Calcium-Binding Generates the Semi-Clathrate Waters on a Type II Antifreeze Protein to Adsorb onto an Ice Crystal Surface

 and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Preparation of Antifreeze Protein from Japanase Smelt and Thermal Hysteresis Measurements
2.2. Crystallization and Structural Analysis of Antifreeze Protein from Japanase Smelt
3. Results and Discussion
3.1. Antifreeze Protein from Japanase Smelt Inhibits Ice Crystal Growth when it Binds Ca2+
3.2. Antifreeze Protein from Japanase Smelt Targets First Prism Plane of a Single Ice Crystal
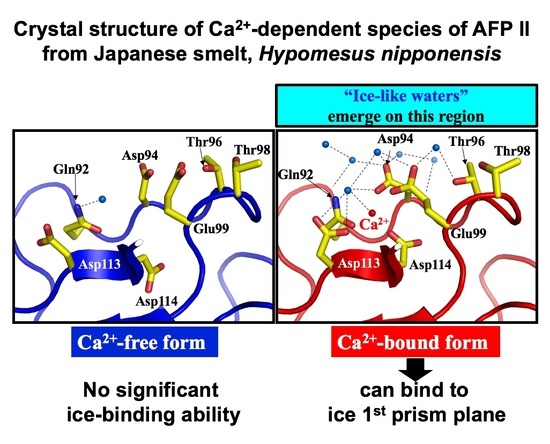
3.3. Crystal Structure of Antifreeze Protein from Japanase Smelt with and without Ca2+
3.4. Ca2+-Bound Antifreeze Protein from Japanase Smelt Locates the Polypentagonal Ice-Like Waters
3.5. Ice-Like Waters can be Incorporated into the Quasi-Liquid Layer
3.6. Rigidity and Hydrophobicity May Relevant to the Ice-Like Water Formation
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Voets, I.K. From ice-binding proteins to bio-inspired antifreeze materials. Soft Matter 2017, 12, 4808–4823. [Google Scholar] [CrossRef] [PubMed]
- Davies, P.L. Ice-binding proteins: A remarkable diversity of structures for stopping and starting ice growth. Trends Biochem. Sci. 2014, 39, 548–555. [Google Scholar] [CrossRef] [PubMed]
- Capicciotti, C.J.; Doshi, M.; Ben, R.N. Ice recrystallization inhibitors: From biological antifreezes to small molecules. In Recent Developments in the Study of Recrystallization; Wilson, P., Ed.; INTECH Open Ltd.: London, UK, 2013; pp. 177–224. [Google Scholar]
- Rahman, A.T.; Arai, T.; Yamauchi, A.; Miura, A.; Kondo, H.; Ohyama, Y.; Tsuda, S. Ice recrystallization is strongly inhibited when antifreeze proteins bind to multiple ice planes. Sci. Rep. 2019, 9, 2212. [Google Scholar] [CrossRef] [PubMed]
- Raymond, J.A.; DeVries, A.L. Adsorption inhibition as a mechanism of freezing resistance in polar fishes. Proc. Natl. Acad. Sci. USA 1977, 74, 2589–2593. [Google Scholar] [CrossRef]
- Barrett, J. Thermal hysteresis proteins. Int. J. Biochem. Cell Biol. 2001, 33, 105–117. [Google Scholar] [CrossRef]
- Hobbs, P.V. Structure of ice Ih. In Ice Physics; Oxford University Press: London, UK, 1974; pp. 18–39. [Google Scholar]
- Scotter, A.J.; Marshall, C.B.; Graham, L.A.; Gilbert, J.A.; Garnham, C.P.; Davies, P.L. The basis for hyperactivity of antifreeze proteins. Cryobiology 2006, 53, 229–239. [Google Scholar] [CrossRef]
- Bar-Dolev, M.; Celik, Y.; Wettlaufer, J.S.; Davies, P.L.; Braslavsky, I. New insights into ice growth and melting modifications by antifreeze proteins. J. R. Soc. Interface 2012, 9, 3249–3259. [Google Scholar] [CrossRef]
- Fukushima, M.; Tsuda, S.; Yoshizawa, Y. Fabrication of highly porous alumina prepared by gelation freezing route with antifreeze protein. J. Am. Chem. Soc. 2013, 96, 1029–1031. [Google Scholar] [CrossRef]
- Kim, H.J.; Lee, J.H.; Hur, Y.B.; Lee, C.W.; Park, S.-H.; Koo, B.-W. Marine antifreeze proteins: Structure, Function, and application to cryopreservation as a potential cryoprotectant. Mar. Drugs 2017, 15, 27. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Lin, F.-H.; Campbell, R.L.; Allingham, J.S.; Davies, P.L. An antifreeze protein folds with an interior network of more than 400 semi-clathrate waters. Science 2014, 343, 795–798. [Google Scholar] [CrossRef]
- Mahatabuddin, S.; Fukami, D.; Arai, T.; Nishimiya, Y.; Shimizu, R.; Shibazaki, C.; Kondo, H.; Adachi, M.; Tsuda, S. Polypentagonal ice-like water networks emerge solely in an activity-improved variant of ice-binding protein. Proc. Natl. Acad. Sci. USA 2018, 115, 5456–5461. [Google Scholar] [CrossRef] [PubMed]
