High-Throughput Screening of Lipidomic Adaptations in Cultured Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Cell Culture
2.3. Lipid Extractions
2.4. Liquid Chromatography-Mass Spectrometry
2.5. Software and Bioinformatics
3. Results
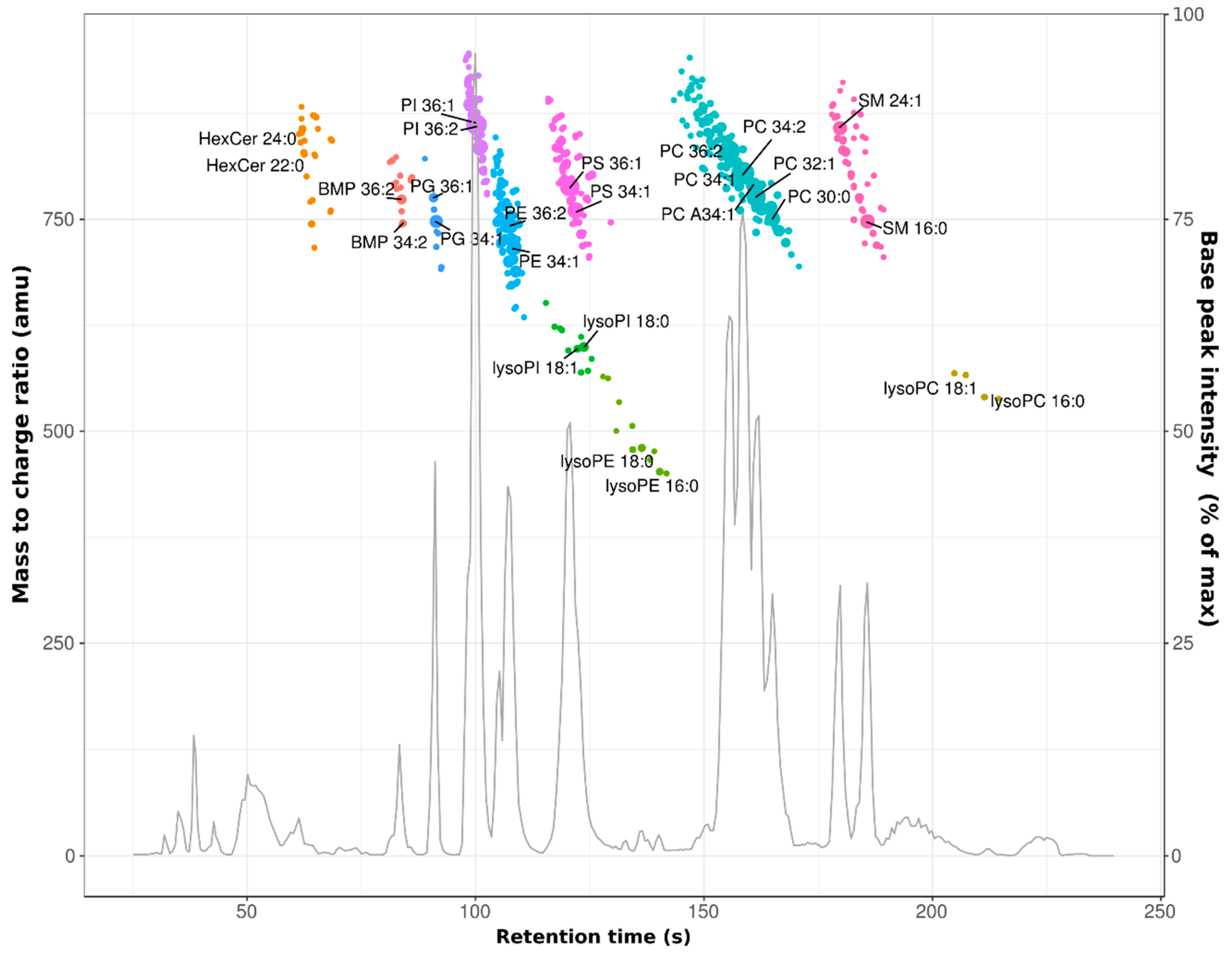
3.1. Profiling of the HeLa Cell Lipidome
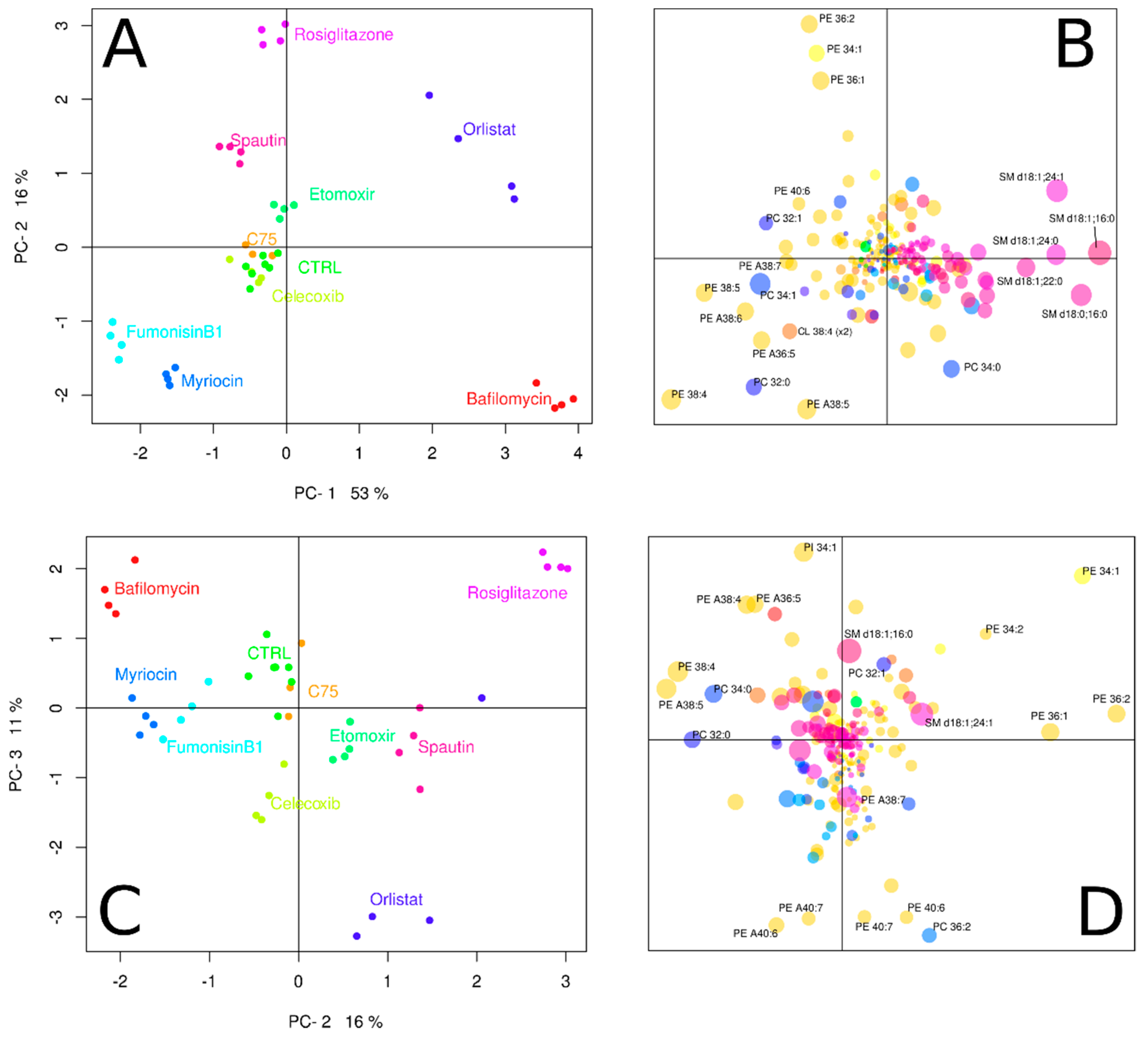
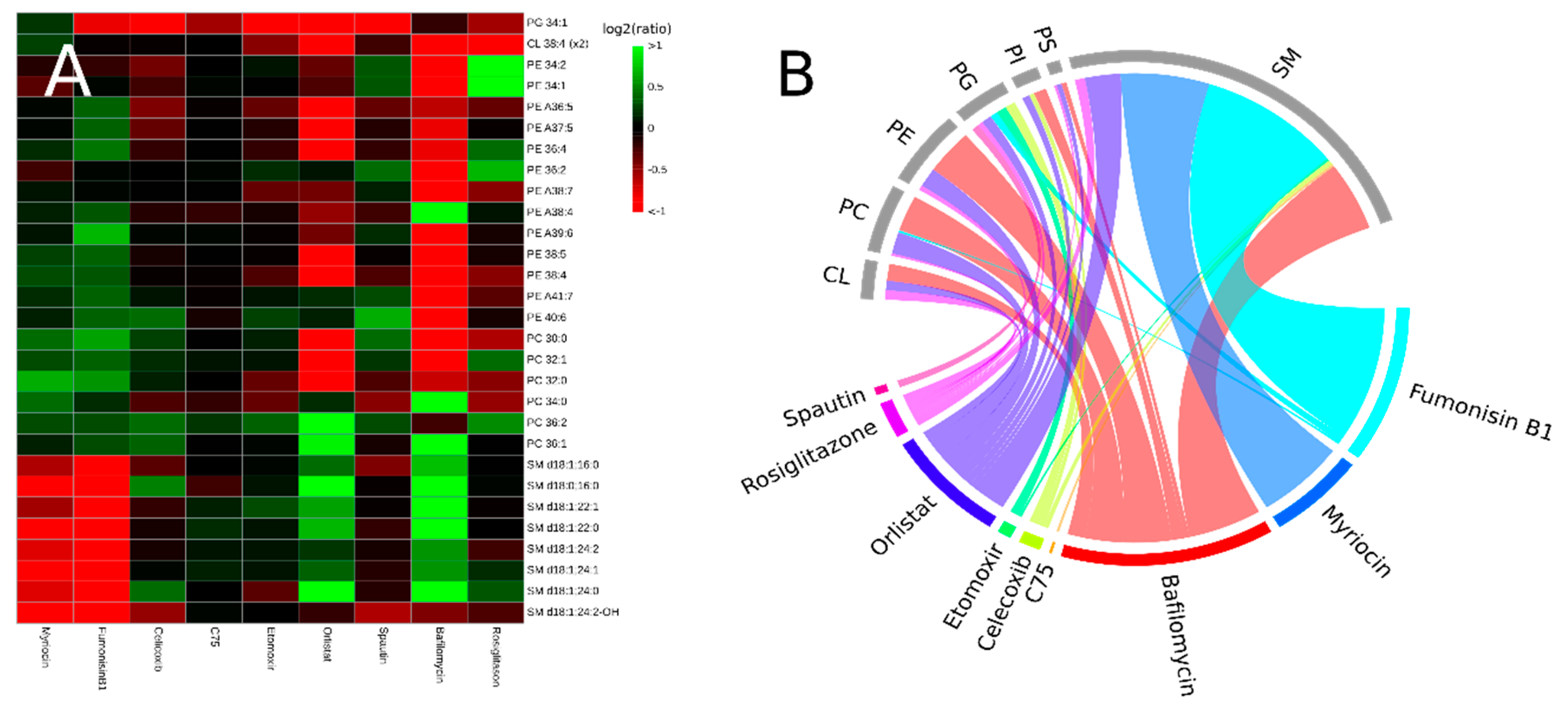
3.2. Inhibitors Induce Specific Alterations of HeLa Lipidomes
3.2.1. Interference with Sphingolipid Biosynthesis by Myriocin and Fumonisinb1
3.2.2. Celicoxib, a cyclooxigenase-2 Inhibitor
3.2.3. Interfering with Fatty Acid Metabolism by Orlistat, and Etomoxir
3.2.4. Inhibitors of Autophagy: Spautin and Bafilomycin
3.2.5. Rosiglitazone, a Thiazolidinone Drug that Acts as a Peroxisome Proliferator Activity Receptor γ Agonist
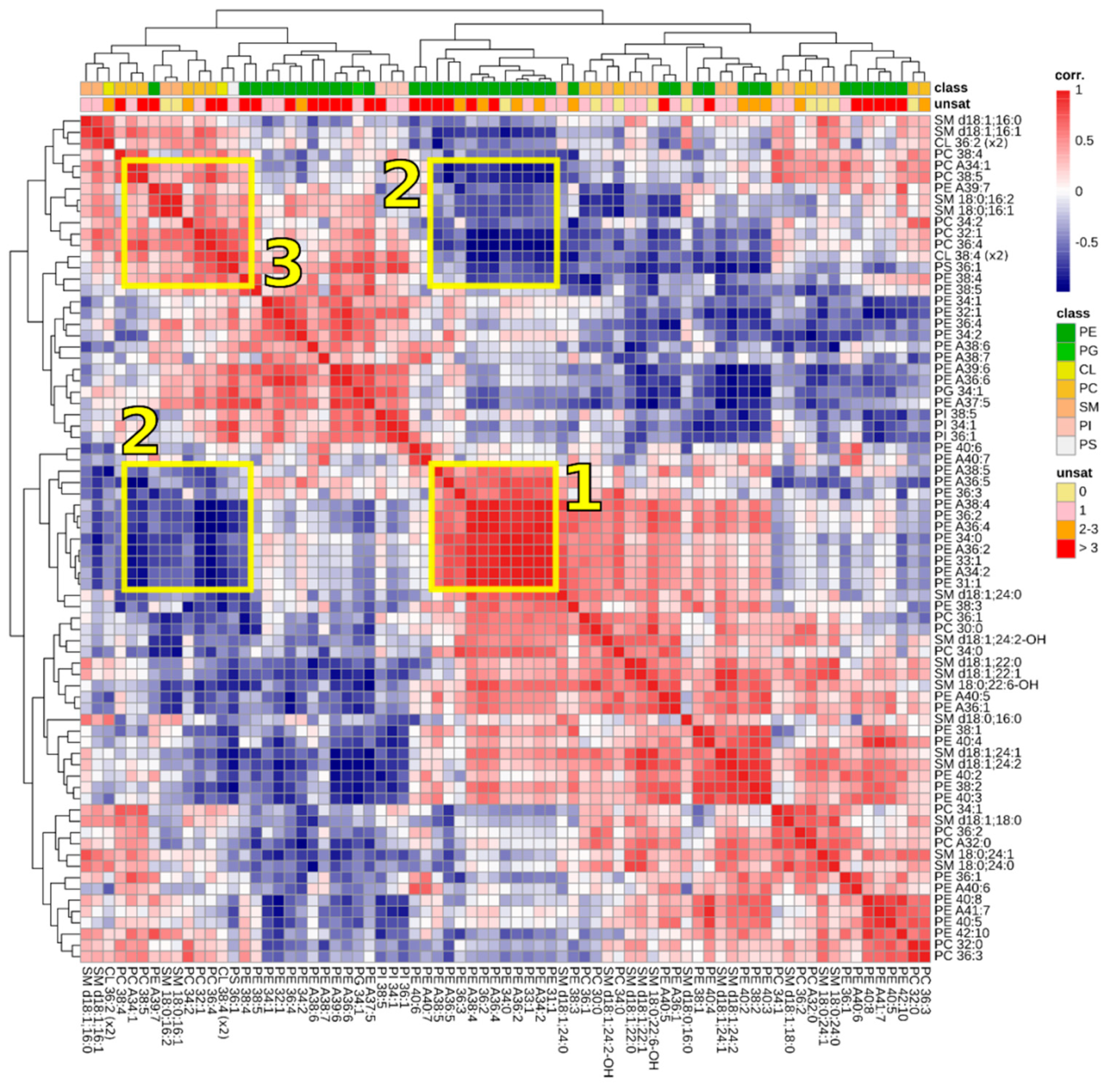
3.3. Correlations between Lipids
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [PubMed]
- Reis, A.; Rudnitskaya, A.; Blackburn, G.J.; Fauzi, N.M.; Pitt, A.R.; Spickett, C.M. A comparison of five lipid extraction solvent systems for lipidomic studies of human LDL. J. Lipid Res. 2013, 54, 1812–1824. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Gross, R.W. Shotgun lipidomics: Electrospray ionization mass spectrometric analysis and quantitation of cellular lipidomes directly from crude extracts of biological samples. Mass Spectrom. Rev. 2005, 24, 367–412. [Google Scholar] [CrossRef] [PubMed]
- Paglia, G.; Kliman, M.; Claude, E.; Geromanos, S.; Astarita, G. Applications of ion-mobility mass spectrometry for lipid analysis. Anal. Bioanal. Chem. 2015, 407, 4995–5007. [Google Scholar] [CrossRef] [PubMed]
