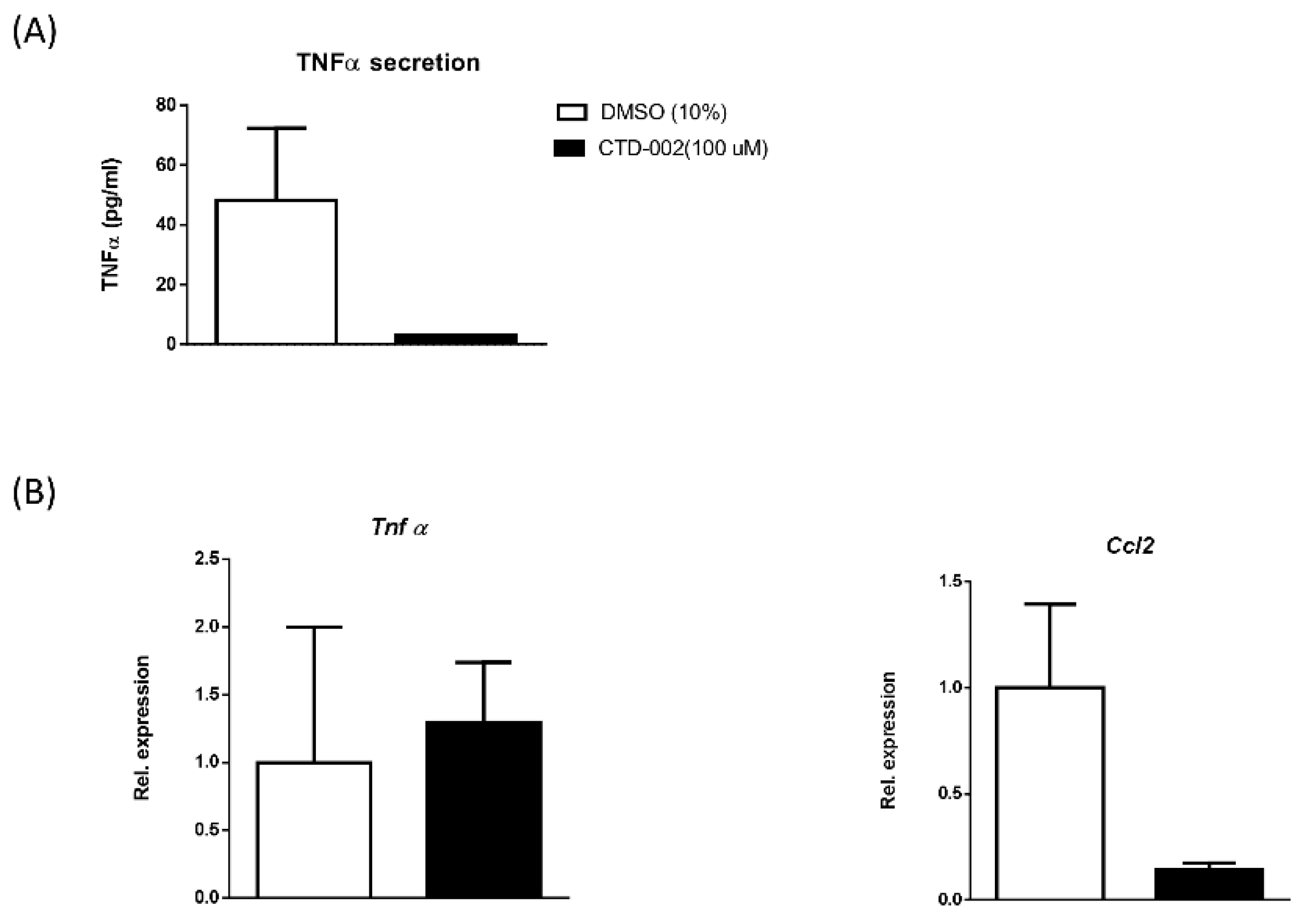
3.2. Inhibition of Macrophage-Derived Extracellular CTSD Reduces Inflammation in HepG2 Cells
3.3. Improved Metabolic Features after Inhibition of Extracellular CTSD in HFD-Fed Sprague–Dawley Rats
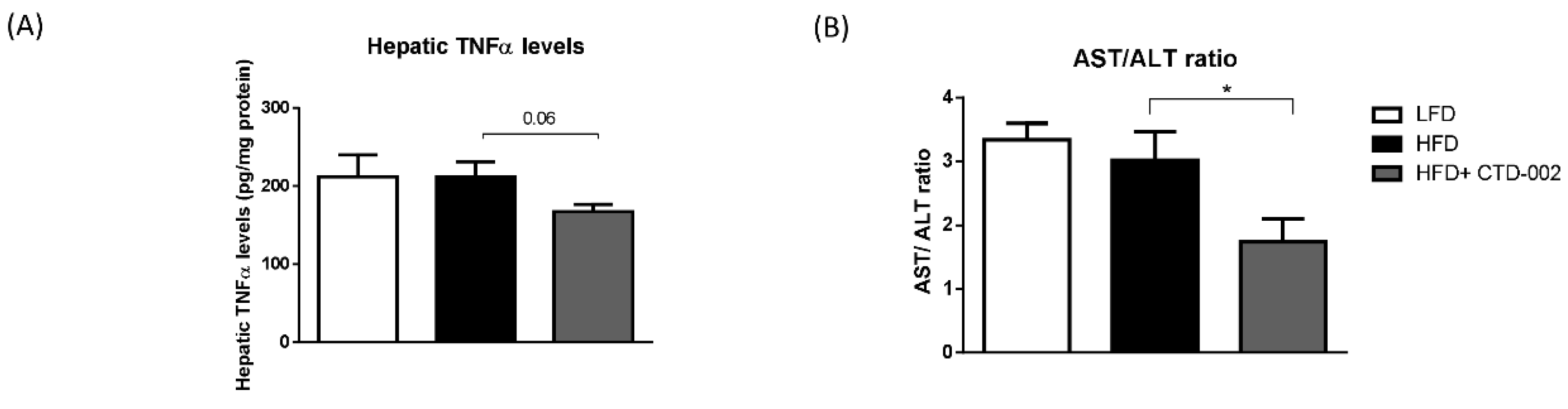
3.4. Reduced Liver Damage after Inhibition of Extracellular CTSD in HFD-Fed Sprague–Dawley Rats
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| NAFLD | non-alcoholic fatty liver disease |
| NASH | non-alcoholic steatohepatitis |
| CTSD | cathepsin D |
| HFD | high-fat diet |
| LFD | low-fat diet |
| BMDMs | Bone Marrow-derived macrophages |
| oxLDL | oxidized low-density lipoprotein |
| Wt | wildtype |
| LDL(R) | low-density lipoprotein (receptor) |
| SD | Sprague–Dawley |
| ELISA | enzyme-linked immune sorbent assay |
| LCM | L929-conditioned medium |
| TNF | tumor necrosis factor |
| LPS | lipopolysaccharide |
| AST | aspartate transaminase |
| ALT | alanine transaminase |
| MetS | metabolic syndrome |
| Pep-A | Pepstatin-A |
References
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef]
- Tandra, S.; Yeh, M.M.; Brunt, E.M.; Vuppalanchi, R.; Cummings, O.W.; Unalp-Arida, A.; Wilson, L.A.; Chalasani, N. Presence and significance of microvesicular steatosis in nonalcoholic fatty liver disease. J. Hepatol. 2011, 55, 654–659. [Google Scholar] [CrossRef]
- Takahashi, Y.; Fukusato, T. Histopathology of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. World J. Gastroenterol. 2014, 20, 15539–15548. [Google Scholar] [CrossRef] [PubMed]
- Contos, M.J.; Choudhury, J.; Mills, A.S.; Sanyal, A.J. The histologic spectrum of nonalcoholic fatty liver disease. Clin. Liver Dis. 2004, 8, 481–500. [Google Scholar] [CrossRef]
- Musso, G.; Cassader, M.; Rosina, F.; Gambino, R. Impact of current treatments on liver disease, glucose metabolism and cardiovascular risk in non-alcoholic fatty liver disease (NAFLD): A systematic review and meta-analysis of randomised trials. Diabetologia 2012, 55, 885–904. [Google Scholar] [CrossRef]
- Cheung, O.; Sanyal, A.J. Abnormalities of lipid metabolism in nonalcoholic fatty liver disease. Semin. Liver Dis. 2008, 28, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Lindstedt, L.; Lee, M.; Oorni, K.; Bromme, D.; Kovanen, P.T. Cathepsins F and S block HDL3-induced cholesterol efflux from macrophage foam cells. Biochem. Biophys. Res. Commun. 2003, 312, 1019–1024. [Google Scholar] [CrossRef] [PubMed]
