A Biophysical Approach to the Identification of Novel ApoE Chemical Probes


Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Mutagenesis, Expression, and Purification of ApoE Isoforms and ApoE4 Amino Terminal Domain
2.3. Expression and Purification of Human Rhino Virus 3C Protease
2.4. Activation of Corning Epic Plates and Immobilization of ApoE4
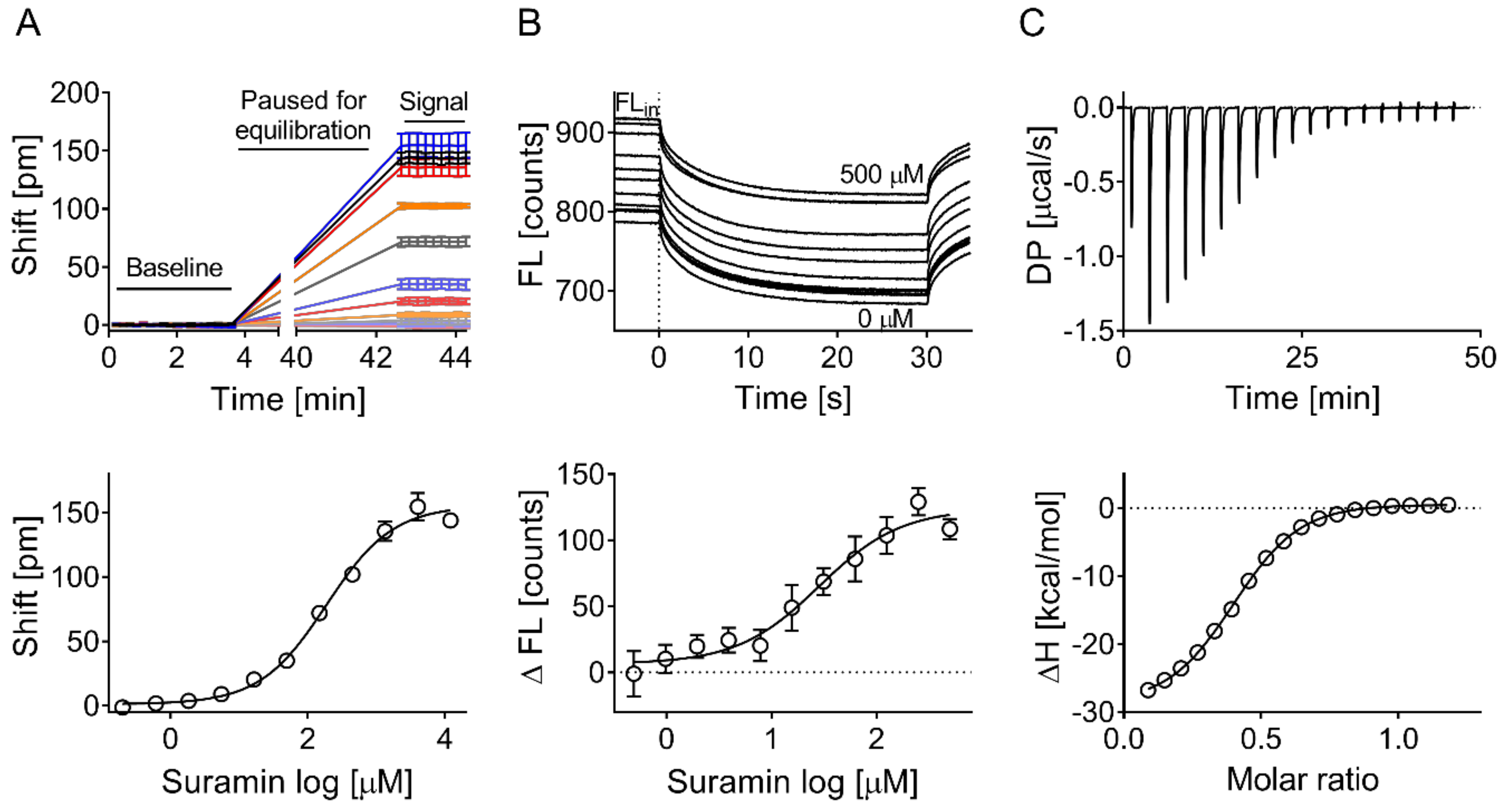
2.5. Suramin-Binding Curve on the Corning Epic
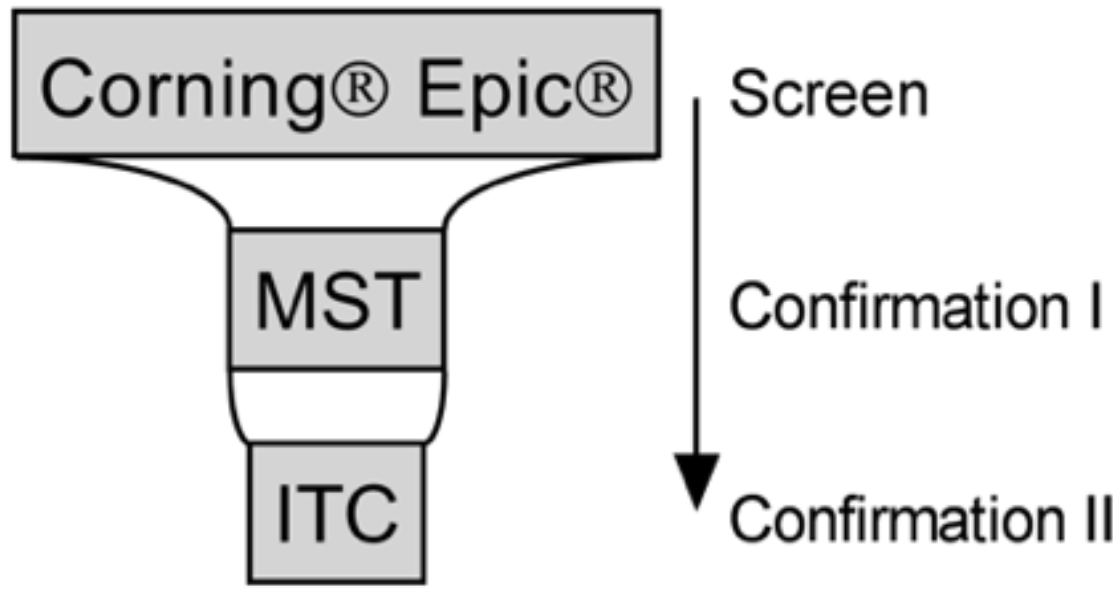
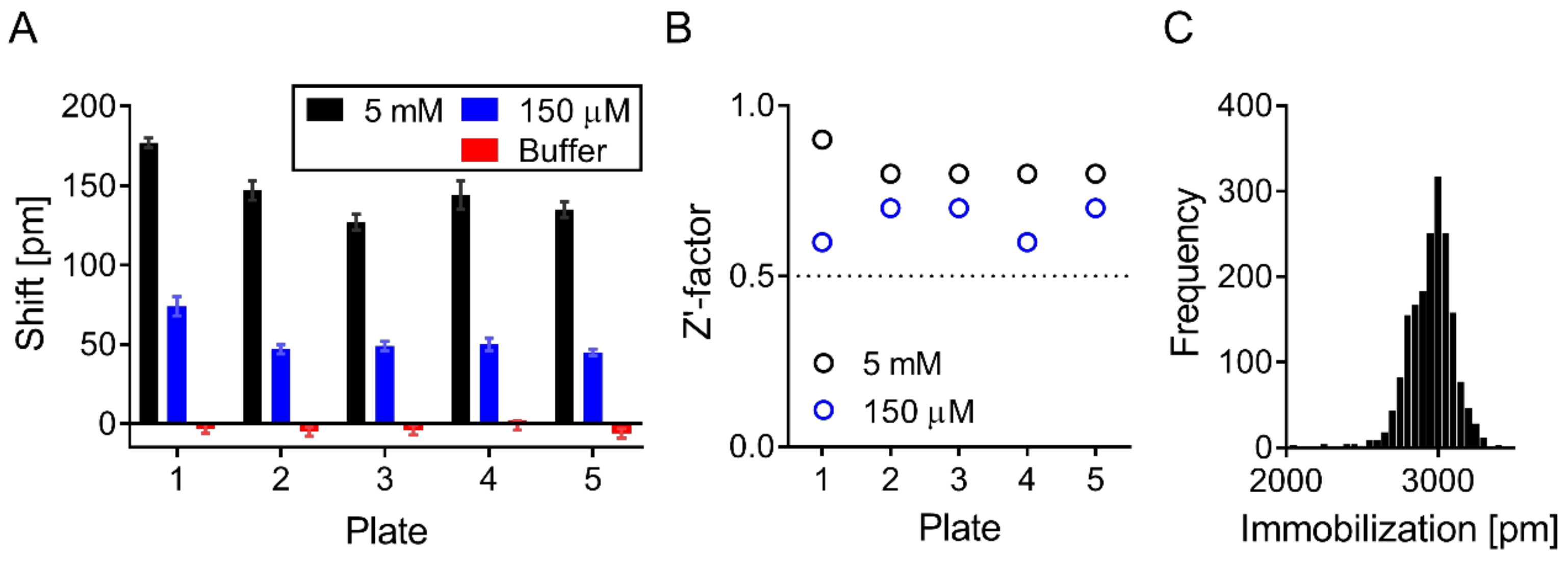
2.6. Screening
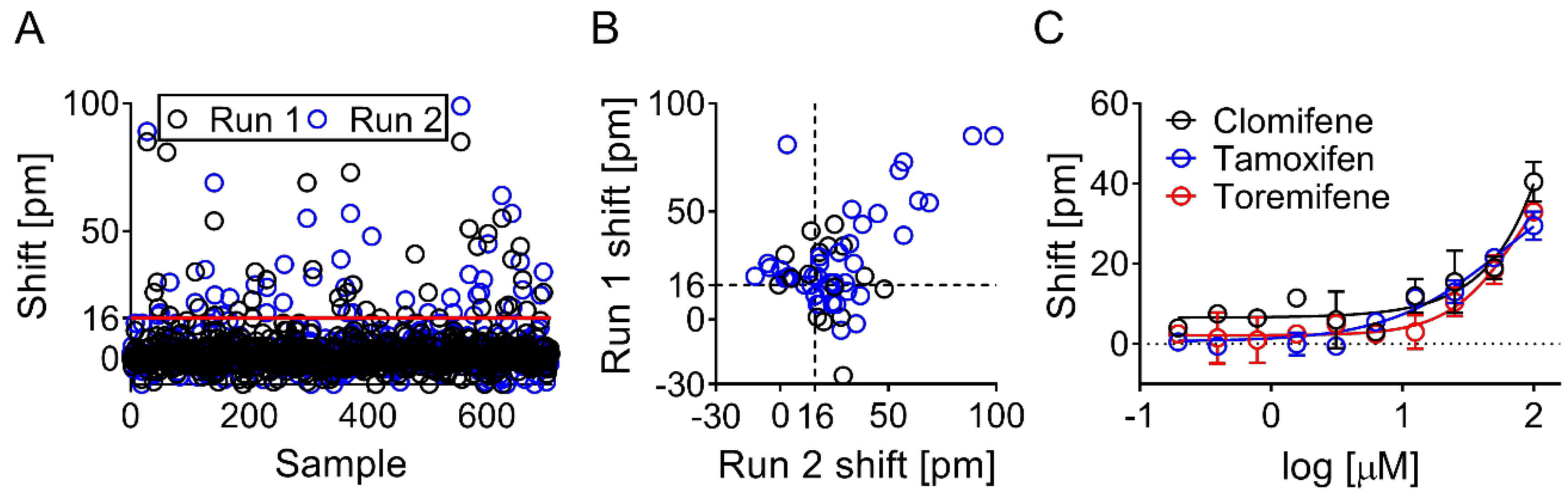
2.7. Screening Data Analysis and Hit Selection
2.8. Microscale Thermophoresis
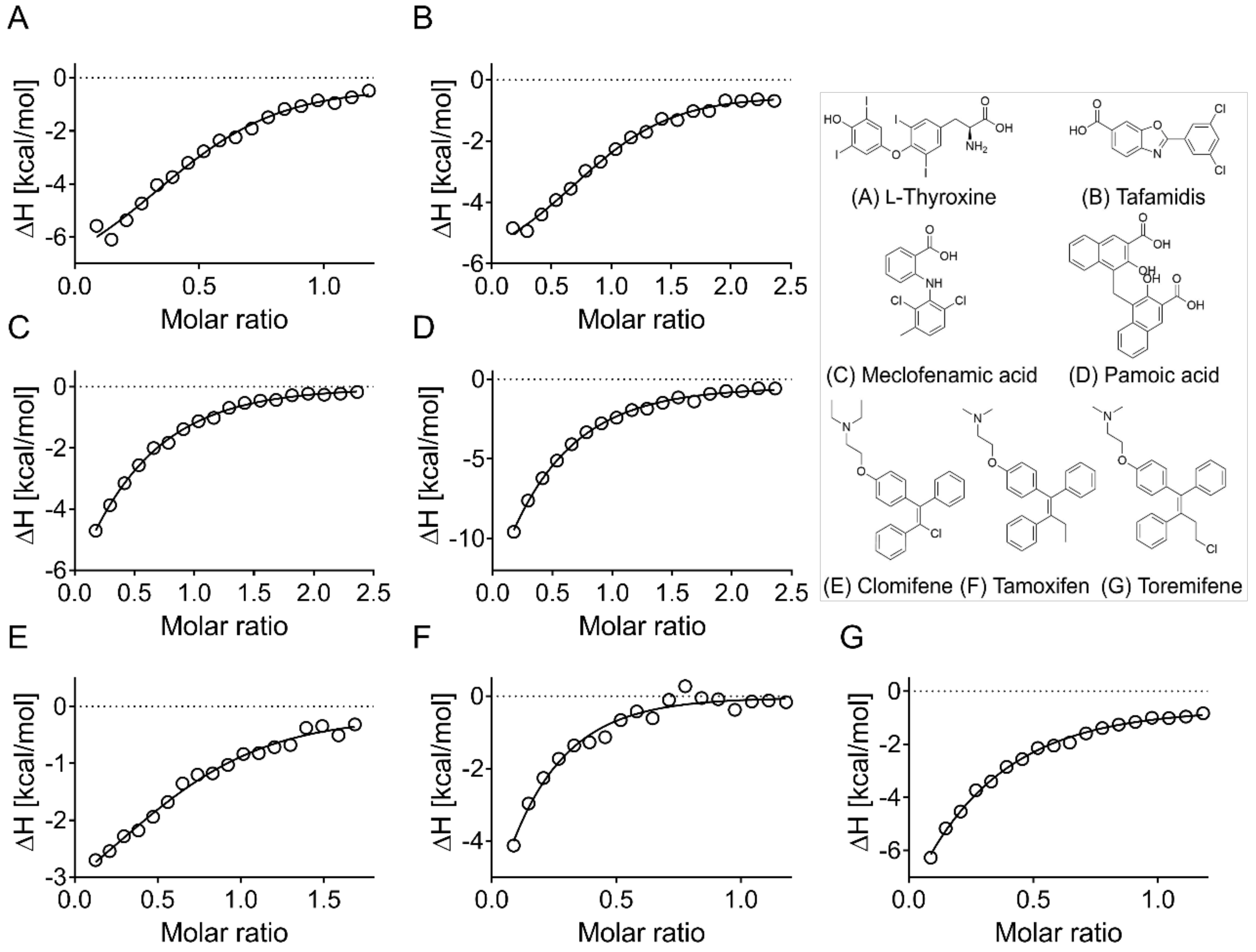
2.9. Isothermal Titration Calorimetry
2.10. Graphical Representations
3. Results
3.1. Expression and Purification of ApoE Isoforms
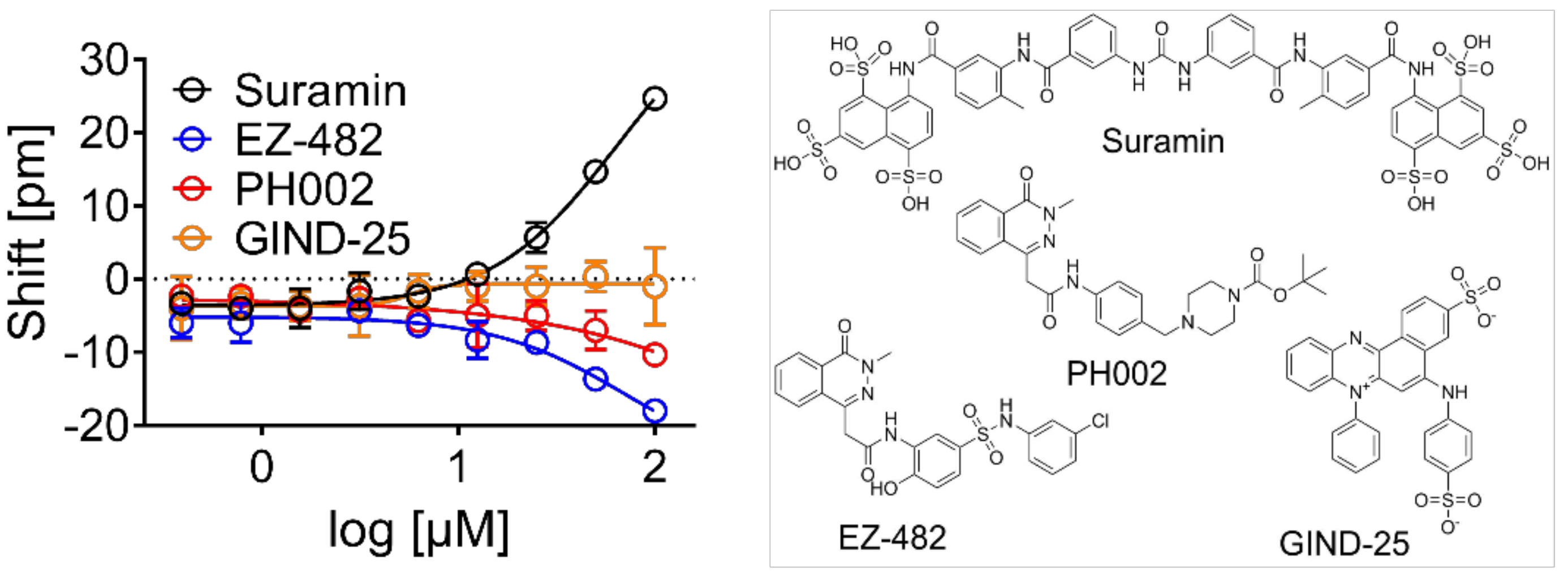
3.2. Validation of Biophysical Assays and Suramin as Positive Control
3.3. Screening Identified Fifty-Nine Compounds
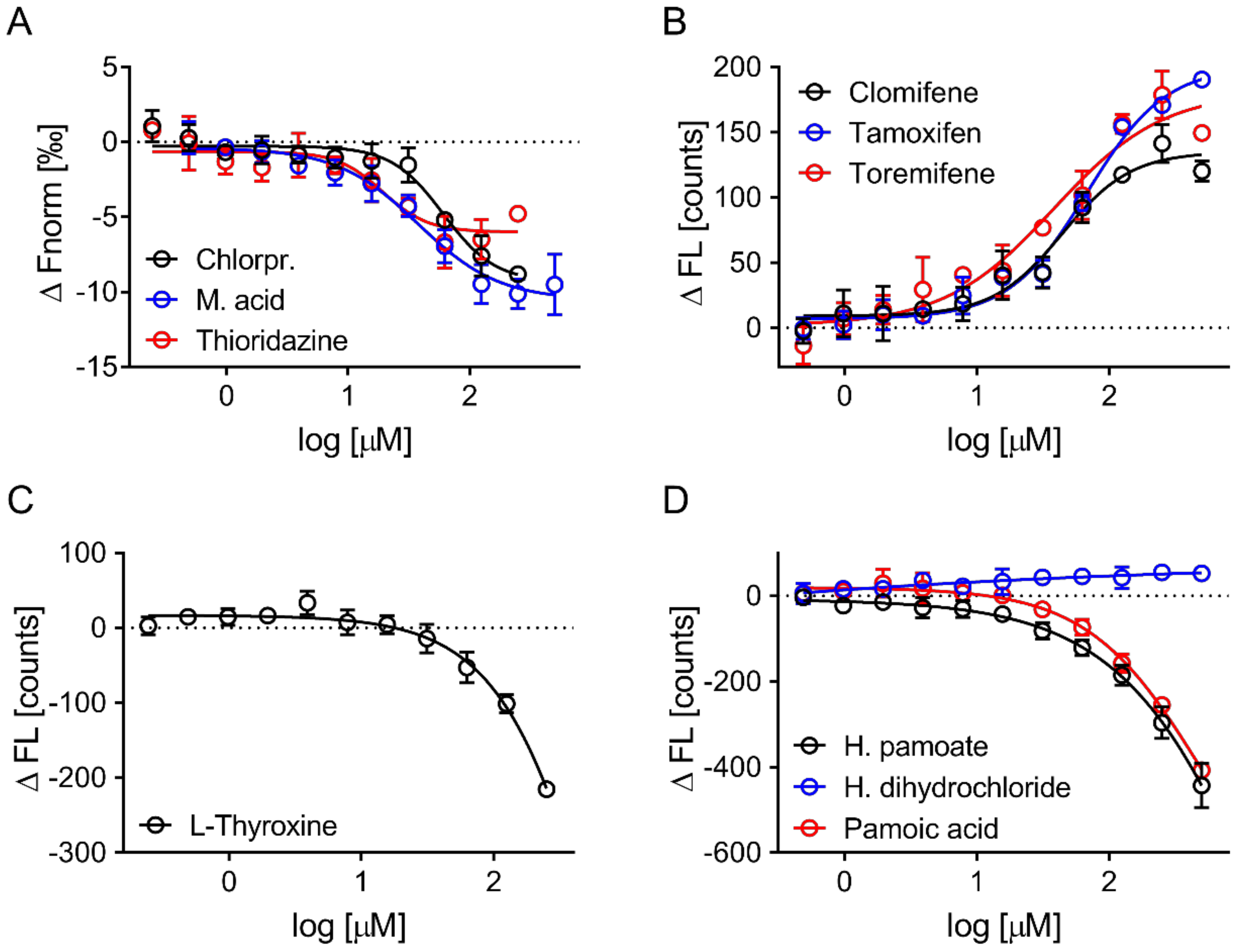
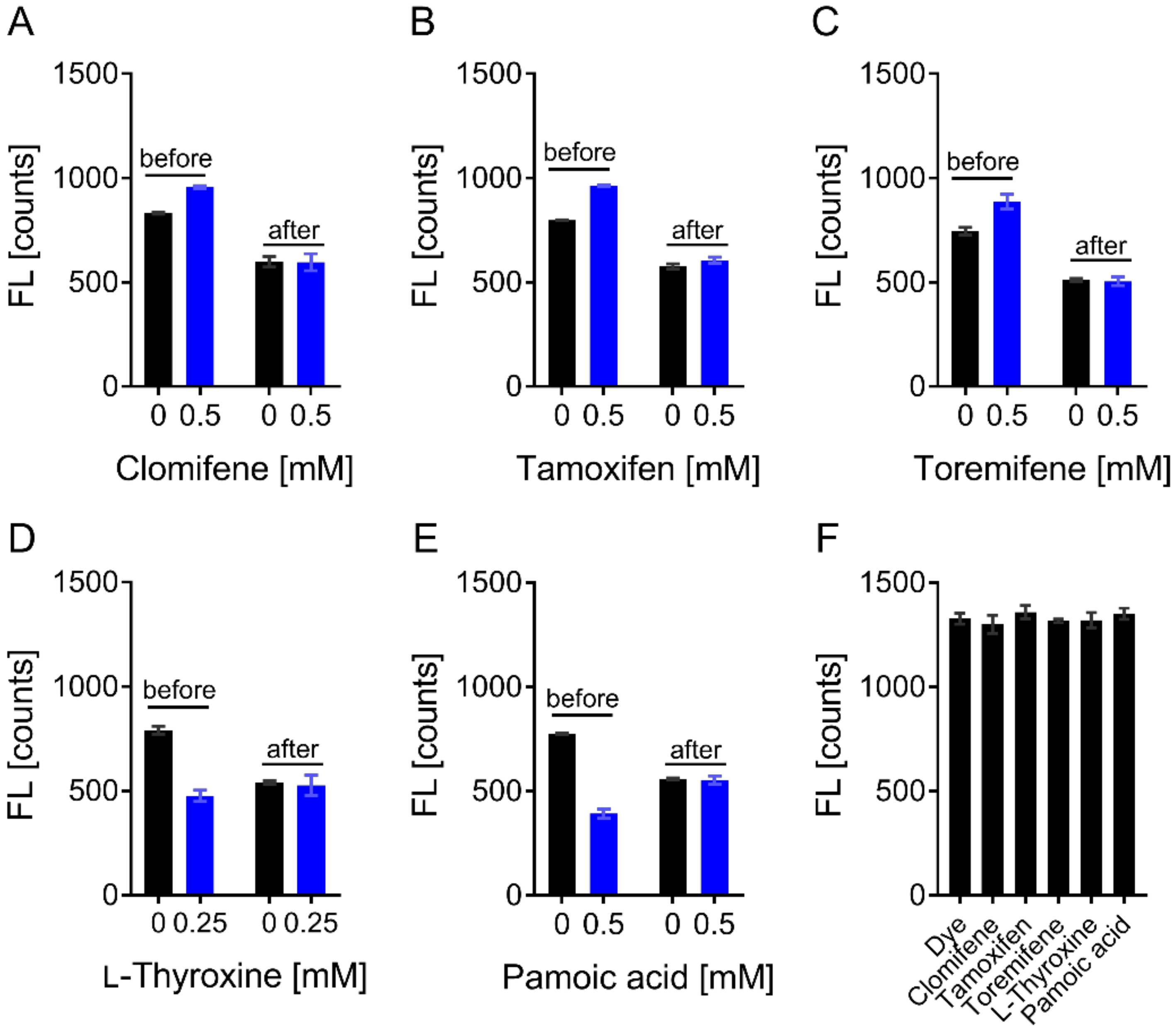
3.4. Evaluation of Hits by Microscale Thermophoresis and Isothermal Titration Calorimetry Confirmed Six Drugs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jonas, A.; Phillips, M.C. Lipoprotein structure. In Biochemistry of Lipids, Lipoproteins and Membranes; Vance, D.E., Vance, J.E., Eds.; Elsevier: Amsterdam, The Netherlands, 2008; Volume 348, pp. 485–506. ISBN 9780444532190. [Google Scholar]