- Ståhlman, M.; Ejsing, C.S.; Tarasov, K.; Perman, J.; Borén, J.; Ekroos, K. High-throughput shotgun lipidomics by quadrupole time-of-flight mass spectrometry. J. Chromatogr. B 2009, 877, 2664–2672. [Google Scholar] [CrossRef] [PubMed]
- Brouwers, J.F. Liquid chromatographic-mass spectrometric analysis of phospholipids. Chromatography, ionization and quantification. Biochim. Biophys. Acta 2011, 1811, 763–775. [Google Scholar] [CrossRef]
- Cajka, T.; Fiehn, O. Comprehensive analysis of lipids in biological systems by liquid chromatography-mass spectrometry. TrAC Trends Anal. Chem. 2014, 61, 192–206. [Google Scholar] [CrossRef]
- Triebl, A.; Trötzmüller, M.; Hartler, J.; Stojakovic, T.; Köfeler, H.C. Lipidomics by ultrahigh performance liquid chromatography-high resolution mass spectrometry and its application to complex biological samples. J. Chromatogr. B 2017, 1053, 72–80. [Google Scholar] [CrossRef]
- Arroyo-Olarte, R.D.; Brouwers, J.F.; Kuchipudi, A.; Helms, J.B.; Biswas, A.; Dunay, I.R.; Lucius, R.; Gupta, N. Phosphatidylthreonine and Lipid-Mediated Control of Parasite Virulence. PLoS Biol. 2015, 13, e1002288. [Google Scholar] [CrossRef]
- Kong, P.; Lehmann, M.J.; Helms, J.B.; Brouwers, J.F.; Gupta, N. Lipid analysis of Eimeria sporozoites reveals exclusive phospholipids, a phylogenetic mosaic of endogenous synthesis, and a host-independent lifestyle. Cell. Discov. 2018, 4, 24. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Ladenson, J.; Turk, J. Algorithms for automatic processing of data from mass spectrometric analyses of lipids. J. Chromatogr. B-Anal. Technol. Biomed. 2009, 877, 2847–2854. [Google Scholar] [CrossRef] [PubMed]
