NP-Scout: Machine Learning Approach for the Quantification and Visualization of the Natural Product-Likeness of Small Molecules




Abstract
1. Introduction
2. Materials and Methods
2.1. Data Preparation
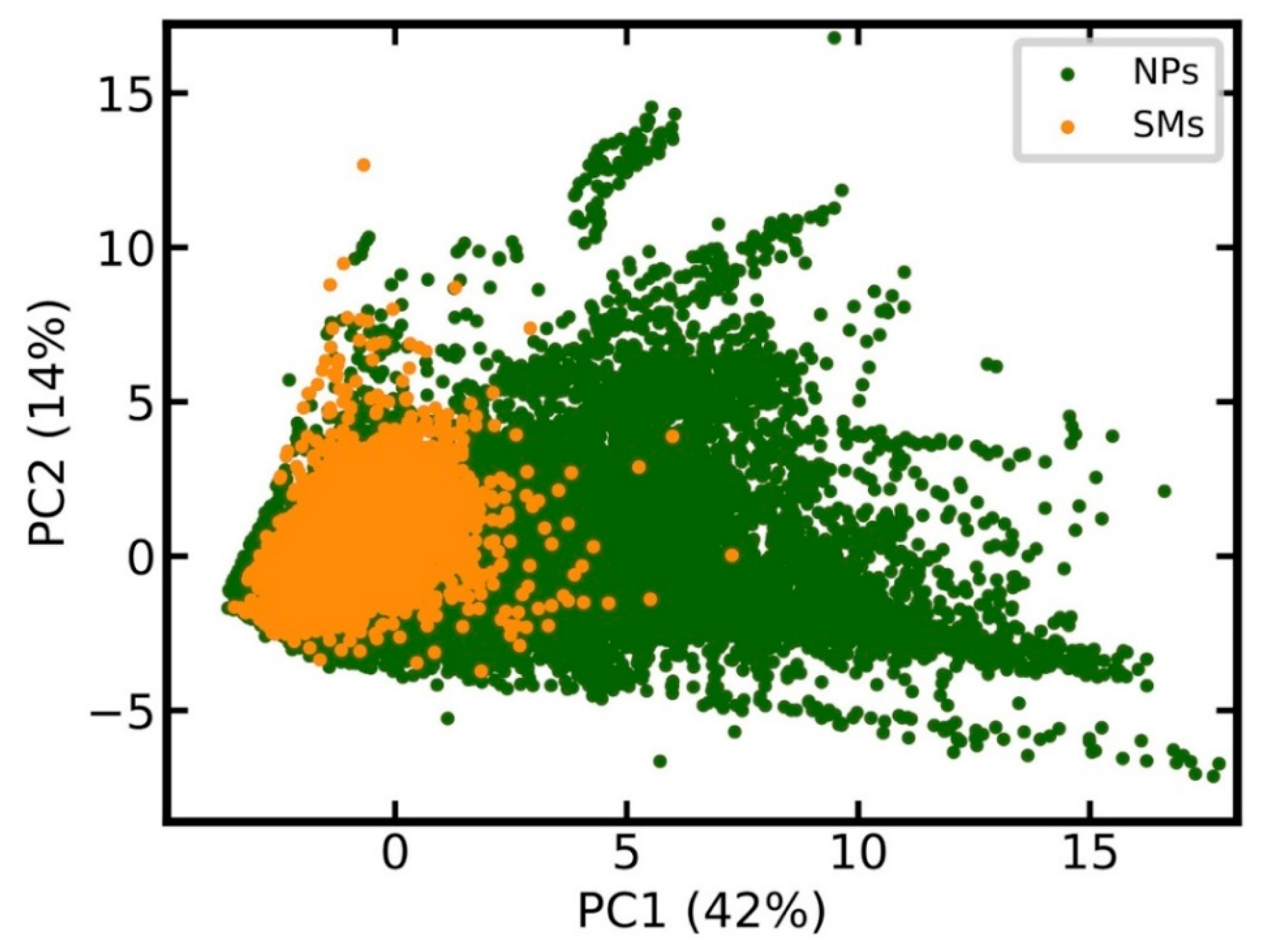
2.2. Principal Component Analysis
2.3. Model Building
2.4. Similarity Maps
3. Results
3.1. Compilation of Data Sets for Model Development
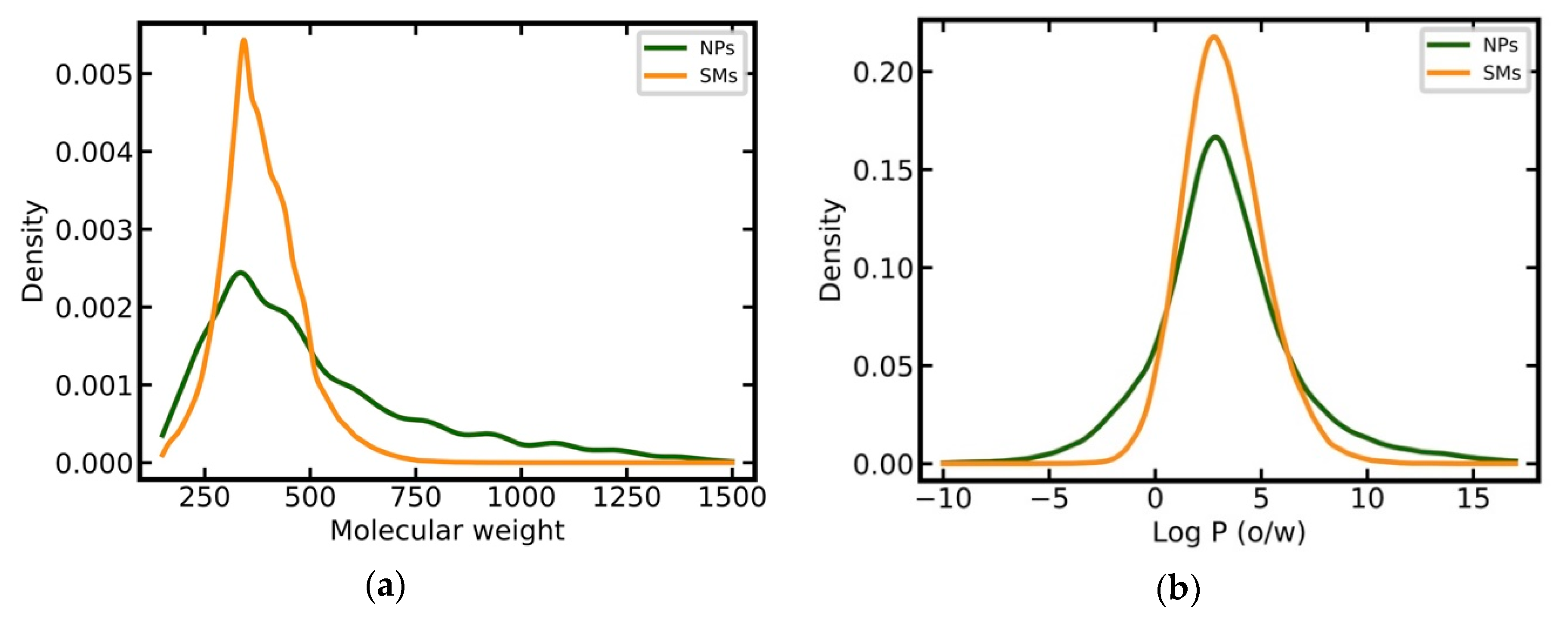
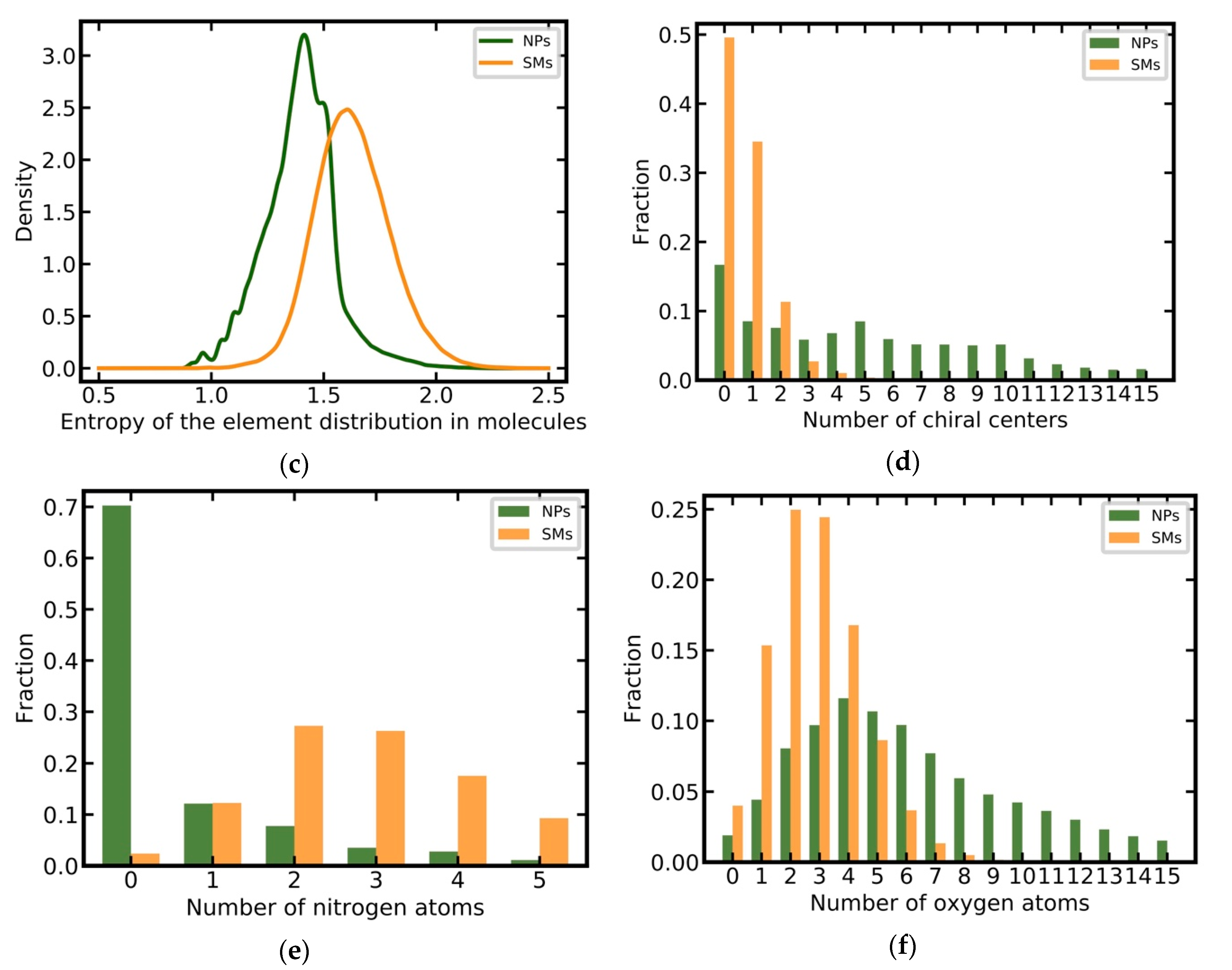
3.2. Analysis of the Physicochemical Properties of Natural Products and Synthetic Molecules
3.3. Model Development and Selection
3.4. Model Validation
3.5. Comparison of Model Performance with the NP-Likeness Calculator
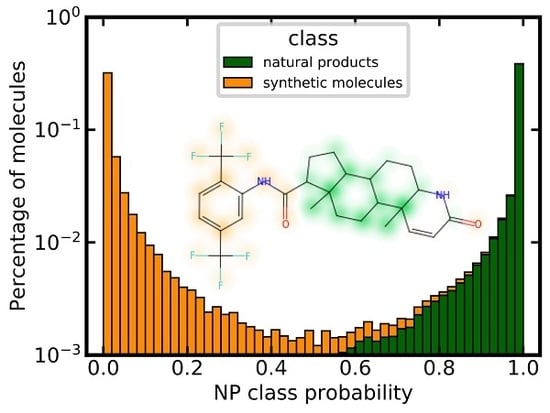
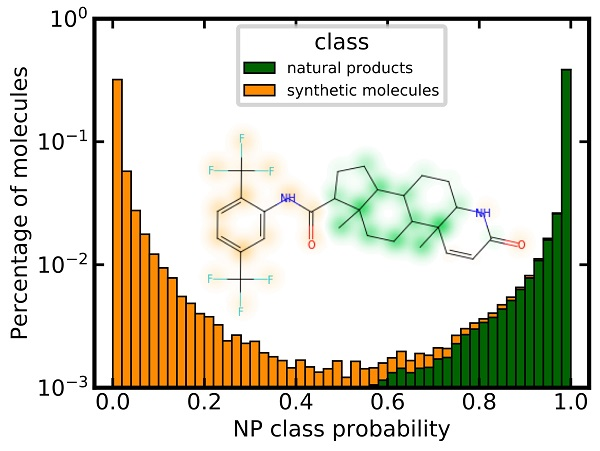
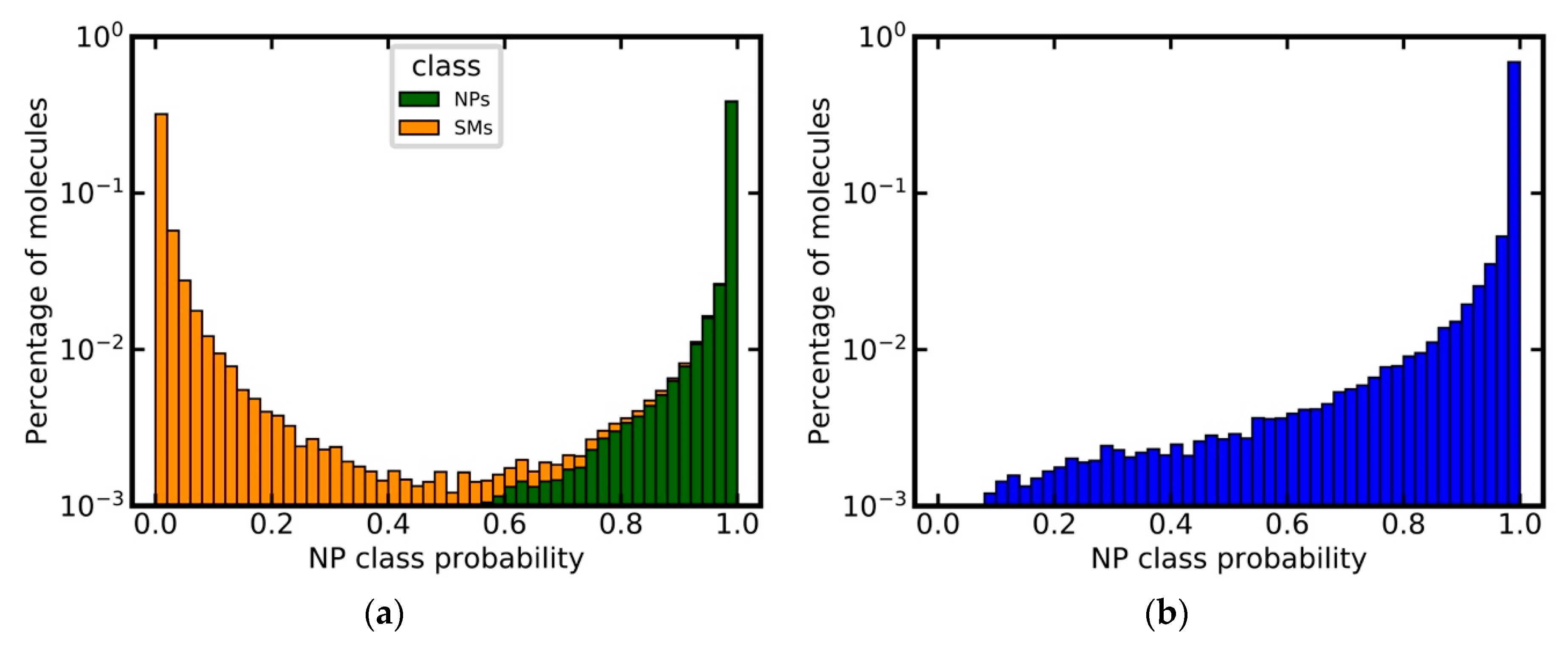
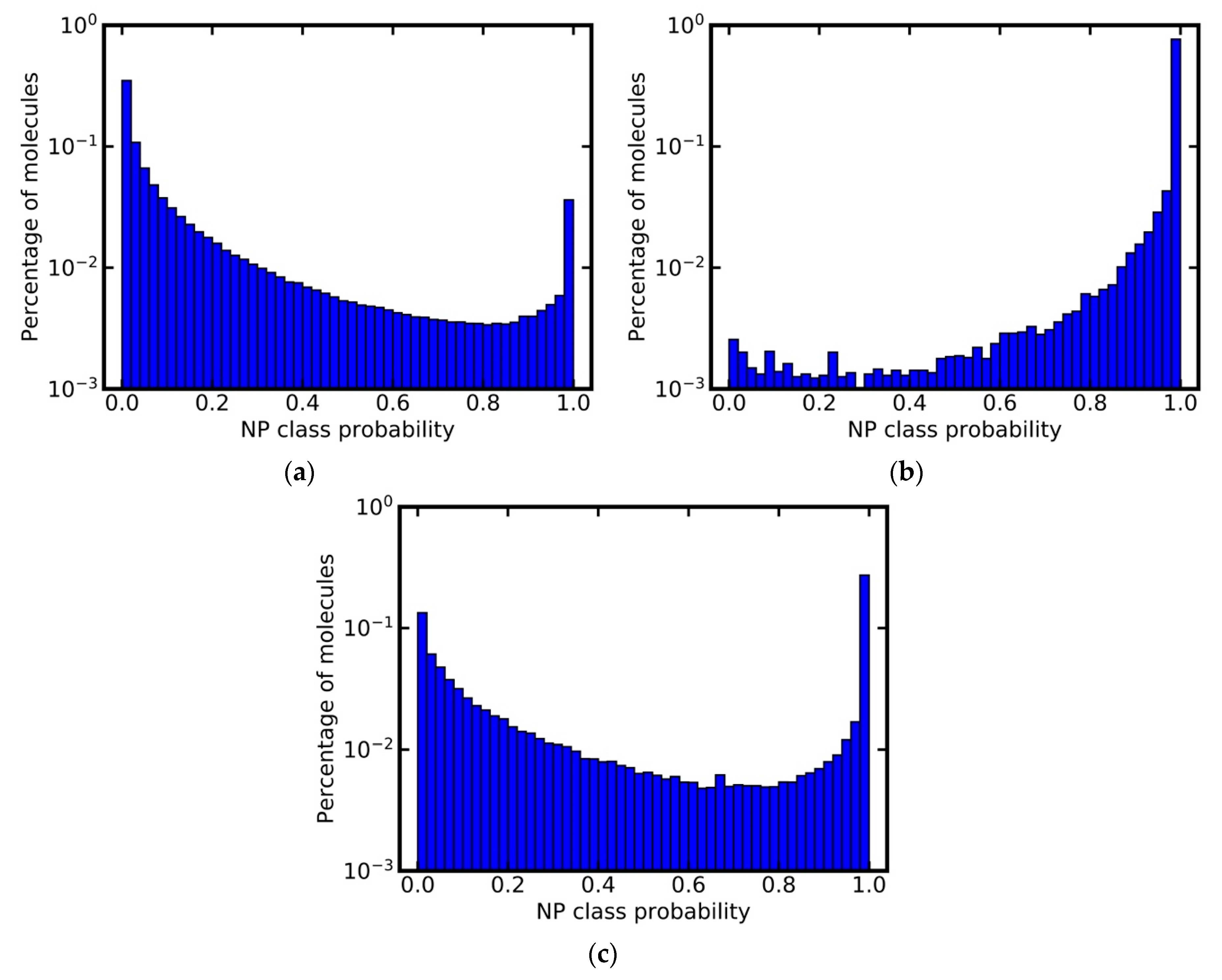
3.6. Analysis of Class Probability Distributions for Different Data Sets
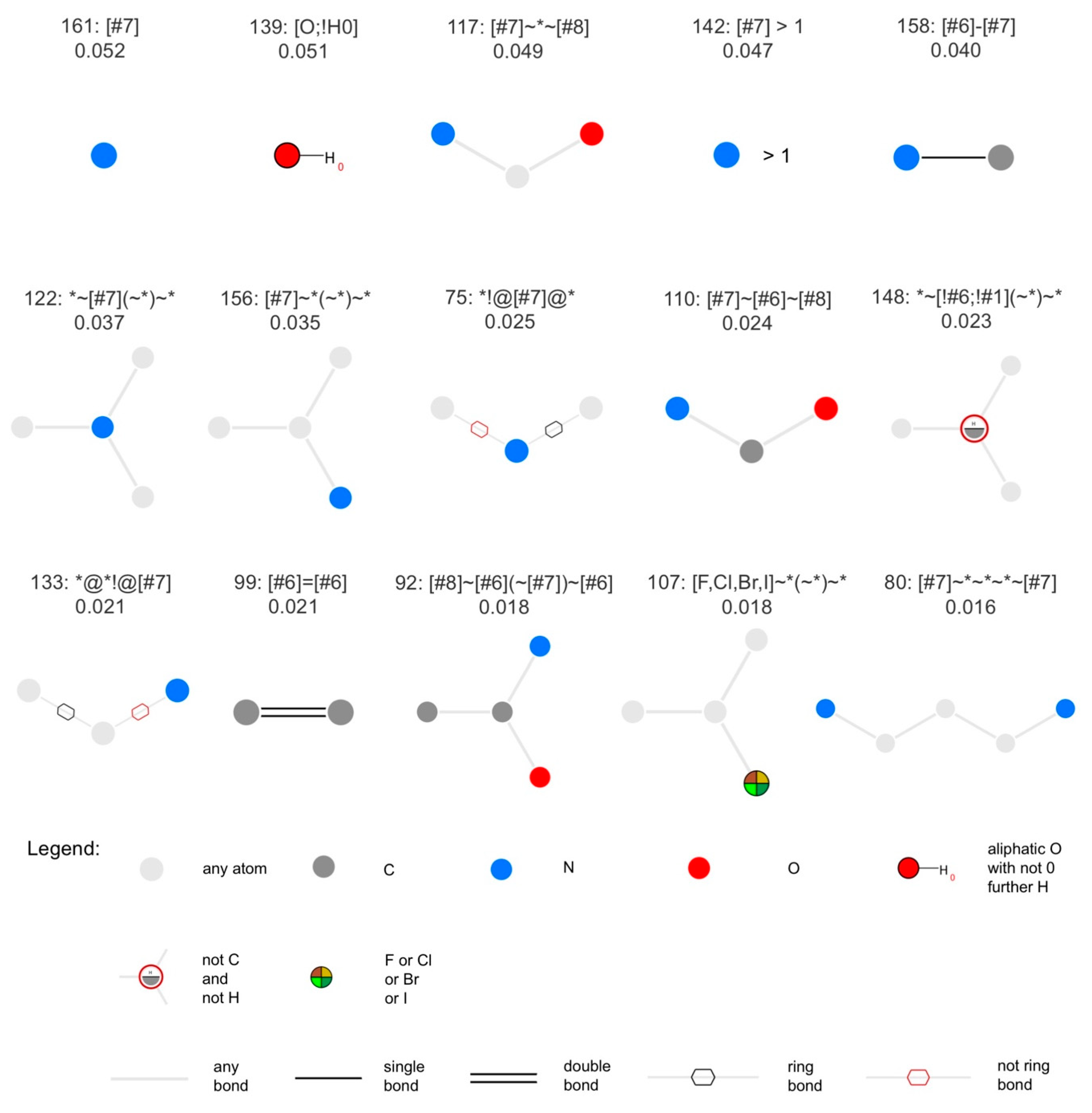
3.7. Analysis of Discriminative Features of Natural Products and Synthetic Molecules
3.8. Similarity Maps
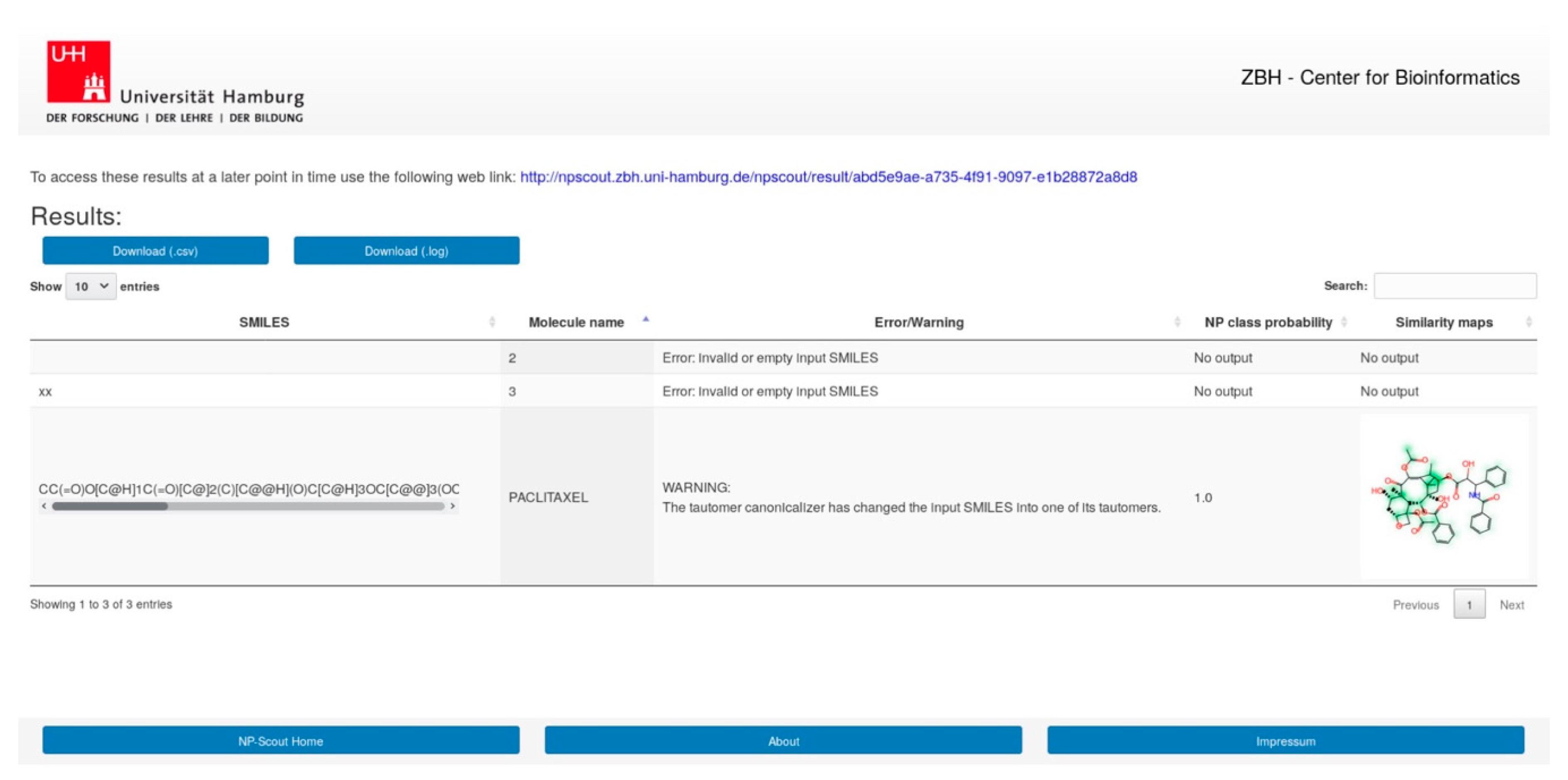
3.9. NP-Scout Web Service
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cragg, G.M.; Newman, D.J. Biodiversity: A continuing source of novel drug leads. J. Macromol. Sci. Part A Pure Appl. Chem. 2005, 77, 7–24. [Google Scholar] [CrossRef]
- Rodrigues, T.; Reker, D.; Schneider, P.; Schneider, G. Counting on natural products for drug design. Nat. Chem. 2016, 8, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Harvey, A.L.; Edrada-Ebel, R.; Quinn, R.J. The re-emergence of natural products for drug discovery in the genomics era. Nat. Rev. Drug Discov. 2015, 14, 111–129. [Google Scholar] [CrossRef]