- Mahley, R.W. Apolipoprotein E: Cholesterol transport protein with expanding role in cell biology. Science 1988, 240, 622–630. [Google Scholar] [CrossRef] [PubMed]
- Stukas, S.; Kulic, I.; Zareyan, S.; Wellington, C.L. Alzheimer’s Disease—Challenges for the Future; Zerr, I., Ed.; InTech: London, UK, 2015; ISBN 978-953-51-2137-4. [Google Scholar]
- Weisgraber, K.H. Apolipoprotein E: Structure-function relationships. Adv. Protein Chem. 1994, 45, 249–302. [Google Scholar] [PubMed]
- Farrer, L.A.; Cupples, L.A.; Haines, J.L.; Hyman, B.; Kukull, W.A.; Mayeux, R.; Myers, R.H.; Pericak-Vance, M.A.; Risch, N.; van Duijn, C.M. Effects of Age, Sex, and Ethnicity on the Association Between Apolipoprotein E Genotype and Alzheimer Disease. JAMA 1997, 278, 1349–1356. [Google Scholar] [CrossRef] [PubMed]
- Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.; George-Hyslop, P.H.; Pericak-Vance, M.A.; Joo, S.H.; Rosi, B.L.; Gusella, J.F.; Crapper-MacLachlan, D.R.; Alberts, M.J. Association of apolipoprotein E allele ε4 with late-onset familial and sporadic Alzheimer’s disease. Neurology 1993, 43, 1467–1472. [Google Scholar] [CrossRef] [PubMed]
- Strittmatter, W.J.; Saunders, A.M.; Schmechel, D.; Pericak-Vance, M.; Enghild, J.; Salvesen, G.S.; Roses, A.D. Apolipoprotein E: High-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 1977–1981. [Google Scholar] [CrossRef] [PubMed]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef] [PubMed]