- Hartler, J.; Trötzmüller, M.; Chitraju, C.; Spener, F.; Köfeler, H.C.; Thallinger, G.G. Lipid Data Analyzer: Unattended Identification and Quantitation of Lipids in LC-MS Data. Bioinformatics 2011, 27, 572–577. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, Z.; Mayr, M.; Zeeshan, S.; Dandekar, T.; Mueller, M.J.; Fekete, A. Lipid-Pro: A computational lipid identification solution for untargeted lipidomics on data-independent acquisition tandem mass spectrometry platforms. Bioinformatics 2015, 31, 1150–1153. [Google Scholar] [CrossRef] [PubMed]
- Koelmel, J.P.; Kroeger, N.M.; Ulmer, C.Z.; Bowden, J.A.; Patterson, R.E.; Cochran, J.A.; Beecher, C.W.W.; Garrett, T.J.; Yost, R.A. LipidMatch: An automated workflow for rule-based lipid identification using untargeted high-resolution tandem mass spectrometry data. BMC Bioinform. 2017, 18, 331. [Google Scholar] [CrossRef] [PubMed]
- Hartler, J.; Triebl, A.; Ziegl, A.; Trötzmüller, M.; Rechberger, G.N.; Zeleznik, O.A.; Zierler, K.A.; Torta, F.; Cazenave-Gassiot, A.; Wenk, M.R.; et al. Deciphering lipid structures based on platform-independent decision rules. Nat. Methods 2017, 14, 1171. [Google Scholar] [CrossRef] [PubMed]
- Cajka, T.; Fiehn, O. LC–MS-Based Lipidomics and Automated Identification of Lipids Using the LipidBlast In-Silico MS/MS Library. In Lipidomics: Methods and Protocols; Bhattacharya, S.K., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2017; pp. 149–170. ISBN 978-1-4939-6996-8. [Google Scholar]