- Shen, B. A new golden age of natural products drug discovery. Cell 2015, 163, 1297–1300. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, K.; Baringhaus, K.-H.; Schneider, G. Scaffold diversity of natural products: Inspiration for combinatorial library design. Nat. Prod. Rep. 2008, 25, 892–904. [Google Scholar] [CrossRef]
- Ertl, P.; Schuffenhauer, A. Cheminformatics analysis of natural products: Lessons from nature inspiring the design of new drugs. Prog. Drug Res. 2008, 66, 219–235. [Google Scholar]
- Chen, Y.; de Lomana, M.G.; Friedrich, N.-O.; Kirchmair, J. Characterization of the chemical space of known and Readily Obtainable Natural Products. J. Chem. Inf. Model. 2018, 58, 1518–1532. [Google Scholar] [CrossRef]
- Chen, H.; Engkvist, O.; Blomberg, N.; Li, J. A comparative analysis of the molecular topologies for drugs, clinical candidates, natural products, human metabolites and general bioactive compounds. Med. Chem. Commun. 2012, 3, 312–321. [Google Scholar] [CrossRef]
- Camp, D.; Garavelas, A.; Campitelli, M. Analysis of physicochemical properties for drugs of natural origin. J. Nat. Prod. 2015, 78, 1370–1382. [Google Scholar] [CrossRef]
- Koch, M.A.; Schuffenhauer, A.; Scheck, M.; Wetzel, S.; Casaulta, M.; Odermatt, A.; Ertl, P.; Waldmann, H. Charting biologically relevant chemical space: A structural classification of natural products (SCONP). Proc. Natl. Acad. Sci. USA 2005, 102, 17272–17277. [Google Scholar] [CrossRef] [PubMed]
- Stratton, C.F.; Newman, D.J.; Tan, D.S. Cheminformatic comparison of approved drugs from natural product versus synthetic origins. Bioorg. Med. Chem. Lett. 2015, 25, 4802–4807. [Google Scholar] [CrossRef] [PubMed]