- Tertov, V.V.; Orekhov, A.N. Metabolism of native and naturally occurring multiple modified low-density lipoprotein in smooth muscle cells of human aortic intima. Exp. Mol. Pathol. 1997, 64, 127–145. [Google Scholar] [CrossRef]
- Thelen, A.M.; Zoncu, R. Emerging Roles for the Lysosome in Lipid Metabolism. Trends Cell Biol. 2017, 27, 833–850. [Google Scholar] [CrossRef] [PubMed]
- Lutgens, S.P.M.; Cleutjens, K.B.J.M.; Daemen, M.J.A.P.; Heeneman, S. Cathepsin cysteine proteases in cardiovascular disease. FASEB J. 2007, 21, 3029–3041. [Google Scholar] [CrossRef]
- Benes, P.; Vetvicka, V.; Fusek, M. Cathepsin D—Many functions of one aspartic protease. Crit. Rev. Oncol. Hematol. 2008, 68, 12–28. [Google Scholar] [CrossRef] [PubMed]
- Moallem, S.A.; Nazemian, F.; Eliasi, S.; Alamdaran, S.A.; Shamsara, J.; Mohammadpour, A.H. Correlation between cathepsin D serum concentration and carotid intima-media thickness in hemodialysis patients. Int. Urol. Nephrol. 2011, 43, 841–848. [Google Scholar] [CrossRef] [PubMed]
- Snir, J.A.; Suchy, M.; St Lawrence, K.; Hudson, R.H.E.; Pasternak, S.H.; Bartha, R. Prolonged In Vivo Retention of a Cathepsin D Targeted Optical Contrast Agent in a Mouse Model of Alzheimer’s Disease. J. Alzheimers Dis. 2015, 48, 73–87. [Google Scholar] [CrossRef] [PubMed]
- Briozzo, P.; Badet, J.; Capony, F.; Pieri, I.; Montcourrier, P.; Barritault, D.; Rochefort, H. MCF7 mammary cancer cells respond to bFGF and internalize it following its release from extracellular matrix: A permissive role of cathepsin D. Exp. Cell Res. 1991, 194, 252–259. [Google Scholar] [CrossRef]
- Porter, K.; Lin, Y.Z.; Liton, P.B. Cathepsin B Is Up-Regulated and Mediates Extracellular Matrix Degradation in Trabecular Meshwork Cells Following Phagocytic Challenge. PLoS ONE 2013, 8, e68668. [Google Scholar] [CrossRef] [PubMed]
- Naseem, R.H.; Hedegard, W.; Henry, T.D.; Lessard, J.; Sutter, K.; Katz, S.A. Plasma cathepsin D isoforms and their active metabolites increase after myocardial infarction and contribute to plasma renin activity. Basic Res. Cardiol. 2005, 100, 139–146. [Google Scholar] [CrossRef]
- Walenbergh, S.M.; Houben, T.; Hendrikx, T.; Jeurissen, M.L.; van Gorp, P.J.; Vreugdenhil, A.C.; Adriaanse, M.P.; Buurman, W.A.; Hofker, M.H.; Mosca, A.; et al. Plasma cathepsin D levels: A novel tool to predict pediatric hepatic inflammation. Am. J. Gastroenterol. 2015, 110, 462–470. [Google Scholar] [CrossRef]
- Walenbergh, S.M.; Houben, T.; Rensen, S.S.; Bieghs, V.; Hendrikx, T.; van Gorp, P.J.; Oligschlaeger, Y.; Jeurissen, M.L.; Gijbels, M.J.; Buurman, W.A.; et al. Plasma cathepsin D correlates with histological classifications of fatty liver disease in adults and responds to intervention. Sci. Rep. 2016, 6, 38278. [Google Scholar] [CrossRef] [PubMed]