- Van Duijn, C.M.; de Knijff, P.; Cruts, M.; Wehnert, A.; Havekes, L.M.; Hofman, A.; Van Broeckhoven, C. Apolipoprotein E4 allele in a population-based study of early-onset Alzheimer’s disease. Nat. Genet. 1994, 7, 74–78. [Google Scholar] [CrossRef]
- Nalbantoglu, J.; Gilfix, B.M.; Bertrand, P.; Robitaille, Y.; Gauthier, S.; Rosenblatt, D.S.; Poirier, J. Predictive value of apolipoprotein E genotyping in Alzheimer’s disease: Results of an autopsy series and an analysis of several combined studies. Ann. Neurol. 1994, 36, 889–895. [Google Scholar] [CrossRef]
- Mahoney-Sanchez, L.; Belaidi, A.A.; Bush, A.I.; Ayton, S. The Complex Role of Apolipoprotein E in Alzheimer’s Disease: An Overview and Update. J. Mol. Neurosci. 2016, 60, 325–335. [Google Scholar] [CrossRef]
- Weisgraber, K.H. Apolipoprotein E distribution among human plasma lipoproteins: Role of the cysteine-arginine interchange at residue 112. J. Lipid Res. 1990, 31, 1503–1511. [Google Scholar]
- Dong, L.M.; Weisgraber, K.H. Human apolipoprotein E4 domain interaction. Arginine 61 and glutamic acid 255 interact to direct the preference for very low-density lipoproteins. J. Biol. Chem. 1996, 271, 19053–19057. [Google Scholar] [CrossRef]
- Dong, L.M.; Wilson, C.; Wardell, M.R.; Simmons, T.; Mahley, R.W.; Weisgraber, K.H.; Agard, D.A. Human apolipoprotein E. Role of arginine 61 in mediating the lipoprotein preferences of the E3 and E4 isoforms. J. Biol. Chem. 1994, 269, 22358–22365. [Google Scholar] [PubMed]