- Wetzel, S.; Schuffenhauer, A.; Roggo, S.; Ertl, P.; Waldmann, H. Cheminformatic analysis of natural products and their chemical space. CHIMIA Int. J. Chem. 2007, 61, 355–360. [Google Scholar] [CrossRef]
- López-Vallejo, F.; Giulianotti, M.A.; Houghten, R.A.; Medina-Franco, J.L. Expanding the medicinally relevant chemical space with compound libraries. Drug Discov. Today 2012, 17, 718–726. [Google Scholar] [CrossRef]
- Feher, M.; Schmidt, J.M. Property distributions: Differences between drugs, natural products, and molecules from combinatorial chemistry. J. Chem. Inf. Comput. Sci. 2003, 43, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Clemons, P.A.; Bodycombe, N.E.; Carrinski, H.A.; Wilson, J.A.; Shamji, A.F.; Wagner, B.K.; Koehler, A.N.; Schreiber, S.L. Small molecules of different origins have distinct distributions of structural complexity that correlate with protein-binding profiles. Proc. Natl. Acad. Sci. USA 2010, 107, 18787–18792. [Google Scholar] [CrossRef] [PubMed]
- Henkel, T.; Brunne, R.M.; Müller, H.; Reichel, F. Statistical investigation into the structural complementarity of natural products and synthetic compounds. Angew. Chem. Int. Ed. Engl. 1999, 38, 643–647. [Google Scholar] [CrossRef]
- Lee, M.L.; Schneider, G. Scaffold architecture and pharmacophoric properties of natural products and trade drugs: Application in the design of natural product-based combinatorial libraries. J. Comb. Chem. 2001, 3, 284–289. [Google Scholar] [CrossRef]
- Chen, Y.; de Bruyn Kops, C.; Kirchmair, J. Data resources for the computer-guided discovery of bioactive natural products. J. Chem. Inf. Model. 2017, 57, 2099–2111. [Google Scholar] [CrossRef]