- Houben, T.; Oligschlaeger, Y.; Hendrikx, T.; Bitorina, A.V.; Walenbergh, S.M.A.; van Gorp, P.J.; Gijbels, M.J.J.; Friedrichs, S.; Plat, J.; Schaap, F.G.; et al. Cathepsin D regulates lipid metabolism in murine steatohepatitis. Sci. Rep. 2017, 7, 3494. [Google Scholar] [CrossRef] [PubMed]
- Marciniszyn, J., Jr.; Hartsuck, J.A.; Tang, J. Mode of inhibition of acid proteases by pepstatin. J. Biol. Chem. 1976, 251, 7088–7094. [Google Scholar]
- Lieber, C.S.; Leo, M.A.; Mak, K.M.; Xu, Y.; Cao, Q.; Ren, C.; Ponomarenko, A.; DeCarli, L.M. Model of nonalcoholic steatohepatitis. Am. J. Clin. Nutr. 2004, 79, 502–509. [Google Scholar] [CrossRef]
- Dixon, S.L.; Smondyrev, A.M.; Knoll, E.H.; Rao, S.N.; Shaw, D.E.; Friesner, R.A. PHASE: A new engine for pharmacophore perception, 3D QSAR model development, and 3D database screening: 1. Methodology and preliminary results. J. Comput. Aided Mol. Des. 2006, 20, 647–671. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.L.; Smondyrev, A.M.; Rao, S.N. PHASE: A novel approach to pharmacophore modeling and 3D database searching. Chem. Biol. Drug Des. 2006, 67, 370–372. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, U.; Redgrave, T.G.; Oates, P.S. Effect of dietary fat to produce non-alcoholic fatty liver in the rat. J. Gastroenterol. Hepatol. 2009, 24, 1463–1471. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Li, J.; Lu, C.; Wang, J.; Ge, J.; Huang, Y.; Zhang, L.; Wang, Y. High-fat emulsion-induced rat model of nonalcoholic steatohepatitis. Life Sci. 2006, 79, 1100–1107. [Google Scholar] [CrossRef]
- Kitade, H.; Chen, G.; Ni, Y.; Ota, T. Nonalcoholic Fatty Liver Disease and Insulin Resistance: New Insights and Potential New Treatments. Nutrients 2017, 9, 387. [Google Scholar] [CrossRef]
- Perry, R.J.; Samuel, V.T.; Petersen, K.F.; Shulman, G.I. The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature 2014, 510, 84–91. [Google Scholar] [CrossRef]
- Samuel, V.T.; Liu, Z.X.; Qu, X.Q.; Elder, B.D.; Bilz, S.; Befroy, D.; Romanelli, A.J.; Shulman, G.I. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J. Biol. Chem. 2004, 279, 32345–32353. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, K.; Nishimura, T.; Ishiba, H.; Seko, Y.; Okajima, A.; Fujii, H.; Tochiki, N.; Umemura, A.; Moriguchi, M.; Sumida, Y.; et al. Blockade of interleukin 6 signalling ameliorates systemic insulin resistance through upregulation of glucose uptake in skeletal muscle and improves hepatic steatosis in high-fat diet fed mice. Liver Int. 2015, 35, 550–561. [Google Scholar] [CrossRef] [PubMed]