- Raffai, R.L.; Dong, L.M.; Farese, R.V.; Weisgraber, K.H. Introduction of human apolipoprotein E4 “domain interaction” into mouse apolipoprotein E. Proc. Natl. Acad. Sci. USA 2001, 98, 11587–11591. [Google Scholar] [CrossRef] [PubMed]
- Zhong, N.; Scearce-Levie, K.; Ramaswamy, G.; Weisgraber, K.H. Apolipoprotein E4 domain interaction: Synaptic and cognitive deficits in mice. Alzheimer’s Dement. 2008, 4, 179–192. [Google Scholar] [CrossRef]
- Zhong, N.; Ramaswamy, G.; Weisgraber, K.H. Apolipoprotein E4 domain interaction induces endoplasmic reticulum stress and impairs astrocyte function. J. Biol. Chem. 2009, 284, 27273–27280. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Liu, X.Q.; Wyss-Coray, T.; Brecht, W.J.; Sanan, D.A.; Mahley, R.W. Apolipoprotein E fragments present in Alzheimer’s disease brains induce neurofibrillary tangle-like intracellular inclusions in neurons. Proc. Natl. Acad. Sci. USA 2001, 98, 8838–8843. [Google Scholar] [CrossRef] [PubMed]
- Harris, F.M.; Brecht, W.J.; Xu, Q.; Tesseur, I.; Kekonius, L.; Wyss-Coray, T.; Fish, J.D.; Masliah, E.; Hopkins, P.C.; Scearce-Levie, K.; et al. Carboxyl-terminal-truncated apolipoprotein E4 causes Alzheimer’s disease-like neurodegeneration and behavioral deficits in transgenic mice. Proc. Natl. Acad. Sci. USA 2003, 100, 10966–10971. [Google Scholar] [CrossRef]