- Rupp, M.; Schroeter, T.; Steri, R.; Zettl, H.; Proschak, E.; Hansen, K.; Rau, O.; Schwarz, O.; Müller-Kuhrt, L.; Schubert-Zsilavecz, M.; et al. From machine learning to natural product derivatives that selectively activate transcription factor PPARγ. ChemMedChem 2010, 5, 191–194. [Google Scholar] [CrossRef]
- Maindola, P.; Jamal, S.; Grover, A. Cheminformatics based machine learning models for AMA1-RON2 abrogators for inhibiting Plasmodium falciparum erythrocyte invasion. Mol. Inform. 2015, 34, 655–664. [Google Scholar] [CrossRef] [PubMed]
- Chagas-Paula, D.A.; Oliveira, T.B.; Zhang, T.; Edrada-Ebel, R.; Da Costa, F.B. Prediction of anti-inflammatory plants and discovery of their biomarkers by machine learning algorithms and metabolomic studies. Planta Med. 2015, 81, 450–458. [Google Scholar] [CrossRef] [PubMed]
- Reker, D.; Perna, A.M.; Rodrigues, T.; Schneider, P.; Reutlinger, M.; Mönch, B.; Koeberle, A.; Lamers, C.; Gabler, M.; Steinmetz, H.; et al. Revealing the macromolecular targets of complex natural products. Nat. Chem. 2014, 6, 1072–1078. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, T.; Sieglitz, F.; Somovilla, V.J.; Cal, P.M.S.D.; Galione, A.; Corzana, F.; Bernardes, G.J.L. Unveiling (−)-englerin A as a modulator of L-type calcium channels. Angew. Chem. Int. Ed. Engl. 2016, 55, 11077–11081. [Google Scholar] [CrossRef] [PubMed]
- Merk, D.; Grisoni, F.; Friedrich, L.; Gelzinyte, E.; Schneider, G. Computer-assisted discovery of retinoid X receptor modulating natural products and isofunctional mimetics. J. Med. Chem. 2018, 61, 5442–5447. [Google Scholar] [CrossRef] [PubMed]