- Pardo, V.; Gonzalez-Rodriguez, A.; Guijas, C.; Balsinde, J.; Valverde, A.M. Opposite cross-talk by oleate and palmitate on insulin signaling in hepatocytes through macrophage activation. J. Biol. Chem. 2015, 290, 11663–11677. [Google Scholar] [CrossRef]
- Melino, M.; Gadd, V.L.; Walker, G.V.; Skoien, R.; Barrie, H.D.; Jothimani, D.; Horsfall, L.; Jones, A.; Sweet, M.J.; Thomas, G.P.; et al. Macrophage secretory products induce an inflammatory phenotype in hepatocytes. World J. Gastroenterol. 2012, 18, 1732–1744. [Google Scholar] [CrossRef]
- Feldstein, A.E.; Werneburg, N.W.; Canbay, A.; Guicciardi, M.E.; Bronk, S.F.; Rydzewski, R.; Burgart, L.J.; Gores, G.J. Free fatty acids promote hepatic lipotoxicity by stimulating TNF-alpha expression via a lysosomal pathway. Hepatology 2004, 40, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.Z.; Berk, M.; McIntyre, T.M.; Gores, G.J.; Feldstein, A.E. The lysosomal-mitochondrial axis in free fatty acid-induced hepatic lipotoxicity. Hepatology 2008, 47, 1495–1503. [Google Scholar] [CrossRef] [PubMed]
- Moles, A.; Tarrats, N.; Fernandez-Checa, J.C.; Mari, M. Cathepsins B and D Drive Hepatic Stellate Cell Proliferation and Promote Their Fibrogenic Potential. Hepatology 2009, 49, 1297–1307. [Google Scholar] [CrossRef]
- Bieghs, V.; Hendrikx, T.; van Gorp, P.J.; Verheyen, F.; Guichot, Y.D.; Walenbergh, S.M.A.; Jeurissen, M.L.J.; Gijbels, M.; Rensen, S.S.; Bast, A.; et al. The Cholesterol Derivative 27-Hydroxycholesterol Reduces Steatohepatitis in Mice. Gastroenterology 2013, 144, 167–178.e1. [Google Scholar] [CrossRef]
- Saftig, P.; Hetman, M.; Schmahl, W.; Weber, K.; Heine, L.; Mossmann, H.; Koster, A.; Hess, B.; Evers, M.; von Figura, K.; et al. Mice deficient for the lysosomal proteinase cathepsin D exhibit progressive atrophy of the intestinal mucosa and profound destruction of lymphoid cells. EMBO J. 1995, 14, 3599–3608. [Google Scholar] [CrossRef] [PubMed]
- Koike, M.; Nakanishi, H.; Saftig, P.; Ezaki, J.; Isahara, K.; Ohsawa, Y.; Schulz-Schaeffer, W.; Watanabe, T.; Waguri, S.; Kametaka, S.; et al. Cathepsin D deficiency induces lysosomal storage with ceroid lipofuscin in mouse CNS neurons. J. Neurosci. 2000, 20, 6898–6906. [Google Scholar] [CrossRef] [PubMed]
- Glondu, M.; Coopman, P.; Laurent-Matha, V.; Garcia, M.; Rochefort, H.; Liaudet-Coopman, E. A mutated cathepsin-D devoid of its catalytic activity stimulates the growth of cancer cells. Oncogene 2001, 20, 6920–6929. [Google Scholar] [CrossRef] [PubMed]
