
p53 Phosphomimetics Preserve Transient Secondary Structure but Reduce Binding to Mdm2 and MdmX



Abstract
1. Introduction
2. Materials and Methods
2.1. Purification of Mdm2, MdmX, KIX, and Labeled p53TAD Constructs
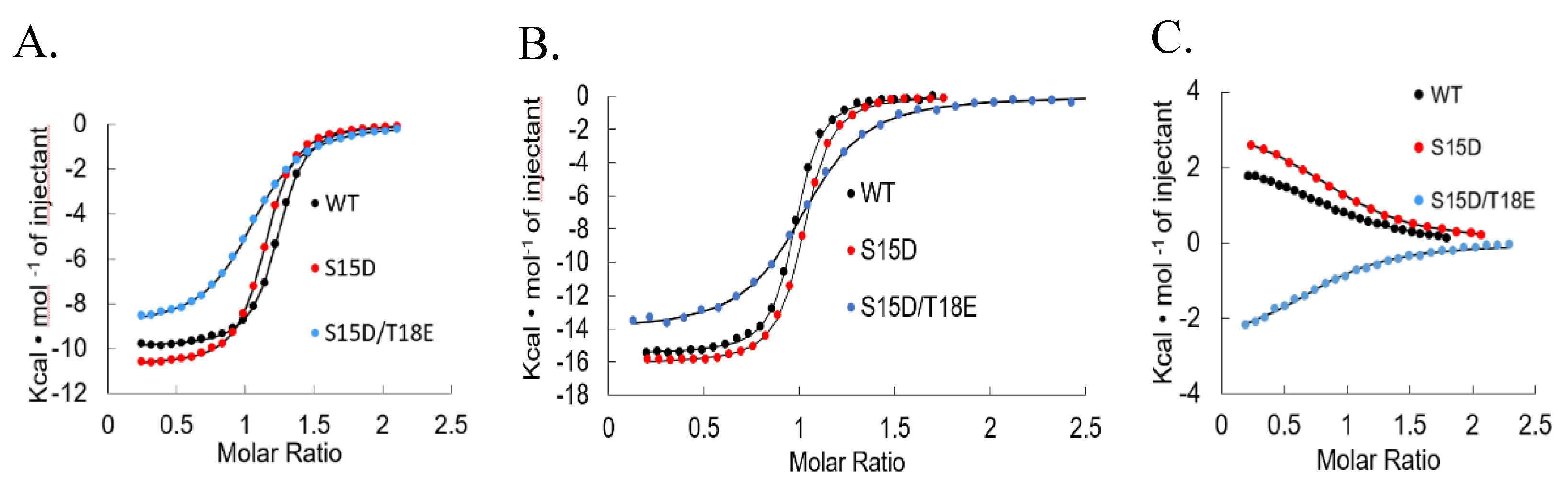
2.2. Isothermal Titration Calorimetry
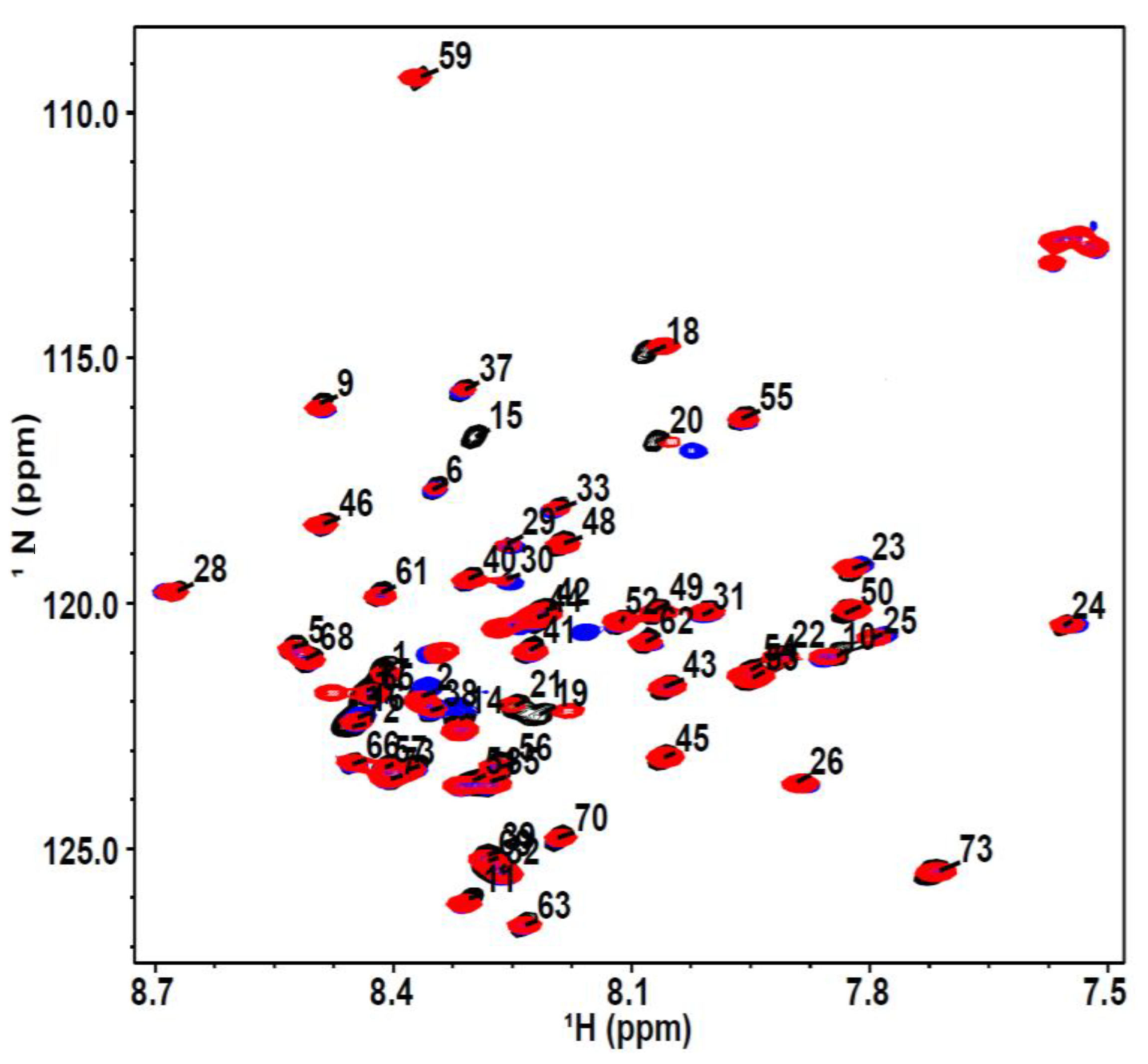
2.3. Nuclear Magnetic Resonance Spectroscopy
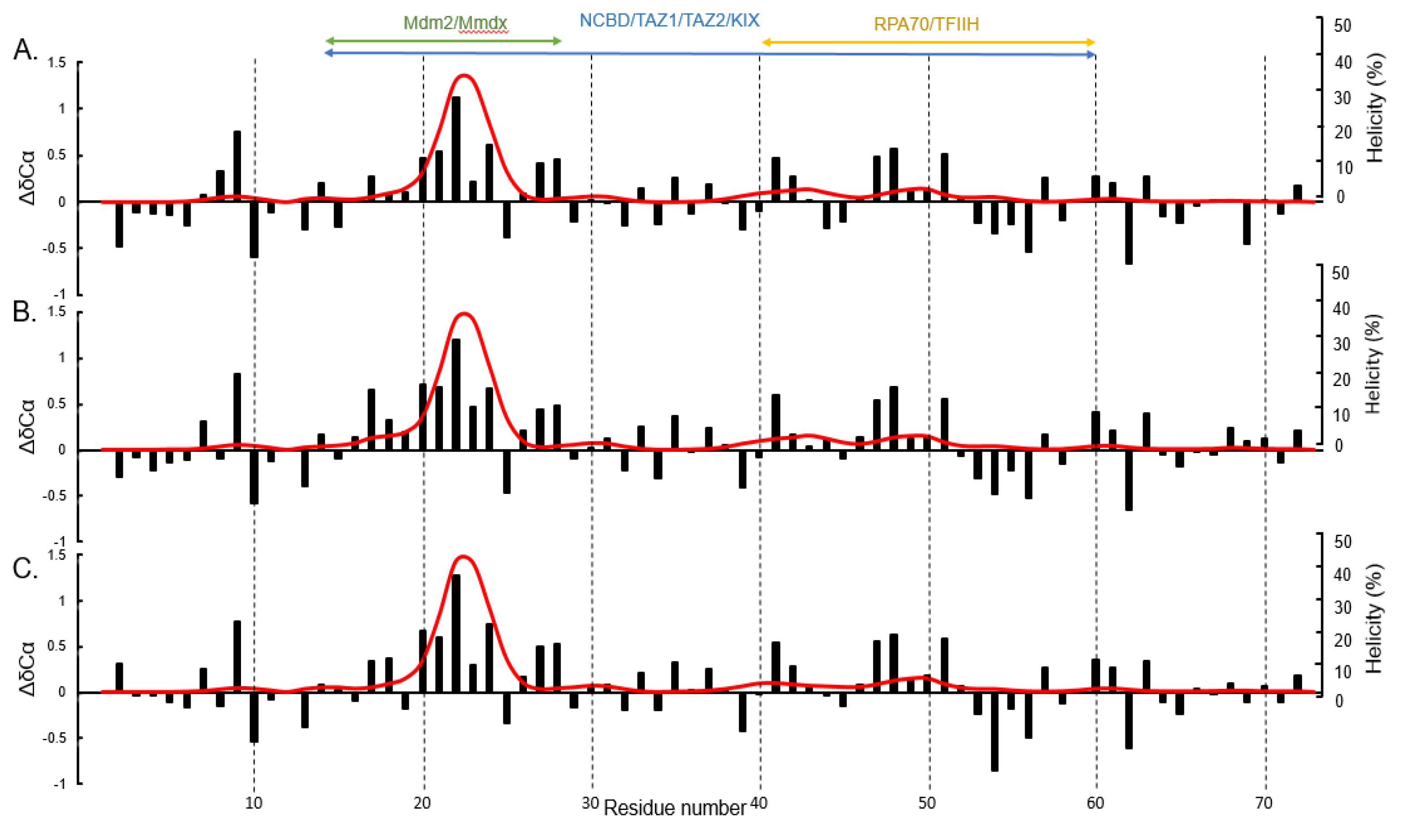
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Blagosklonny, M.V. P53: An ubiquitous target of anticancer drugs. Int. J. Cancer 2002, 98, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Chene, P. Targeting p53 in cancer. Curr. Med. Chem. Anticancer Agents 2001, 1, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Hainaut, P.; Hollstein, M. P53 and human cancer: The first ten thousand mutations. Adv. Cancer Res. 2000, 77, 81–137. [Google Scholar] [PubMed]
- Sigal, A.; Rotter, V. Oncogenic mutations of the p53 tumor suppressor: The demons of the guardian of the genome. Cancer Res. 2000, 60, 6788–6793. [Google Scholar] [PubMed]
- Dunker, A.K.; Brown, C.J.; Lawson, J.D.; Iakoucheva, L.M.; Obradovic, Z. Intrinsic disorder and protein function. Biochemistry 2002, 41, 6573–6582. [Google Scholar] [CrossRef] [PubMed]
- Matas, D.; Sigal, A.; Stambolsky, P.; Milyavsky, M.; Weisz, L.; Schwartz, D.; Goldfinger, N.; Rotter, V. Integrity of the n-terminal transcription domain of p53 is required for mutant p53 interference with drug-induced apoptosis. EMBO J. 2001, 20, 4163–4172. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Silman, I.; Uversky, V.N.; Sussman, J.L. Function and structure of inherently disordered proteins. Curr. Opin. Struct. Biol. 2008, 18, 756–764. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Intrinsically disordered proteins and novel strategies for drug discovery. Expert Opin. Drug Discov. 2012, 7, 475–488. [Google Scholar] [CrossRef] [PubMed]
- van der Lee, R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T.; et al. Classification of intrinsically disordered regions and proteins. Chem. Rev. 2014, 114, 6589–6631. [Google Scholar] [CrossRef] [PubMed]
- Ferreon, J.C.; Lee, C.W.; Arai, M.; Martinez-Yamout, M.A.; Dyson, H.J.; Wright, P.E. Cooperative regulation of p53 by modulation of ternary complex formation with cbp/p300 and hdm2. Proc. Natl. Acad. Sci. USA 2009, 106, 6591. [Google Scholar] [CrossRef] [PubMed]
- Oda, K.; Arakawa, H.; Tanaka, T.; Matsuda, K.; Tanikawa, C.; Mori, T.; Nishimori, H.; Tamai, K.; Tokino, T.; Nakamura, Y.; et al. P53aip1, a potential mediator of p53-dependent apoptosis, and its regulation by Ser-46-phosphorylated p53. Cell 2000, 102, 849–862. [Google Scholar] [CrossRef]