- Schneider, P.; Schneider, G. De-orphaning the marine natural product (±)-marinopyrrole A by computational target prediction and biochemical validation. Chem. Commun. 2017, 53, 2272–2274. [Google Scholar]
- Merk, D.; Grisoni, F.; Friedrich, L.; Schneider, G. Tuning artificial intelligence on the de novo design of natural-product-inspired retinoid X receptor modulators. Commun. Chem. 2018, 1, 68. [Google Scholar]
- Friedrich, L.; Rodrigues, T.; Neuhaus, C.S.; Schneider, P.; Schneider, G. From complex natural products to simple synthetic mimetics by computational de novo design. Angew. Chem. Int. Ed. Engl. 2016, 55, 6789–6792. [Google Scholar] [CrossRef]
- Grisoni, F.; Merk, D.; Consonni, V.; Hiss, J.A.; Tagliabue, S.G.; Todeschini, R.; Schneider, G. Scaffold hopping from natural products to synthetic mimetics by holistic molecular similarity. Commun. Chem. 2018, 1, 44. [Google Scholar]
- Ertl, P.; Roggo, S.; Schuffenhauer, A. Natural product-likeness score and its application for prioritization of compound libraries. J. Chem. Inf. Model. 2008, 48, 68–74. [Google Scholar] [CrossRef]
- Jayaseelan, K.V.; Moreno, P.; Truszkowski, A.; Ertl, P.; Steinbeck, C. Natural product-likeness score revisited: An open-source, open-data implementation. BMC Bioinform. 2012, 13, 106. [Google Scholar] [CrossRef] [PubMed]
- Jayaseelan, K.V.; Steinbeck, C. Building blocks for automated elucidation of metabolites: Natural product-likeness for candidate ranking. BMC Bioinform. 2014, 15, 234. [Google Scholar] [CrossRef] [PubMed]
- RDKit NP_Score. Available online: https://github.com/rdkit/rdkit/tree/master/Contrib/NP_Score (accessed on 27 November 2018).