- Ulmer, C.Z.; Patterson, R.E.; Koelmel, J.P.; Garrett, T.J.; Yost, R.A. A. A Robust Lipidomics Workflow for Mammalian Cells, Plasma, and Tissue Using Liquid-Chromatography High-Resolution Tandem Mass Spectrometry. In Lipidomics: Methods and Protocols; Bhattacharya, S.K., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2017; pp. 91–106. ISBN 978-1-4939-6996-8. [Google Scholar]
- Aloulou, A.; Rahier, R.; Arhab, Y.; Noiriel, A.; Abousalham, A. Phospholipases: An Overview. In Lipases and Phospholipases: Methods and Protocols; Sandoval, G., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2018; pp. 69–105. ISBN 978-1-4939-8672-9. [Google Scholar]
- van Gestel, R.A.; Brouwers, J.F.; Ultee, A.; Helms, J.B.; Gadella, B.M. Ultrastructure and lipid composition of detergent-resistant membranes derived from mammalian sperm and two types of epithelial cells. Cell Tissue Res. 2015, 363, 129–145. [Google Scholar] [CrossRef]
- Simons, K.; Sampaio, J.L. Membrane organization and lipid rafts. Cold Spring Harb. Perspect. Biol. 2011, 3, a004697. [Google Scholar] [CrossRef]
- Omta, W.A.; van Heesbeen, R.G.; Pagliero, R.J.; van der Velden, L.M.; Lelieveld, D.; Nellen, M.; Kramer, M.; Yeong, M.; Saeidi, A.M.; Medema, R.H.; et al. HC StratoMineR: A Web-Based Tool for the Rapid Analysis of High-Content Datasets. ASSAY Drug Dev. Technol. 2016, 14, 439–452. [Google Scholar] [CrossRef]
- R Development Core Team. R: A Language and Environment for Statistical Computing; Scientific Research Publishing Inc.: Vienna, Austria, 2011. [Google Scholar]
- Smith, C.A.; Want, E.J.; O’Maille, G.; Abagyan, R.; Siuzdak, G. XCMS: Processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal. Chem. 2006, 78, 779–787. [Google Scholar] [CrossRef]