- Brecht, W.J.; Harris, F.M.; Chang, S.; Tesseur, I.; Yu, G.-Q.; Xu, Q.; Dee Fish, J.; Wyss-Coray, T.; Buttini, M.; Mucke, L.; et al. Neuron-specific apolipoprotein ε4 proteolysis is associated with increased tau phosphorylation in brains of transgenic mice. J. Neurosci. 2004, 24, 2527–2534. [Google Scholar] [CrossRef]
- Chen, H.-K.; Ji, Z.-S.; Dodson, S.E.; Miranda, R.D.; Rosenblum, C.I.; Reynolds, I.J.; Freedman, S.B.; Weisgraber, K.H.; Huang, Y.; Mahley, R.W. Apolipoprotein E4 Domain Interaction Mediates Detrimental Effects on Mitochondria and Is a Potential Therapeutic Target for Alzheimer Disease. J. Biol. Chem. 2011, 286, 5215–5221. [Google Scholar] [CrossRef]
- Mahley, R.W.; Huang, Y. Small-Molecule Structure Correctors Target Abnormal Protein Structure and Function: Structure Corrector Rescue of Apolipoprotein E4–Associated Neuropathology. J. Med. Chem. 2012, 55, 8997–9008. [Google Scholar] [CrossRef]
- Mahley, R.W.; Weisgraber, K.H.; Huang, Y. Apolipoprotein E4: A causative factor and therapeutic target in neuropathology, including Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2006, 103, 5644–5651. [Google Scholar] [CrossRef] [PubMed]
- Ye, S.; Huang, Y.; Müllendorff, K.; Dong, L.; Giedt, G.; Meng, E.C.; Cohen, F.E.; Kuntz, I.D.; Weisgraber, K.H.; Mahley, R.W. Apolipoprotein (Apo) E4 enhances amyloid beta peptide production in cultured neuronal cells: ApoE structure as a potential therapeutic target. Proc. Natl. Acad. Sci. USA 2005, 102, 18700–18705. [Google Scholar] [CrossRef] [PubMed]