- Yu, M.J. Natural product-like virtual libraries: Recursive atom-based enumeration. J. Chem. Inf. Model. 2011, 51, 541–557. [Google Scholar] [CrossRef] [PubMed]
- Zaid, H.; Raiyn, J.; Nasser, A.; Saad, B.; Rayan, A. Physicochemical properties of natural based products versus synthetic chemicals. Open Nutraceuticals J. 2010, 3, 194–202. [Google Scholar] [CrossRef]
- Riniker, S.; Landrum, G.A. Similarity maps—A visualization strategy for molecular fingerprints and machine-learning methods. J. Cheminform. 2013, 5, 43. [Google Scholar] [CrossRef] [PubMed]
- RDKit Version 2017.09.3: Open-source cheminformatics software. Available online: http://www.rdkit.org (accessed on 22 May 2018).
- Stork, C.; Wagner, J.; Friedrich, N.-O.; de Bruyn Kops, C.; Šícho, M.; Kirchmair, J. Hit Dexter: A machine-learning model for the prediction of frequent hitters. ChemMedChem 2018, 13, 564–571. [Google Scholar] [CrossRef]
- MolVs Version 0.1.1. Available online: https://github.com/mcs07/MolVS (accessed on 12 July 2018).
- Sterling, T.; Irwin, J.J. ZINC 15-Ligand discovery for everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- ZINC “in-stock” subset. ZINC15. Available online: http://zinc15.docking.org/ (accessed on 21 August 2018).
- Dictionary of Natural Products, version 19.1; Chapman & Hall/CRC: London, UK, 2010.
- Bento, A.P.; Gaulton, A.; Hersey, A.; Bellis, L.J.; Chambers, J.; Davies, M.; Krüger, F.A.; Light, Y.; Mak, L.; McGlinchey, S.; et al. The ChEMBL bioactivity database: An update. Nucleic Acids Res. 2014, 42, D1083–D1090. [Google Scholar] [CrossRef]
- ChEMBL Version 24_1. Available online: https://www.ebi.ac.uk/chembl/ (accessed on 30 July 2018).
- ChEMBL Version 23. Available online: https://www.ebi.ac.uk/chembl (accessed on 6 June 2017).
- Natural products subset of ZINC. ZINC15. Available online: http://zinc15.docking.org/substances/subsets/ (accessed on 7 November 2018).
- Molecular Operating Environment (MOE), version 2016.08; Chemical Computing Group: Montreal, QC, Canada, 2016.
- Morgan, H.L. The generation of a unique machine description for chemical structures-A technique developed at Chemical Abstracts Service. J. Chem. Doc. 1965, 5, 107–113. [Google Scholar] [CrossRef]
- Rogers, D.; Hahn, M. Extended-connectivity fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Scikit-Learn: Machine Learning in Python. version 0.19.1.
- Natural Product Likeness Calculator Version 2.1. Available online: https://sourceforge.net/projects/np-likeness/ (accessed on 5 October 2018).
- Natural Products Atlas. Available online: https://www.npatlas.org/ (accessed on 20 August 2018).
- Gu, J.; Gui, Y.; Chen, L.; Yuan, G.; Lu, H.-Z.; Xu, X. Use of natural products as chemical library for drug discovery and network pharmacology. PLoS ONE 2013, 8, e62839. [Google Scholar] [CrossRef]
- Universal Natural Products Database (UNPD). Available online: http://pkuxxj.pku.edu.cn/UNPD (accessed on 17 October 2016).
- Chen, C.Y.-C. TCM Database@Taiwan: The world’s largest traditional Chinese medicine database for drug screening in silico. PLoS ONE 2011, 6, e15939. [Google Scholar] [CrossRef] [PubMed]
- TCM Database@Taiwan. Available online: http://tcm.cmu.edu.tw (accessed on 17 October 2016).
- Xue, R.; Fang, Z.; Zhang, M.; Yi, Z.; Wen, C.; Shi, T. TCMID: Traditional Chinese medicine integrative database for herb molecular mechanism analysis. Nucleic Acids Res. 2013, 41, D1089–D1095. [Google Scholar] [CrossRef] [PubMed]
- Traditional Chinese Medicine Integrated Database (TCMID). Available online: www.megabionet.org/tcmid (accessed on 19 October 2016).
- Lin, Y.-C.; Wang, C.-C.; Chen, I.-S.; Jheng, J.-L.; Li, J.-H.; Tung, C.-W. TIPdb: A database of anticancer, antiplatelet, and antituberculosis phytochemicals from indigenous plants in Taiwan. Sci. World J. 2013, 2013, 736386. [Google Scholar] [CrossRef]
- Tung, C.-W.; Lin, Y.-C.; Chang, H.-S.; Wang, C.-C.; Chen, I.-S.; Jheng, J.-L.; Li, J.-H. TIPdb-3D: The three-dimensional structure database of phytochemicals from Taiwan indigenous plants. Database 2014, 2014, bau055. [Google Scholar] [CrossRef] [PubMed]
- Taiwan Indigenous Plant Database (TIPdb). Available online: http://cwtung.kmu.edu.tw/tipdb (accessed on 19 October 2016).