- Scoumanne, A.; Harms, K.L.; Chen, X. Structural basis for gene activation by p53 family members. Cancer Biol. Ther. 2005, 4, 1178–1185. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Kim, D.H.; Han, J.J.; Cha, E.J.; Lim, J.E.; Cho, Y.J.; Lee, C.; Han, K.H. Understanding pre-structured motifs (presmos) in intrinsically unfolded proteins. Curr. Protein Pept. Sci. 2012, 13, 34–54. [Google Scholar] [CrossRef] [PubMed]
- Oldfield, C.J.; Dunker, A.K. Intrinsically disordered proteins and intrinsically disordered protein regions. Annu. Rev. Biochem. 2014, 83, 553–584. [Google Scholar] [CrossRef] [PubMed]
- Crabtree, M.D.; Borcherds, W.; Poosapati, A.; Shammas, S.L.; Daughdrill, G.W.; Clarke, J. Conserved helix-flanking prolines modulate intrinsically disordered protein:Target affinity by altering the lifetime of the bound complex. Biochemistry 2017, 56, 2379–2384. [Google Scholar] [CrossRef] [PubMed]
- Bah, A.; Forman-Kay, J.D. Modulation of intrinsically disordered protein function by post-translational modifications. J. Biol. Chem. 2016, 291, 6696–6705. [Google Scholar] [CrossRef] [PubMed]
- Bah, A.; Vernon, R.M.; Siddiqui, Z.; Krzeminski, M.; Muhandiram, R.; Zhao, C.; Sonenberg, N.; Kay, L.E.; Forman-Kay, J.D. Folding of an intrinsically disordered protein by phosphorylation as a regulatory switch. Nature 2015, 519, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Oren, M. Regulation of the p53 tumor suppressor protein. J. Biol. Chem. 1999, 274, 36031–36034. [Google Scholar] [CrossRef] [PubMed]
- Donehower, L.A.; Harvey, M.; Slagle, B.L.; McArthur, M.J.; Montgomery, C.A., Jr.; Butel, J.S.; Bradley, A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992, 356, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Oliner, J.D.; Kinzler, K.W.; Meltzer, P.S.; George, D.L.; Vogelstein, B. Amplification of a gene encoding a p53-associated protein in human sarcomas. Nature 1992, 358, 80–83. [Google Scholar] [CrossRef] [PubMed]
- Badciong, J.C.; Haas, A.L. Mdmx is a ring finger ubiquitin ligase capable of synergistically enhancing Mdm2 ubiquitination. J. Biol. Chem. 2002, 277, 49668–49675. [Google Scholar] [CrossRef] [PubMed]
- Picksley, S.M.; Lane, D.P. The p53-Mdm2 autoregulatory feedback loop: A paradigm for the regulation of growth control by p53? Bioessays 1993, 15, 689–690. [Google Scholar] [CrossRef] [PubMed]
- Wienken, M.; Dickmanns, A.; Nemajerova, A.; Kramer, D.; Najafova, Z.; Weiss, M.; Karpiuk, O.; Kassem, M.; Zhang, Y.; Lozano, G.; et al. Mdm2 associates with polycomb repressor complex 2 and enhances stemness-promoting chromatin modifications independent of p53. Mol. Cell 2016, 61, 68–83. [Google Scholar] [CrossRef] [PubMed]