- Mahley, R.W.; Weisgraber, K.H.; Huang, Y. ApoE4 Domain Interaction Inhibitors and Methods of Use Thereof. U.S. Patent 7,964,598, 21 June 2011. [Google Scholar]
- Brodbeck, J.; McGuire, J.; Liu, Z.; Meyer-Franke, A.; Balestra, M.E.; Jeong, D.E.; Pleiss, M.; McComas, C.; Hess, F.; Witter, D.; et al. Structure-dependent impairment of intracellular apolipoprotein E4 trafficking and its detrimental effects are rescued by small-molecule structure correctors. J. Biol. Chem. 2011, 286, 17217–17226. [Google Scholar] [CrossRef]
- Chen, H.-K.; Liu, Z.; Meyer-Franke, A.; Brodbeck, J.; Miranda, R.D.; McGuire, J.G.; Pleiss, M.A.; Ji, Z.S.; Balestra, M.E.; Walker, D.W.; et al. Small molecule structure correctors abolish detrimental effects of apolipoprotein E4 in cultured neurons. J. Biol. Chem. 2012, 287, 5253–5266. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Najm, R.; Xu, Q.; Jeong, D.; Walker, D.; Balestra, M.E.; Yoon, S.Y.; Yuan, H.; Li, G.; Miller, Z.A.; et al. Gain of Toxic Apolipoprotein E4 Effects in Human iPSC-derived Neurons is Ameliorated by a Small-Molecule Structure Corrector. Nat. Med. 2018, 24, 647. [Google Scholar] [CrossRef]
- Mahley, R.W. Apolipoprotein E: From cardiovascular disease to neurodegenerative disorders. J. Mol. Med. 2016, 94, 739–746. [Google Scholar] [CrossRef]