- Beaglehole, D.; Wilson, P. Thickness and anisotropy of the ice-water interface. J. Phys. Chem. 1993, 97, 11053–11055. [Google Scholar] [CrossRef]
- Mantz, Y.A.; Geiger, F.M.; Molina, L.T.; Molina, M.J.; Trout, B.L. First-principles molecular-dynamics study of surface disordering of the (0001) face of hexagonal ice. J. Chem. Phys. 2000, 113, 10733–10743. [Google Scholar] [CrossRef]
- Hayward, J.A.; Haymet, A.D.J. The ice/water interface: Molecular dynamics simulations of the basal, prism, {20-21}, and {2-1-10} interfaces of Ice Ih. J. Chem. Phys. 2001, 114, 3713–3726. [Google Scholar] [CrossRef]
- Garnham, C.P.; Campbell, R.L.; Davies, P.L. Anchored clathrate waters bind antifreeze proteins to ice. Proc. Natl. Acad. Sci. USA 2011, 108, 7363–7367. [Google Scholar] [CrossRef]
- Sun, T.; Gauthier, S.Y.; Campbell, R.L.; Davies, P.L. Revealing surface waters on an antifreeze protein by fusion protein crystallography combined with molecular dynamic simulations. J. Phys. Chem. B 2015, 119, 12808–12815. [Google Scholar] [CrossRef] [PubMed]
- Mahatabuddin, S.; Tsuda, S. Applications of antifreeze proteins: Practical use of the quality products from Japanese fishes. In Survival Strategies in Extreme Cold and Desiccation; Iwaya-Inoue, M., Sakurai, M., Uemura, M., Eds.; Springer: Singapore, 2018; pp. 321–337. [Google Scholar] [CrossRef]
- Gauthier, S.Y.; Scotter, A.J.; Lin, F.-H.; Baardsnes, J.; Fletcher, G.L.; Davies, P.L. A re-evaluation of the role of type IV antifreeze protein. Cryobiology 2008, 57, 292–296. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, Y.; Miura, R.; Takemoto, Y.; Tsuda, S.; Kawahara, H.; Obata, H. Type II antifreeze protein from a mid-latitude freshwater fish, Japanese smelt (Hypomesus nipponensis). Biosci. Biotechnol. Biochem. 2003, 67, 461–466. [Google Scholar] [CrossRef]
- Ewart, K.V.; Fletcher, G.L. Isolation and characterization of antifreeze proteins from smelt (Osmerus mordax) and Atlantic herring (Clupea harengus harengus). Can. J. Zool. 1990, 68, 1652–1658. [Google Scholar] [CrossRef]
- Ng, N.F.; Trinh, K.-Y.; Hew, C.L. Structure of an antifreeze polypeptide precursor from the Sea Raven, Hemitripterus americanus. J. Biol. Chem. 1986, 261, 15690–15695. [Google Scholar]
- Nishimiya, Y.; Kondo, H.; Takamichi, M.; Sugimoto, H.; Suzuki, M.; Miura, A.; Tsuda, S. Crystal structure and mutational analysis of Ca2+-independent type II antifreeze protein from Longsnout poacher, Brachyopsis rostratus. J. Mol. Biol. 2008, 382, 734–746. [Google Scholar] [CrossRef]
- Ewart, K.V.; Li, Z.; Yang, D.S.C.; Fletcher, G.L.; Hew, C.L. The ice-binding site of Atlantic herring antifreeze protein corresponds to the carbohydrate-binding site of C-type lectins. Biochemistry 1998, 37, 4080–4085. [Google Scholar] [CrossRef] [PubMed]
- Yasui, M.; Takamichi, M.; Miura, A.; Nishimiya, Y.; Kondo, H.; Tsuda, S. Hydroxyl groups of threonines contribute to the activity of Ca2+-dependent type II antifreeze protein. Cryobiol. Cryotech. 2008, 54, 1–8. [Google Scholar]
- Liu, Y.; Li, Z.; Lin, Q.; Kosinski, J.; Seetharaman, J.; Bujnicki, J.M.; Sivaramnan, J.; Hew, C.L. Structure and evolutionary origin of Ca2+-dependent herring type II antifreeze protein. PLoS ONE 2007, 2, e548. [Google Scholar] [CrossRef]
- Nishimiya, Y.; Kondo, H.; Yasui, M.; Sugimoto, H.; Noro, N.; Sato, R.; Suzuki, M.; Miura, A.; Tsuda, S. Crystallization and preliminary X-ray crystallographic analysis of Ca2+-independent and Ca2+-dependent species of the type II antifreeze protein. Acta Cryst. 2006, 62, 538–541. [Google Scholar] [CrossRef] [PubMed]