- Stacklies, W.; Redestig, H.; Scholz, M.; Walther, D.; Selbig, J. pcaMethods—A Bioconductor Package Providing PCA Methods for Incomplete Data. Bioinformatics 2007, 23, 1164–1167. [Google Scholar] [CrossRef] [PubMed]
- Miyake, Y.; Kozutsumi, Y.; Nakamura, S.; Fujita, T.; Kawasaki, T. Serine Palmitoyltransferase Is the Primary Target of a Sphingosine-like Immunosuppressant, ISP-1/Myriocin. Biochem. Biophys. Res. Commun. 1995, 211, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Merrill, A.H., Jr.; Wang, E.; Gilchrist, D.G.; Riley, R.T. Fumonisins and other inhibitors of de novo sphingolipid biosynthesis. Adv. Lipid Res. 1993, 26, 215–234. [Google Scholar] [PubMed]
- Hashemi Goradel, N.; Najafi, M.; Salehi, E.; Farhood, B.; Mortezaee, K. Cyclooxygenase-2 in cancer: A review. J. Cell. Physiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Nandakishore, R.; Yalavarthi, P.R.; Kiran, Y.R.; Rajapranathi, M. Selective cyclooxygenase inhibitors: Current status. Curr. Drug Discov. Technol. 2014, 11, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Kirkby, N.S.; Zaiss, A.K.; Urquhart, P.; Jiao, J.; Austin, P.J.; Al-Yamani, M.; Lundberg, M.H.; MacKenzie, L.S.; Warner, T.D.; Nicolaou, A.; et al. LC-MS/MS confirms that COX-1 drives vascular prostacyclin whilst gene expression pattern reveals non-vascular sites of COX-2 expression. PLoS ONE 2013, 8, e69524. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Wang, L.; Xu, C.; Han, S. Effect of photodynamic therapy combined with Celecoxib on expression of cyclooxygenase-2 protein in HeLa cells. Oncol. Lett. 2018, 15, 6599–6603. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Lupu, R. Fatty acid synthase (FASN) as a therapeutic target in breast cancer. Expert Opin. Ther. Targets 2017, 21, 1001–1016. [Google Scholar] [CrossRef]