- Vetvicka, V.; Vetvickova, J.; Fusek, M. Effect of procathepsin D and its activation peptide on prostate cancer cells. Cancer Lett. 1998, 129, 55–59. [Google Scholar] [CrossRef]
- Vetvicka, V.; Vetvickova, J.; Benes, P. Role of enzymatically inactive procathepsin D in lung cancer. Anticancer Res. 2004, 24, 2739–2743. [Google Scholar]
- Dubey, V.; Luqman, S. Cathepsin D as a Promising Target for the Discovery of Novel Anticancer Agents. Curr. Cancer Drug Targets 2017, 17, 404–422. [Google Scholar] [CrossRef]
- Ruan, H.; Hao, S.; Young, P.; Zhang, H. Targeting Cathepsin B for Cancer Therapies. Horiz. Cancer Res. 2015, 56, 23–40. [Google Scholar] [PubMed]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Garcia Arencibia, M.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB links autophagy to lysosomal biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, M.G.; Kreibich, G.; Popov, D.; Kato, K.; Sabatini, D.D. Biosynthesis of lysosomal hydrolases: Their synthesis in bound polysomes and the role of co- and post-translational processing in determining their subcellular distribution. J. Cell Biol. 1982, 93, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Ungewickell, A.J.; Majerus, P.W. Increased levels of plasma lysosomal enzymes in patients with Lowe syndrome. Proc. Natl. Acad. Sci. USA 1999, 96, 13342–13344. [Google Scholar] [CrossRef] [PubMed]
- Hultberg, B.; Isaksson, A.; Sjoblad, S.; Ockerman, P.A. Acid hydrolases in serum from patients with lysosomal disorders. Clin. Chim. Acta 1980, 100, 33–38. [Google Scholar] [CrossRef]
- Hoppe, G.; O’Neil, J.; Hoff, H.F.; Sears, J. Products of lipid peroxidation induce missorting of the principal lysosomal protease in retinal pigment epithelium. BBA-Mol. Basis Dis. 2004, 1689, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Yuan, X.M.; Olsson, A.G.; Brunk, U.T. Uptake of oxidized LDL by macrophages results in partial lysosomal enzyme inactivation and relocation. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 177–184. [Google Scholar] [CrossRef]
- Reddy, V.Y.; Zhang, Q.Y.; Weiss, S.J. Pericellular mobilization of the tissue-destructive cysteine proteinases, cathepsins B, L, and S, by human monocyte-derived macrophages. Proc. Natl. Acad. Sci. USA 1995, 92, 3849–3853. [Google Scholar] [CrossRef]