- Takamichi, M.; Nishimiya, Y.; Miura, A.; Tsuda, S. Effect of annealing time of an ice crystal on the activity of type III antifreeze protein. FEBS J. 2007, 274, 6469–6476. [Google Scholar] [CrossRef] [PubMed]
- Chavas, L.M.; Matsugaki, N.; Yamada, Y.; Hiraki, M.; Igarashi, N.; Suzuki, M.; Wakatsuki, S. Beamline AR-NW12A: High-throughput beamline for macromolecular crystallography at the Photon Factory. J. Synchrotron Radiat. 2012, 19, 450–454. [Google Scholar] [CrossRef]
- Otwinowski, Z.; Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997, 276, 307–326. [Google Scholar] [CrossRef]
- Collaborative Computational Project, Number 4. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 1994, 50, 760–763. [Google Scholar] [CrossRef]
- Vagin, A.; Teplyakov, A. MOLREP: An automated program for molecular replacement. J. Appl. Crystallogr. 1997, 30, 1022–1025. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef]
- Brünger, A.T. Version 1.2 of the crystallography and NMR system. Nat. Protoc. 2007, 2, 2728–2733. [Google Scholar] [CrossRef]
- Murshudov, G.N.; Vagin, A.A.; Dodson, E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 1997, 53, 240–255. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Cheng, J.; Hanada, Y.; Miura, A.; Tsuda, S.; Kondo, H. Hydrophobic ice-binding sites confer hyperactivity of an antifreeze protein from a snow mold fungus. Biochem. J. 2016, 473, 4011–4026. [Google Scholar] [CrossRef] [PubMed]
- Momma, K.; Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Basu, K.; Garnham, C.P.; Nishimiya, Y.; Tsuda, S.; Braslavsky, I.; Davies, P.L. Determining the ice-binding planes of antifreeze proteins by fluorescence-based ice plane affinity. J. Vis. Exp. 2014, 83, e51185. [Google Scholar] [CrossRef]
- Brünger, A.T. Free R value: A novel statistical quantity for assessing the accuracy of crystal structures. Nature 1992, 355, 472–475. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.J.; Headd, J.J.; Moriarty, N.W.; Prisant, M.G.; Videau, L.L.; Deis, L.N.; Verma, V.; Keedy, D.A.; Hintze, B.J.; Chen, V.B.; et al. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 2018, 27, 293–315. [Google Scholar] [CrossRef] [PubMed]
- Nielbo, S.; Thomsen, J.K.; Graversen, J.H.; Jensen, P.H.; Etzerodt, M.; Poulsen, F.M.; Thøgersen, H.C. Structure of the plasminogen Kringle 4 binding calcium-free form of the C-type lectin-like domain of tetranectin. Biochemistry 2004, 43, 8636–8643. [Google Scholar] [CrossRef]
- Ng, K.K.S.; Park-Snyder, S.; Weis, W.I. Ca2+-dependent structural changes in C-type mannose-binding proteins. Biochemistry 1998, 37, 17965–17976. [Google Scholar] [CrossRef]
- Graether, S.P.; Sykes, B.D. Cold survival in freeze-intolerant insects: The structure and function of β-helical antifreeze proteins. Eur. J. Biochem. 2004, 271, 3285–3296. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Sharp, K.A. The mechanism of the type III antifreeze protein action: A computational study. Biophys. Chem. 2004, 109, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, K.R.; Sharp, K.A. Analysis of thermal hysteresis protein hydration using the random network model. Biophys. Chem. 2003, 105, 195–209. [Google Scholar] [CrossRef]
- Huang, W.; Blinov, N.; Wishart, D.S.; Kovalenko, A. Role of water in ligand binding to maltose-binding protein: Insight from a new docking protocol based on the 3D-RISM-KH molecular theory of solvation. J. Chem. Inf. Model. 2015, 55, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Gauto, D.F.; Petruk, A.A.; Modenutti, C.P.; Blanco, J.I.; Di Lella, S.; Martí, M.A. Solvent structure improves docking prediction in lectin–carbohydrate complexes. Glycobiology 2012, 23, 241–258. [Google Scholar] [CrossRef] [PubMed]