- Ambinter. Available online: www.ambinter.com (accessed on 2 June 2017).
- GreenPharma. Available online: www.greenpharma.com (accessed on 2 June 2017).
- AnalytiCon Discovery. Available online: www.ac-discovery.com (accessed on 14 November 2017).
- Ntie-Kang, F.; Telukunta, K.K.; Döring, K.; Simoben, C.V.; A Moumbock, A.F.; Malange, Y.I.; Njume, L.E.; Yong, J.N.; Sippl, W.; Günther, S. NANPDB: A resource for natural products from Northern African sources. J. Nat. Prod. 2017, 80, 2067–2076. [Google Scholar] [CrossRef]
- Northern African Natural Products Database (NANPDB). Available online: www.african-compounds.org/nanpdb (accessed on 5 April 2017).
- Klementz, D.; Döring, K.; Lucas, X.; Telukunta, K.K.; Erxleben, A.; Deubel, D.; Erber, A.; Santillana, I.; Thomas, O.S.; Bechthold, A.; et al. StreptomeDB 2.0—An extended resource of natural products produced by streptomycetes. Nucleic Acids Res. 2015, 44, D509–D514. [Google Scholar] [CrossRef]
- StreptomeDB. Available online: http://132.230.56.4/streptomedb2/ (accessed on 13 April 2017).
- Ming, H.; Tiejun, C.; Yanli, W.; Stephen, B.H. Web search and data mining of natural products and their bioactivities in PubChem. Sci. China Chem. 2013, 56, 1424–1435. [Google Scholar]
- Natural products subset. PubChem Substance Database. Available online: http://ncbi.nlm.nih.gov/pcsubstance (accessed on 7 April 2017).
- Pilon, A.C.; Valli, M.; Dametto, A.C.; Pinto, M.E.F.; Freire, R.T.; Castro-Gamboa, I.; Andricopulo, A.D.; Bolzani, V.S. NuBBE: An updated database to uncover chemical and biological information from Brazilian biodiversity. Sci. Rep. 2017, 7, 7215. [Google Scholar] [CrossRef]
- Núcleo de Bioensaios, Biossíntese e Ecofisiologia de Produtos Naturais (NuBBE). Available online: http://nubbe.iq.unesp.br/portal/nubbedb.html (accessed on 19 April 2017).
- PI Chemicals. Available online: www.pipharm.com (accessed on 5 May 2017).
- Choi, H.; Cho, S.Y.; Pak, H.J.; Kim, Y.; Choi, J.-Y.; Lee, Y.J.; Gong, B.H.; Kang, Y.S.; Han, T.; Choi, G.; et al. NPCARE: Database of natural products and fractional extracts for cancer regulation. J. Cheminform. 2017, 9, 2. [Google Scholar] [CrossRef] [PubMed]
- Database of Natural Products for Cancer Gene Regulation (NPCARE). Available online: http://silver.sejong.ac.kr/npcare (accessed on 20 February 2017).
- Mangal, M.; Sagar, P.; Singh, H.; Raghava, G.P.S.; Agarwal, S.M. NPACT: Naturally Occurring Plant-based Anti-cancer Compound-Activity-Target database. Nucleic Acids Res. 2013, 41, D1124–D1129. [Google Scholar] [CrossRef] [PubMed]
- Naturally Occurring Plant-based Anti-cancer Compound-Activity-Target database (NPACT). Available online: http://crdd.osdd.net/raghava/npact (accessed on 13 April 2017).
- InterBioScreen. Available online: www.ibscreen.com (accessed on 14 November 2017).
- Ntie-Kang, F.; Zofou, D.; Babiaka, S.B.; Meudom, R.; Scharfe, M.; Lifongo, L.L.; Mbah, J.A.; Mbaze, L.M.; Sippl, W.; Efange, S.M.N. AfroDb: A select highly potent and diverse natural product library from African medicinal plants. PLoS ONE 2013, 8, e78085. [Google Scholar] [CrossRef] [PubMed]
- AfroDb. Available online: http://african-compounds.org/about/afrodb (accessed on 18 October 2016).
- TargetMol. Available online: www.targetmol.com (accessed on 17 May 2017).
- Kang, H.; Tang, K.; Liu, Q.; Sun, Y.; Huang, Q.; Zhu, R.; Gao, J.; Zhang, D.; Huang, C.; Cao, Z. HIM-herbal ingredients in-vivo metabolism database. J. Cheminform. 2013, 5, 28. [Google Scholar] [CrossRef] [PubMed]
- Herbal Ingredients In-Vivo Metabolism database (HIM). Available online: http://binfo.shmtu.edu.cn:8080/him (accessed on 13 April 2017).
- Hatherley, R.; Brown, D.K.; Musyoka, T.M.; Penkler, D.L.; Faya, N.; Lobb, K.A.; Tastan Bishop, Ö. SANCDB: A South African natural compound database. J. Cheminform. 2015, 7, 29. [Google Scholar] [CrossRef] [PubMed]
- South African Natural Compound Database (SANCDB). Available online: http://sancdb.rubi.ru.ac.za (accessed on 8 February 2017).
- UEFS Natural Products Catalog. ZINC15. Available online: http://zinc15.docking.org (accessed on 26 May 2017).
- Ntie-Kang, F.; Amoa Onguéné, P.; Fotso, G.W.; Andrae-Marobela, K.; Bezabih, M.; Ndom, J.C.; Ngadjui, B.T.; Ogundaini, A.O.; Abegaz, B.M.; Meva’a, L.M. Virtualizing the p-ANAPL library: A step towards drug discovery from African medicinal plants. PLoS ONE 2014, 9, e90655. [Google Scholar] [CrossRef]
- Natural Products Set IV of the Developmental Therapeutic Program of the National Cancer Institute/National Institutes of Health. Available online: http://dtp.cancer.gov/organization/dscb/obtaining/available_plates.htm (accessed on 20 October 2016).
- Ye, H.; Ye, L.; Kang, H.; Zhang, D.; Tao, L.; Tang, K.; Liu, X.; Zhu, R.; Liu, Q.; Chen, Y.Z.; et al. HIT: Linking herbal active ingredients to targets. Nucleic Acids Res. 2011, 39, D1055–D1059. [Google Scholar] [CrossRef]
- Herbal Ingredients’ Targets database (HIT). Available online: http://lifecenter.sgst.cn/hit (accessed on 13 April 2017).
- Ntie-Kang, F.; Nwodo, J.N.; Ibezim, A.; Simoben, C.V.; Karaman, B.; Ngwa, V.F.; Sippl, W.; Adikwu, M.U.; Mbaze, L.M. Molecular modeling of potential anticancer agents from African medicinal plants. J. Chem. Inf. Model. 2014, 54, 2433–2450. [Google Scholar] [CrossRef]
- AfroCancer. Available online: http://african-compounds.org/about/afrocancer (accessed on 10 February 2017).