- Neurath, H. Evolution of proteolytic enzymes. Science 1984, 224, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Dubrac, S.; Lear, S.R.; Ananthanarayanan, M.; Balasubramaniyan, N.; Bollineni, J.; Shefer, S.; Hyogo, H.; Cohen, D.E.; Blanche, P.J.; Krauss, R.M.; et al. Role of CYP27A in cholesterol and bile acid metabolism. J. Lipid Res. 2005, 46, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Oram, J.F. HDL apolipoproteins and ABCA1: Partners in the removal of excess cellular cholesterol. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 720–727. [Google Scholar] [CrossRef]
- Oldoni, F.; van Capelleveen, J.C.; Dalila, N.; Wolters, J.C.; Heeren, J.; Sinke, R.J.; Hui, D.Y.; Dallinga-Thie, G.M.; Frikke-Schmidt, R.; Hovingh, K.G.; et al. Naturally Occurring Variants in LRP1 (Low-Density Lipoprotein Receptor-Related Protein 1) Affect HDL (High-Density Lipoprotein) Metabolism Through ABCA1 (ATP-Binding Cassette A1) and SR-B1 (Scavenger Receptor Class B Type 1) in Humans. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1440–1453. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.S.; Goldstein, J.L. The SREBP pathway: Regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 1997, 89, 331–340. [Google Scholar] [CrossRef]
- Widenmaier, S.B.; Snyder, N.A.; Nguyen, T.B.; Arduini, A.; Lee, G.Y.; Arruda, A.P.; Saksi, J.; Bartelt, A.; Hotamisligil, G.S. NRF1 Is an ER Membrane Sensor that Is Central to Cholesterol Homeostasis. Cell 2017, 171, 1094–1109. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khurana, P.; Yadati, T.; Goyal, S.; Dolas, A.; Houben, T.; Oligschlaeger, Y.; Agarwal, A.K.; Kulkarni, A.; Shiri-Sverdlov, R. Inhibiting Extracellular Cathepsin D Reduces Hepatic Steatosis in Sprague–Dawley Rats. Biomolecules 2019, 9, 171. https://doi.org/10.3390/biom9050171
Khurana P, Yadati T, Goyal S, Dolas A, Houben T, Oligschlaeger Y, Agarwal AK, Kulkarni A, Shiri-Sverdlov R. Inhibiting Extracellular Cathepsin D Reduces Hepatic Steatosis in Sprague–Dawley Rats. Biomolecules. 2019; 9(5):171. https://doi.org/10.3390/biom9050171
Chicago/Turabian StyleKhurana, Princy, Tulasi Yadati, Sandeep Goyal, Atul Dolas, Tom Houben, Yvonne Oligschlaeger, Anil K. Agarwal, Aditya Kulkarni, and Ronit Shiri-Sverdlov. 2019. "Inhibiting Extracellular Cathepsin D Reduces Hepatic Steatosis in Sprague–Dawley Rats" Biomolecules 9, no. 5: 171. https://doi.org/10.3390/biom9050171
APA StyleKhurana, P., Yadati, T., Goyal, S., Dolas, A., Houben, T., Oligschlaeger, Y., Agarwal, A. K., Kulkarni, A., & Shiri-Sverdlov, R. (2019). Inhibiting Extracellular Cathepsin D Reduces Hepatic Steatosis in Sprague–Dawley Rats. Biomolecules, 9(5), 171. https://doi.org/10.3390/biom9050171