- Mahley, R.W. Apolipoprotein E: Remarkable Protein Sheds Light on Cardiovascular and Neurological Diseases. Clin. Chem. 2017, 63, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Mondal, T.; Wang, H.; DeKoster, G.T.; Baban, B.; Gross, M.L.; Frieden, C. ApoE: In Vitro Studies of a Small Molecule Effector. Biochemistry 2016, 55, 2613–2621. [Google Scholar] [CrossRef]
- Burgess, J.W.; Gould, D.R.; Marcel, Y.L. The HepG2 extracellular matrix contains separate heparinase- and lipid-releasable pools of ApoE. Implications for hepatic lipoprotein metabolism. J. Biol. Chem. 1998, 273, 5645–5654. [Google Scholar] [CrossRef]
- Burgess, J.W.; Liang, P.; Vaidyanath, C.; Marcel, Y.L. ApoE of the HepG2 cell surface includes a major pool associated with chondroitin sulfate proteoglycans. Biochemistry 1999, 38, 524–531. [Google Scholar] [CrossRef]
- Burgess, J.W.; Marcel, Y.L. Dynamic and stable pools of apoE differ functionally at the HepG2 cell surface. J. Lipid Res. 2001, 42, 1413–1420. [Google Scholar] [PubMed]
- Ganesh, V.K.; Muthuvel, S.K.; Smith, S.A.; Kotwal, G.J.; Murthy, K.H.M. Structural basis for antagonism by suramin of heparin binding to vaccinia complement protein. Biochemistry 2005, 44, 10757–10765. [Google Scholar] [CrossRef] [PubMed]
- Prince, M.; Wimo, A.; Guerchet, M.; Ali, G.-C.; Wu, Y.-T.; Prina, M. World Alzheimer Report 2015—The Global Impact of Dementia; Alzheimer’s Disease International: London, UK, 2015. [Google Scholar]
- WHO Dementia Fact Sheet N°362. Available online: http://www.who.int/mediacentre/factsheets/fs362/en/ (accessed on 2 April 2018).