- Shvarts, A.; Steegenga, W.T.; Riteco, N.; van Laar, T.; Dekker, P.; Bazuine, M.; van Ham, R.C.; van der Houven van Oordt, W.; Hateboer, G.; van der Eb, A.J.; et al. Mdmx: A novel p53-binding protein with some functional properties of Mdm2. EMBO J. 1996, 15, 5349–5357. [Google Scholar] [CrossRef] [PubMed]
- Stad, R.; Little, N.A.; Xirodimas, D.P.; Frenk, R.; van der Eb, A.J.; Lane, D.P.; Saville, M.K.; Jochemsen, A.G. Mdmx stabilizes p53 and Mdm2 via two distinct mechanisms. EMBO Rep. 2001, 2, 1029–1034. [Google Scholar] [CrossRef] [PubMed]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Kubbutat, M.H.; Jones, S.N.; Vousden, K.H. Regulation of p53 stability by Mdm2. Nature 1997, 387, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Francoz, S.; Froment, P.; Bogaerts, S.; De Clercq, S.; Maetens, M.; Doumont, G.; Bellefroid, E.; Marine, J.C. Mdm4 and Mdm2 cooperate to inhibit p53 activity in proliferating and quiescent cells in vivo. Proc. Natl. Acad. Sci. USA 2006, 103, 3232–3237. [Google Scholar] [CrossRef] [PubMed]
- Xiong, S.; Van Pelt, C.S.; Elizondo-Fraire, A.C.; Liu, G.; Lozano, G. Synergistic roles of Mdm2 and Mdm4 for p53 inhibition in central nervous system development. Proc. Natl. Acad. Sci. USA 2006, 103, 3226–3231. [Google Scholar] [CrossRef] [PubMed]
- Jabbur, J.R.; Tabor, A.D.; Cheng, X.; Wang, H.; Uesugi, M.; Lozano, G.; Zhang, W. Mdm-2 binding and TAFII31 recruitment is regulated by hydrogen bond disruption between the p53 residues Thr18 and Asp21. Oncogene 2002, 21, 7100–7113. [Google Scholar] [CrossRef] [PubMed]
- Serrano, M.A.; Li, Z.; Dangeti, M.; Musich, P.R.; Patrick, S.; Roginskaya, M.; Cartwright, B.; Zou, Y. DNA-PK, ATM and ATR collaboratively regulate p53-RPA interaction to facilitate homologous recombination DNA repair. Oncogene 2013, 32, 2452–2462. [Google Scholar] [CrossRef] [PubMed]
- Shieh, S.Y.; Ikeda, M.; Taya, Y.; Prives, C. DNA damage-induced phosphorylation of p53 alleviates inhibition by Mdm2. Cell 1997, 91, 325–334. [Google Scholar] [CrossRef]
- Lee, C.W.; Ferreon, J.C.; Ferreon, A.C.; Arai, M.; Wright, P.E. Graded enhancement of p53 binding to CREB-binding protein (CBP) by multisite phosphorylation. Proc. Natl. Acad. Sci. USA 2010, 107, 19290–19295. [Google Scholar] [CrossRef] [PubMed]
- Bottger, V.; Bottger, A.; Garcia-Echeverria, C.; Ramos, Y.F.; van der Eb, A.J.; Jochemsen, A.G.; Lane, D.P. Comparative study of the p53-Mdm2 and p53-Mmdmx interfaces. Oncogene 1999, 18, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Baresova, P.; Musilova, J.; Pitha, P.M.; Lubyova, B. P53 tumor suppressor protein stability and transcriptional activity are targeted by Kaposi’s Sarcoma-associated herpesvirus-encoded viral interferon regulatory factor 3. Mol. Cell. Biol. 2014, 34, 386–399. [Google Scholar] [CrossRef] [PubMed]