- Menendez, J.A.; Lupu, R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 2007, 7, 763–777. [Google Scholar] [CrossRef] [PubMed]
- Rendina, A.R.; Cheng, D. Characterization of the inactivation of rat fatty acid synthase by C75: Inhibition of partial reactions and protection by substrates. Biochem. J. 2005, 388, 895–903. [Google Scholar] [CrossRef]
- Ho, T.-S.; Ho, Y.-P.; Wong, W.-Y.; Chi-Ming Chiu, L.; Wong, Y.-S.; Eng-Choon Ooi, V. Fatty acid synthase inhibitors cerulenin and C75 retard growth and induce caspase-dependent apoptosis in human melanoma A-375 cells. Biomed. Pharmacother. 2007, 61, 578–587. [Google Scholar] [CrossRef]
- Puig, T.; Vázquez-Martín, A.; Relat, J.; Pétriz, J.; Menéndez, J.A.; Porta, R.; Casals, G.; Marrero, P.F.; Haro, D.; Brunet, J.; et al. Fatty acid metabolism in breast cancer cells: Differential inhibitory effects of epigallocatechin gallate (EGCG) and C75. Breast Cancer Res. Treat. 2008, 109, 471–479. [Google Scholar] [CrossRef] [PubMed]
- McGarry, J.D.; Leatherman, G.F.; Foster, D.W. Carnitine palmitoyltransferase I. The site of inhibition of hepatic fatty acid oxidation by malonyl-CoA. J. Biol. Chem. 1978, 253, 4128–4136. [Google Scholar]
- Pemble, C.W.; Johnson, L.C.; Kridel, S.J.; Lowther, W.T. Crystal structure of the thioesterase domain of human fatty acid synthase inhibited by Orlistat. Nat. Struct. Mol. Biol. 2007, 14, 704–709. [Google Scholar] [CrossRef] [PubMed]