- Tanzi, R.E. The Genetics of Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2012, 2, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Blacker, D.; Haines, J.L.; Rodes, L.; Terwedow, H.; Go, R.C.; Harrell, L.E.; Perry, R.T.; Bassett, S.S.; Chase, G.; Meyers, D.; et al. ApoE-4 and age at onset of Alzheimer’s disease: The NIMH genetics initiative. Neurology 1997, 48, 139–147. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Painter, M.M.; Bu, G.; Kanekiyo, T. Apolipoprotein E as a Therapeutic Target in Alzheimer’s Disease: A Review of Basic Research and Clinical Evidence. CNS Drugs 2016, 30, 773–789. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.-T.; Tan, L.; Hardy, J. Apolipoprotein E in Alzheimer’s Disease: An Update. Annu. Rev. Neurosci. 2014, 37, 79–100. [Google Scholar] [CrossRef] [PubMed]
- Renaud, J.-P.; Chung, C.-W.; Danielson, U.H.; Egner, U.; Hennig, M.; Hubbard, R.E.; Nar, H. Biophysics in drug discovery: Impact, challenges and opportunities. Nat. Rev. Drug Discov. 2016, 15, 679–698. [Google Scholar] [CrossRef]
- Fang, Y. Optical Guided-Wave Chemical and Biosensors II; Zourob, M., Lakhtakia, A., Eds.; Springer Series on Chemical Sensors and Biosensors; Springer: Berlin/Heidelberg, Germany, 2010; Volume 8, ISBN 978-3-642-02826-7. [Google Scholar]
- Jerabek-Willemsen, M.; André, T.; Wanner, R.; Roth, H.M.; Duhr, S.; Baaske, P.; Breitsprecher, D. MicroScale Thermophoresis: Interaction analysis and beyond. J. Mol. Struct. 2014, 1077, 101–113. [Google Scholar] [CrossRef]
- Morrow, J.A.; Arnold, K.S.; Weisgraber, K.H. Functional characterization of apolipoprotein E isoforms overexpressed in Escherichia coli. Protein Expr. Purif. 1999, 16, 224–230. [Google Scholar] [CrossRef]
- Vogel, T.; Weisgraber, K.H.; Zeevi, M.I.; Ben-Artzi, H.; Levanon, A.Z.; Rall, S.C.; Innerarity, T.L.; Hui, D.Y.; Taylor, J.M.; Kanner, D. Human apolipoprotein E expression in Escherichia coli: Structural and functional identity of the bacterially produced protein with plasma apolipoprotein E. Proc. Natl. Acad. Sci. USA 1985, 82, 8696–8700. [Google Scholar] [CrossRef]
- Bojanowski, K.; Lelievre, S.; Markovits, J.; Couprie, J.; Jacquemin-Sablon, A.; Larsen, A.K. Suramin is an inhibitor of DNA topoisomerase II in vitro and in Chinese hamster fibrosarcoma cells. Proc. Natl. Acad. Sci. USA 1992, 89, 3025–3029. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-L.; Keng, Y.-F.; Zhao, Y.; Wu, L.; Zhang, Z.-Y. Suramin Is an Active Site-directed, Reversible, and Tight-binding Inhibitor of Protein-tyrosine Phosphatases. J. Biol. Chem. 1998, 273, 12281–12287. [Google Scholar] [CrossRef] [PubMed]
- Kloen, P.; Jennings, C.L.; Gebhardt, M.C.; Springfield, D.S.; Mankin, H.J. Suramin inhibits growth and transforming growth factor-beta 1 (TGF-β 1) binding in osteosarcoma cell lines. Eur. J. Cancer 1994, 30A, 678–682. [Google Scholar] [CrossRef]
- Zhao, P.; Sharir, H.; Kapur, A.; Cowan, A.; Geller, E.B.; Adler, M.W.; Seltzman, H.H.; Reggio, P.H.; Heynen-Genel, S.; Sauer, M.; et al. Targeting of the orphan receptor GPR35 by pamoic acid: A potent activator of extracellular signal-regulated kinase and β-arrestin2 with antinociceptive activity. Mol. Pharmacol. 2010, 78, 560–568. [Google Scholar] [CrossRef]
- Neubig, R.R. Mind your salts: When the inactive constituent isn’t. Mol. Pharmacol. 2010, 78, 558–559. [Google Scholar] [CrossRef] [PubMed]
- Benvenga, S.; Cahnmann, H.J.; Rader, D.; Kindt, M.; Robbins, J. Thyroxine binding to the apolipoproteins of high density lipoproteins HDL2 and HDL3. Endocrinology 1992, 131, 2805–2811. [Google Scholar] [CrossRef] [PubMed]
- Benvenga, S.; Cahnmann, H.J.; Robbins, J. Characterization of thyroid hormone binding to apolipoprotein-E: Localization of the binding site in the exon 3-coded domain. Endocrinology 1993, 133, 1300–1305. [Google Scholar] [CrossRef]
- Henderson, V.W. The Role of Sex Steroids in Alzheimer’s Disease: Prevention and Treatment. In Treatment of the Postmenopausal Woman; Elsevier: Amsterdam, The Netherlands, 2007; pp. 295–306. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ApoE4 | ApoE2 | ApoE3 | ||
|---|---|---|---|---|
| MST (μM) | ITC (μM) | ITC (μM) | ITC (μM) | |
| l-Thyroxine | * | 18 ± 12 | 33 ± 10 | 20 ± 3 |
| Tafamidis | - | 8 ± 2 | 6 ± 2 | 8 ± 1 |
| Meclofenamic acid | 51 ± 12 | 45 ± 13 | 47 ± 32 | 47 ± 9 |
| Pamoic acid | * | 38 ± 4 | 55 ± 14 | 119 ± 50 |
| Clomifene | 44 ± 2 | 15 ± 10 | 67 ± 36 | 24 ± 9 |
| Tamoxifen | 67 ± 19 | * | * | * |
| Toremifene | 44 ± 6 | * | * | * |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kraft, L.; Serpell, L.C.; Atack, J.R. A Biophysical Approach to the Identification of Novel ApoE Chemical Probes. Biomolecules 2019, 9, 48. https://doi.org/10.3390/biom9020048
Kraft L, Serpell LC, Atack JR. A Biophysical Approach to the Identification of Novel ApoE Chemical Probes. Biomolecules. 2019; 9(2):48. https://doi.org/10.3390/biom9020048
Chicago/Turabian StyleKraft, Lucas, Louise C. Serpell, and John R. Atack. 2019. "A Biophysical Approach to the Identification of Novel ApoE Chemical Probes" Biomolecules 9, no. 2: 48. https://doi.org/10.3390/biom9020048
APA StyleKraft, L., Serpell, L. C., & Atack, J. R. (2019). A Biophysical Approach to the Identification of Novel ApoE Chemical Probes. Biomolecules, 9(2), 48. https://doi.org/10.3390/biom9020048