- Dumaz, N.; Milne, D.M.; Meek, D.W. Protein kinase CK1 is a p53-threonine 18 kinase which requires prior phosphorylation of serine 15. FEBS Lett. 1999, 463, 312–316. [Google Scholar] [CrossRef]
- Saito, S.; Yamaguchi, H.; Higashimoto, Y.; Chao, C.; Xu, Y.; Fornace, A.J., Jr.; Appella, E.; Anderson, C.W. Phosphorylation site interdependence of human p53 post-translational modifications in response to stress. J. Biol. Chem. 2003, 278, 37536–37544. [Google Scholar] [CrossRef] [PubMed]
- McKinney, K.; Prives, C. Efficient specific DNA binding by p53 requires both its central and C-terminal domains as revealed by studies with high-mobility group 1 protein. Mol. Cell. Biol. 2002, 22, 6797–6808. [Google Scholar] [CrossRef] [PubMed]
- Dumaz, N.; Meek, D.W. Serine15 phosphorylation stimulates p53 transactivation but does not directly influence interaction with HDM2. EMBO J. 1999, 18, 7002–7010. [Google Scholar] [CrossRef] [PubMed]
- Grossman, S.R.; Perez, M.; Kung, A.L.; Joseph, M.; Mansur, C.; Xiao, Z.X.; Kumar, S.; Howley, P.M.; Livingston, D.M. P300/Mdm2 complexes participate in Mdm2-mediated p53 degradation. Mol. Cell 1998, 2, 405–415. [Google Scholar] [CrossRef]
- Kruse, J.P.; Gu, W. Modes of p53 regulation. Cell 2009, 137, 609–622. [Google Scholar] [CrossRef] [PubMed]
- Teufel, D.P.; Freund, S.M.; Bycroft, M.; Fersht, A.R. Four domains of p300 each bind tightly to a sequence spanning both transactivation subdomains of p53. Proc. Natl. Acad. Sci. USA 2007, 104, 7009–7014. [Google Scholar] [CrossRef] [PubMed]
- Teufel, D.P.; Bycroft, M.; Fersht, A.R. Regulation by phosphorylation of the relative affinities of the N-terminal transactivation domains of p53 for p300 domains and Mdm2. Oncogene 2009, 28, 2112–2118. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Jenkins, L.M.; Durell, S.R.; Hayashi, R.; Mazur, S.J.; Cherry, S.; Tropea, J.E.; Miller, M.; Wlodawer, A.; Appella, E.; et al. Structural basis for p300 Taz2-p53 TAD1 binding and modulation by phosphorylation. Structure 2009, 17, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Gao, M.; Yang, F.; Zhang, L.; Su, Z. Deciphering the promiscuous interactions between intrinsically disordered transactivation domains and the KIX domain. Proteins 2017, 85, 2088–2095. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.W.; Arai, M.; Martinez-Yamout, M.A.; Dyson, H.J.; Wright, P.E. Mapping the interactions of the p53 transactivation domain with the KIX domain of CBP. Biochemistry 2009, 48, 2115–2124. [Google Scholar] [CrossRef] [PubMed]
- Stock, J.; Da Re, S. Signal transduction: Response regulators on and off. Curr. Biol. 2000, 10, R420–R424. [Google Scholar] [CrossRef]