- Ewart, K.V.; Yang, D.S.C.; Ananthanarayanan, V.S.; Fletcher, G.L.; Hew, C.L. Ca2+-dependent antifreeze proteins: Modulation of conformation and activity by divalent metal ions. J. Biol. Chem. 1996, 271, 16627–16632. [Google Scholar] [CrossRef] [PubMed]
- Meister, K.; Ebbinghaus, S.; Xu, Y.; Duman, J.G.; DeVries, A.L.; Gruebele, M.; Leitner, D.M.; Havenith, M. Long-range protein–water dynamics in hyperactive insect antifreeze proteins. Proc. Natl. Acad. Sci. USA 2013, 110, 1617–1622. [Google Scholar] [CrossRef] [PubMed]
- Midya, U.S.; Bandyopadhyay, S. Hydration behavior at the ice-binding surface of the Tenebrio molitor antifreeze protein. J. Phys. Chem. B 2014, 118, 4743–4752. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Lin, Q.; Yang, D.S.C.; Ewart, Y.K.; Hew, C.L. The role of Ca2+-coordinating residues of herring antifreeze protein in antifreeze activity. Biochemistry 2004, 43, 14547–14554. [Google Scholar] [CrossRef]
- Yang, D.S.C.; Hon, W.-C.; Bubanko, S.; Xue, Y.; Seetharaman, J.; Hew, C.L.; Sicheri, F. Identification of the ice-binding surface on a type III antifreeze protein with a “flatness function” algorithm. Biophys. J. 1998, 74, 2142–2151. [Google Scholar] [CrossRef]
- Talon, R.; Coquelle, N.; Madern, D.; Girard, E. An experimental point of view on hydration/solvation in halophilic proteins. Front. Microbiol. 2014, 5, 1–8. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Crystal | Ca2+-Bound Form (pH7) | Ca2+-Free Form (pH3) |
|---|---|---|
| Data Collection | ||
| Space group | P3121 | |
| Unit-cell parameters (Å) | a = b = 65.95, c = 49.79 | a = b = 66.03, c = 50.30 |
| Beam line | Photon Factory AR-NW12A | |
| Wavelength (Å) | 1.0 | |
| Resolution range a (Å) | 21.6–1.06 (1.12–1.06) | 16.4–1.25 (1.32–1.25) |
| Rmerge a, b | 0.045 (0.229) | 0.052 (0.234) |
| Completeness a (%) | 99.7 (100) | 98.3 (100) |
| Multiplicity a (%) | 10.5 (10.3) | 10.1 (10.4) |
| <I/σ(I)> a | 10.1 (3.4) | 8.5 (3.2) |
| Refinement Statistics | ||
| Resolution range a (Å) | 19.0–1.06 (1.087–1.06) | 16.4–1.25 (1.282–1.25) |
| R factor a, c | 0.154 (0.213) | 0.180 (0.247) |
| Free R factor a, c, d | 0.174 (0.227) | 0.212 (282) |
| R.M.S bond length (Å) | 0.017 | 0.016 |
| R.M.S bond angles (°) | 1.931 | 1.853 |
| Residues | 126 | 125 |
| Number of non-hydrogen atoms | ||
| Protein | 993 | 975 |
| Water | 289 | 237 |
| Other | 1 (Ca2+) | 5 (SO42−) |
| Ramachandran plot e (%) | ||
| Residues in favored regions | 93 | 95 |
| Residues in allowed regions | 7 | 5 |
| Residues in outliner regions | 0 | 0 |
| Average B factor (Å2) | 11.0 | 17.0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arai, T.; Nishimiya, Y.; Ohyama, Y.; Kondo, H.; Tsuda, S. Calcium-Binding Generates the Semi-Clathrate Waters on a Type II Antifreeze Protein to Adsorb onto an Ice Crystal Surface. Biomolecules 2019, 9, 162. https://doi.org/10.3390/biom9050162
Arai T, Nishimiya Y, Ohyama Y, Kondo H, Tsuda S. Calcium-Binding Generates the Semi-Clathrate Waters on a Type II Antifreeze Protein to Adsorb onto an Ice Crystal Surface. Biomolecules. 2019; 9(5):162. https://doi.org/10.3390/biom9050162
Chicago/Turabian StyleArai, Tatsuya, Yoshiyuki Nishimiya, Yasushi Ohyama, Hidemasa Kondo, and Sakae Tsuda. 2019. "Calcium-Binding Generates the Semi-Clathrate Waters on a Type II Antifreeze Protein to Adsorb onto an Ice Crystal Surface" Biomolecules 9, no. 5: 162. https://doi.org/10.3390/biom9050162
APA StyleArai, T., Nishimiya, Y., Ohyama, Y., Kondo, H., & Tsuda, S. (2019). Calcium-Binding Generates the Semi-Clathrate Waters on a Type II Antifreeze Protein to Adsorb onto an Ice Crystal Surface. Biomolecules, 9(5), 162. https://doi.org/10.3390/biom9050162