- Onguéné, P.A.; Ntie-Kang, F.; Mbah, J.A.; Lifongo, L.L.; Ndom, J.C.; Sippl, W.; Mbaze, L.M. The potential of anti-malarial compounds derived from African medicinal plants, part III: An in silico evaluation of drug metabolism and pharmacokinetics profiling. Org. Med. Chem. Lett. 2014, 4, 6. [Google Scholar] [CrossRef]
- AfroMalariaDB. Available online: http://african-compounds.org/about/afromalariadb (accessed on 10 February 2017).
- Natural products subset of AK Scientific. AK Scientific. Available online: www.aksci.com (accessed on 19 April 2017).
- Natural products of Selleck Chemicals. Selleck Chemicals. Available online: www.selleckchem.com (accessed on 14 November 2017).
- Breiman, L. Random forests. Machine Learning 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Matthews, B.W. Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochim. Biophys. Acta 1975, 405, 442–451. [Google Scholar] [CrossRef]
- Schomburg, K.; Ehrlich, H.-C.; Stierand, K.; Rarey, M. From structure diagrams to visual chemical patterns. J. Chem. Inf. Model. 2010, 50, 1529–1535. [Google Scholar] [CrossRef] [PubMed]
- SMARTSview. Available online: http://smartsview.zbh.uni-hamburg.de/ (accessed on 30 November 2018).
- Bienfait, B.; Ertl, P. JSME: A free molecule editor in JavaScript. J. Cheminform. 2013, 5, 24. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name 1 | Number of Molecules in SMILES Notation Successfully Parsed with RDKit | Number of Unique Molecules After Data Preprocessing | Scientific Literature and/or Online Presence |
|---|---|---|---|
| UNPD | 229,140 | 161,228 | [54,55] |
| TCM Database@Taiwan | 56,325 | 45,422 | [56,57] |
| NP Atlas | 20,018 | 18,358 | [53] |
| TCMID | 13,188 | 10,918 | [58,59] |
| TIPdb | 8838 | 7620 | [60,61,62] |
| Ambinter and Greenpharma NPs | 7905 | 6680 | [63,64] |
| AnalytiCon Discovery MEGx | 4315 | 4063 | [65] |
| NANPDB | 6841 | 3734 | [66,67] |
| StreptomeDB | 3990 | 3353 | [68,69] |
| NPs of PubChem Substance Database | 3533 | 2638 | [70,71] |
| NuBBE | 1856 | 1637 | [72,73] |
| Pi Chemicals NPs | 1783 | 1511 | [74] |
| NPCARE | 1613 | 1479 | [75,76] |
| NPACT | 1516 | 1376 | [77,78] |
| InterBioScreen NPs | 1359 | 1116 | [79] |
| AfroDb | 954 | 865 | [80,81] |
| TargetMol Natural Compound Library | 850 | 745 | [82] |
| HIM | 1284 | 641 | [83,84] |
| SANCDB | 623 | 588 | [85,86] |
| UEFS Natural Products | 493 | 469 | via ZINC [40,87] |
| p-ANAPL | 538 | 456 | [88] |
| NCI/NIH DTP NP set IV | 419 | 394 | [89] |
| HIT | 707 | 362 | [90,91] |
| AfroCancer | 388 | 352 | [92,93] |
| AfroMalariaDB | 265 | 250 | [94,95] |
| AK Scientific NPs | 242 | 177 | [96] |
| Selleck Chemicals NPs | 173 | 163 | [97] |
| NP data set TOTAL | - | 201761 |
| Test Method | Metric 1 | MOE Two-Dimensional Descriptors | Morgan2 Fingerprints (1024 Bits) | MACCS Keys | NP-Likeness Calculator |
|---|---|---|---|---|---|
| 10-fold cross-validation | AUC | 0.997 | 0.997 | 0.996 | / |
| MCC | 0.953 | 0.958 | 0.950 | / | |
| Independent test set | AUC | 0.997 | 0.997 | 0.997 | 0.997 |
| MCC | 0.954 | 0.960 | 0.960 | 0.959 |
| Identifier Used by MOE | Feature Importance 1 | Description |
|---|---|---|
| a_nN | 0.103 | Number of nitrogen atoms. |
| a_ICM | 0.051 | Entropy of the element distribution in the molecule. |
| chiral_u | 0.045 | Number of unconstrained chiral centers. |
| GCUT_SLOGP_0 | 0.045 | Descriptor derived from graph distance adjacency matrices utilizing atomic contribution to log P. |
| SlogP_VSA0 | 0.044 | Surface area descriptor taking into account the contributions of individual atoms to log P. |
| chiral | 0.042 | Number of chiral centers. |
| GCUT_SLOGP_3 | 0.036 | Descriptor derived from graph distance adjacency matrices utilizing atomic contribution to log P. |
| a_nO | 0.025 | The number of oxygen atoms. |
| GCUT_PEOE_0 | 0.025 | Descriptor derived from graph distance adjacency matrices utilizing partial equalization of orbital electronegativities charges. |
| SlogP_VSA1 | 0.024 | Surface area descriptor taking into account the contributions of individual atoms to log P. |
| Similarity Map 1 | Name | Source 2 | NP Class Probability | Disease Indication | Year Introduced |
|---|---|---|---|---|---|
 | arglabin | N | 1.0 | anticancer | 1999 |
 | cefonicid sodium | ND | 0.34 | antibacterial | 1984 |
 | dutaseride | ND | 0.18 | benign prostatic hypertrophy | 2001 |
 | vorapaxar | ND | 0.30 | coronary artery disease | 2014 |
 | empagliflozin | S*/NM | 0.67 | antidiabetic (diabetes 2) | 2014 |
 | belinostat | S*/NM | 0.09 | anticancer | 2014 |
 | febuxostat | S/NM | 0.19 | hyperuricemia | 2009 |
 | zalcitabine | S* | 0.46 | antiviral | 1992 |
 | bilastine | S | 0.17 | antihistamine | 2011 |
 | perampanel | S | 0.16 | antiepileptic | 2012 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.; Stork, C.; Hirte, S.; Kirchmair, J. NP-Scout: Machine Learning Approach for the Quantification and Visualization of the Natural Product-Likeness of Small Molecules. Biomolecules 2019, 9, 43. https://doi.org/10.3390/biom9020043
Chen Y, Stork C, Hirte S, Kirchmair J. NP-Scout: Machine Learning Approach for the Quantification and Visualization of the Natural Product-Likeness of Small Molecules. Biomolecules. 2019; 9(2):43. https://doi.org/10.3390/biom9020043
Chicago/Turabian StyleChen, Ya, Conrad Stork, Steffen Hirte, and Johannes Kirchmair. 2019. "NP-Scout: Machine Learning Approach for the Quantification and Visualization of the Natural Product-Likeness of Small Molecules" Biomolecules 9, no. 2: 43. https://doi.org/10.3390/biom9020043
APA StyleChen, Y., Stork, C., Hirte, S., & Kirchmair, J. (2019). NP-Scout: Machine Learning Approach for the Quantification and Visualization of the Natural Product-Likeness of Small Molecules. Biomolecules, 9(2), 43. https://doi.org/10.3390/biom9020043