- Johnson, L.N.; Lewis, R.J. Structural basis for control by phosphorylation. Chem. Rev. 2001, 101, 2209–2242. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Chen, Y.; Mooney, S.M.; Rajagopalan, K.; Bhargava, A.; Sacho, E.; Weninger, K.; Bryan, P.N.; Kulkarni, P.; Orban, J. Phosphorylation-induced conformational ensemble switching in an intrinsically disordered cancer/testis antigen. J. Biol. Chem. 2015, 290, 25090–25102. [Google Scholar] [CrossRef] [PubMed]
- Kussie, P.H.; Gorina, S.; Marechal, V.; Elenbaas, B.; Moreau, J.; Levine, A.J.; Pavletich, N.P. Structure of the mdm2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 1996, 274, 948–953. [Google Scholar] [CrossRef] [PubMed]
- Mavinahalli, J.N.; Madhumalar, A.; Beuerman, R.W.; Lane, D.P.; Verma, C. Differences in the transactivation domains of p53 family members: A computational study. BMC Genom. 2010, 11 (Suppl. 1), S5. [Google Scholar] [CrossRef] [PubMed]
- Hol, W.G. The role of the α-helix dipole in protein function and structure. Prog. Biophys. Mol. Biol. 1985, 45, 149–195. [Google Scholar] [CrossRef]
- Sheridan, R.P.; Levy, R.M.; Salemme, F.R. α-helix dipole model and electrostatic stabilization of 4-α-helical proteins. Proc. Natl. Acad. Sci. USA 1982, 79, 4545–4549. [Google Scholar] [CrossRef] [PubMed]
- Guerra-Castellano, A.; Diaz-Moreno, I.; Velazquez-Campoy, A.; De la Rosa, M.A.; Diaz-Quintana, A. Structural and functional characterization of phosphomimetic mutants of cytochrome c at Threonine 28 and Serine 47. Biochim. Biophys. Acta 2016, 1857, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Luwang, J.W.; Natesh, R. Phosphomimetic mutation destabilizes the central core domain of human p53. IUBMB Life 2018, 70, 1023–1031. [Google Scholar] [CrossRef] [PubMed]
- Pecina, P.; Borisenko, G.G.; Belikova, N.A.; Tyurina, Y.Y.; Pecinova, A.; Lee, I.; Samhan-Arias, A.K.; Przyklenk, K.; Kagan, V.E.; Huttemann, M. Phosphomimetic substitution of cytochrome C Tyrosine 48 decreases respiration and binding to cardiolipin and abolishes ability to trigger downstream caspase activation. Biochemistry 2010, 49, 6705–6714. [Google Scholar] [CrossRef] [PubMed]
- Loughery, J.; Cox, M.; Smith, L.M.; Meek, D.W. Critical role for p53-serine 15 phosphorylation in stimulating transactivation at p53-responsive promoters. Nucleic Acids Res. 2014, 42, 7666–7680. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.A. Using nmrview to visualize and analyze the NMR spectra of macromolecules. Methods Mol. Biol. 2004, 278, 313–352. [Google Scholar] [PubMed]
- Johnson, B.A.; Blevins, R.A. NMR view: A computer program for the visualization and analysis of NMR data. J. Biomol. NMR 1994, 4, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, J.T.; Mulder, F.A.A. Potenci: Prediction of temperature, neighbor and pH-corrected chemical shifts for intrinsically disordered proteins. J. Biomol. NMR 2018, 70, 141–165. [Google Scholar] [CrossRef] [PubMed]
- Camilloni, C.; De Simone, A.; Vranken, W.F.; Vendruscolo, M. Determination of secondary structure populations in disordered states of proteins using nuclear magnetic resonance chemical shifts. Biochemistry 2012, 51, 2224–2231. [Google Scholar] [CrossRef] [PubMed]
- Theillet, F.X.; Rose, H.M.; Liokatis, S.; Binolfi, A.; Thongwichian, R.; Stuiver, M.; Selenko, P. Site-specific nmr mapping and time-resolved monitoring of serine and threonine phosphorylation in reconstituted kinase reactions and mammalian cell extracts. Nat. Protoc. 2013, 8, 1416–1432. [Google Scholar] [CrossRef] [PubMed]
- Gokirmak, T.; Denison, F.C.; Laughner, B.J.; Paul, A.L.; Ferl, R.J. Phosphomimetic mutation of a conserved serine residue in arabidopsis thaliana 14–3-3omega suggests a regulatory role of phosphorylation in dimerization and target interactions. Plant Physiol. Biochem. 2015, 97, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Chen, L.; Li, Z.; Lane, W.S.; Chen, J. ATM activates p53 by regulating Mdm2 oligomerization and E3 processivity. EMBO J. 2009, 28, 3857–3867. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Cross, B.; Li, B.; Chen, L.; Li, Z.; Chen, J. Regulation of Mdm2 E3 ligase activity by phosphorylation after DNA damage. Mol. Cell. Biol. 2011, 31, 4951–4963. [Google Scholar] [CrossRef] [PubMed]
- Nakamizo, A.; Amano, T.; Zhang, W.; Zhang, X.Q.; Ramdas, L.; Liu, T.J.; Bekele, B.N.; Shono, T.; Sasaki, T.; Benedict, W.F.; et al. Phosphorylation of Thr18 and Ser20 of p53 in AD-p53-induced apoptosis. Neuro Oncol. 2008, 10, 275–291. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Kamura, T.; Shirata, N.; Murata, T.; Kudoh, A.; Iwahori, S.; Nakayama, S.; Isomura, H.; Nishiyama, Y.; Tsurumi, T. Degradation of phosphorylated p53 by viral protein-ECS E3 ligase complex. PLoS Pathog. 2009, 5, e1000530. [Google Scholar] [CrossRef] [PubMed]
- Jane Dyson, H.; Ewright, P. Insights into the structure and dynamics of unfolded proteins from nuclear magnetic resonance. In Advances in Protein Chemistry; Academic Press: Cambridge, MA, USA, 2002; Volume 62, pp. 311–340. [Google Scholar]
- Borcherds, W.M.; Daughdrill, G.W. Using nmr chemical shifts to determine residue-specific secondary structure populations for intrinsically disordered proteins. Methods Enzymol. 2018, 611, 101–136. [Google Scholar] [PubMed]
- Bista, M.; Wolf, S.; Khoury, K.; Kowalska, K.; Huang, Y.; Wrona, E.; Arciniega, M.; Popowicz, G.M.; Holak, T.A.; Domling, A. Transient protein states in designing inhibitors of the Mdm2-p53 interaction. Structure 2013, 21, 2143–2151. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.V.; Ping Koh, D.X.; Liu, Y.; Joseph, T.L.; Lane, D.P.; Verma, C.S.; Tan, Y.S. Role of the N-terminal lid in regulating the interaction of phosphorylated Mmdmx with p53. Oncotarget 2017, 8, 112825–112840. [Google Scholar] [CrossRef] [PubMed]
- McCoy, M.A.; Gesell, J.J.; Senior, M.M.; Wyss, D.F. Flexible lid to the p53-binding domain of human Mdm2: Implications for p53 regulation. Proc. Natl. Acad. Sci. USA 2003, 100, 1645–1648. [Google Scholar] [CrossRef] [PubMed]
- Pierce, M.M.; Raman, C.S.; Nall, B.T. Isothermal titration calorimetry of protein-protein interactions. Methods 1999, 19, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Rajarathnam, K.; Rosgen, J. Isothermal titration calorimetry of membrane proteins—Progress and challenges. Biochim. Biophys. Acta 2014, 1838, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Borcherds, W.; Theillet, F.-X.; Katzer, A.; Finzel, A.; Mishall, K.M.; Powell, A.T.; Wu, H.; Manieri, W.; Dieterich, C.; Selenko, P.; et al. Disorder and residual helicity alter p53-Mdm2 binding affinity and signaling in cells. Nat. Chem. Biol. 2014, 10, 1000. [Google Scholar] [CrossRef] [PubMed]
- Fraser, J.A.; Madhumalar, A.; Blackburn, E.; Bramham, J.; Walkinshaw, M.D.; Verma, C.; Hupp, T.R. A novel p53 phosphorylation site within the Mdm2 ubiquitination signal: II. A model in which phosphorylation at ser269 induces a mutant conformation to p53. J. Biol. Chem. 2010, 285, 37773–37786. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Breyssens, H.; Salter, V.; Zhong, S.; Hu, Y.; Baer, C.; Ratnayaka, I.; Sullivan, A.; Brown, N.R.; Endicott, J.; et al. Restoring p53 function in human melanoma cells by inhibiting MDM2 and cyclin B1/CDK1-phosphorylated nuclear iASPP. Cancer Cell 2016, 30, 822–823. [Google Scholar] [CrossRef] [PubMed]
- Freyer, M.W.; Lewis, E.A. Isothermal titration calorimetry: Experimental design, data analysis, and probing macromolecule/ligand binding and kinetic interactions. Methods Cell Biol. 2008, 84, 79–113. [Google Scholar] [PubMed]
- Pise-Masison, C.A.; Radonovich, M.; Sakaguchi, K.; Appella, E.; Brady, J.N. Phosphorylation of p53: A novel pathway for p53 inactivation in human T-cell lymphotropic virus type 1-transformed cells. J. Virol. 1998, 72, 6348–6355. [Google Scholar] [PubMed]
- Sakaguchi, K.; Saito, S.; Higashimoto, Y.; Roy, S.; Anderson, C.W.; Appella, E. Damage-mediated phosphorylation of human p53 threonine 18 through a cascade mediated by a casein 1-like kinase. Effect on Mdm2 binding. J. Biol. Chem. 2000, 275, 9278–9283. [Google Scholar] [CrossRef] [PubMed]
- Schon, O.; Friedler, A.; Bycroft, M.; Freund, S.M.; Fersht, A.R. Molecular mechanism of the interaction between Mdm2 and p53. J. Mol. Biol. 2002, 323, 491–501. [Google Scholar] [CrossRef]
- Yadahalli, S.; Neira, J.L.; Johnson, C.M.; Tan, Y.S.; Rowling, P.J.E.; Chattopadhyay, A.; Verma, C.S.; Itzhaki, L.S. Kinetic and thermodynamic effects of phosphorylation on p53 binding to Mdm2. Sci. Rep. 2019, 9, 693. [Google Scholar] [CrossRef] [PubMed]
- Vega, F.M.; Sevilla, A.; Lazo, P.A. P53 stabilization and accumulation induced by human vaccinia-related kinase 1. Mol. Cell. Biol. 2004, 24, 10366–10380. [Google Scholar] [CrossRef] [PubMed]
- Dornan, D.; Hupp, T.R. Inhibition of p53-dependent transcription by BOX-I phospho-peptide mimetics that bind to p300. EMBO Rep. 2001, 2, 139–144. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| p53TAD | S15D | S15D/T18E | ||
|---|---|---|---|---|
| Mdm2 | Kd (nM) | 219.5 ± 0.012 | 392.5 ± 0.015 | 1.001 ± 0.016 |
| ΔG (Kcal/mol) | −9.1 | −8.75 ± 0.071 | −8.2 | |
| ΔH (Kcal/mol) | −9.721 ± 0.202 | −11.335 ± 0.827 | −8.173 ± 1.203 | |
| TΔS (Kcaal/mol/deg) | −0.634 ± 0.236 | −2.499 ± 0.977 | −0.964 ± 0.188 | |
| MdmX | Kd (nM) | 29 ± 0.004 | 30 ± 0.002 | 108 ± 12.5 |
| ΔG (Kcal/mol) | −10.3 ± 0.100 | −10.27 ± 0.058 | −9.5 ± 0.07 | |
| ΔH (Kcal/mol) | −16.260 ± 0.913 | −16.99 ± 1.405 | −14.27 ± 0.19 | |
| TΔS (Kcal/mol/deg) | −5.960 ± 0.988 | −6.794 ± 1.55 | −4.75 ± 0.16 | |
| KIX | Kd (nM) | 11,000 ± 2.700 | 8200 ± 1870 | 8380 ± 969 |
| ΔG (Kcal/mol) | −6.77 ± 0.157 | −6.95 ± 0.127 | −6.92 ± 0.056 | |
| ΔH (Kcal/mol) | 2.606 ± 0.308 | 2.926 ± 0.416 | −2.822 ± 0.079 | |
| TΔS (Kcal/mol/deg) | 9.377 ±0.165 | 9.874 ± 0.308 | 3.963 ± 0.270 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Levy, R.; Gregory, E.; Borcherds, W.; Daughdrill, G. p53 Phosphomimetics Preserve Transient Secondary Structure but Reduce Binding to Mdm2 and MdmX. Biomolecules 2019, 9, 83. https://doi.org/10.3390/biom9030083
Levy R, Gregory E, Borcherds W, Daughdrill G. p53 Phosphomimetics Preserve Transient Secondary Structure but Reduce Binding to Mdm2 and MdmX. Biomolecules. 2019; 9(3):83. https://doi.org/10.3390/biom9030083
Chicago/Turabian StyleLevy, Robin, Emily Gregory, Wade Borcherds, and Gary Daughdrill. 2019. "p53 Phosphomimetics Preserve Transient Secondary Structure but Reduce Binding to Mdm2 and MdmX" Biomolecules 9, no. 3: 83. https://doi.org/10.3390/biom9030083
APA StyleLevy, R., Gregory, E., Borcherds, W., & Daughdrill, G. (2019). p53 Phosphomimetics Preserve Transient Secondary Structure but Reduce Binding to Mdm2 and MdmX. Biomolecules, 9(3), 83. https://doi.org/10.3390/biom9030083