- Sankaranarayanapillai, M.; Zhang, N.; Baggerly, K.A.; Gelovani, J.G. Metabolic Shifts Induced by Fatty Acid Synthase Inhibitor Orlistat in Non-small Cell Lung Carcinoma Cells Provide Novel Pharmacodynamic Biomarkers for Positron Emission Tomography and Magnetic Resonance Spectroscopy. Mol. Imaging Biol. 2013, 15, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Bialecka-Florjanczyk, E.; Fabiszewska, A.U.; Krzyczkowska, J.; Kurylowicz, A. Synthetic and Natural Lipase Inhibitors. Mini. Rev. Med. Chem. 2018, 18, 672–683. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Xia, H.; Kim, M.; Xu, L.; Li, Y.; Zhang, L.; Cai, Y.; Norberg, H.V.; Zhang, T.; Furuya, T.; et al. Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell 2011, 147, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Vakifahmetoglu-Norberg, H.; Xia, H.; Yuan, J. Pharmacologic agents targeting autophagy. J. Clin. Investig. 2015, 125, 5–13. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Codogno, P.; Levine, B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat. Rev. Drug Discov. 2012, 11, 709–730. [Google Scholar] [CrossRef]
- Strzyz, P. Autophagy: Mitochondria encaged. Nat. Rev. Mol. Cell Biol. 2018, 19, 212. [Google Scholar] [CrossRef]
- Bowman, E.J.; Graham, L.A.; Stevens, T.H.; Bowman, B.J. The Bafilomycin/Concanamycin Binding Site in Subunit c of the V-ATPases from Neurospora crassa and Saccharomyces cerevisiae. J. Biol. Chem. 2004, 279, 33131–33138. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, A.; Tagawa, Y.; Yoshimori, T.; Moriyama, Y.; Masaki, R.; Tashiro, Y. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct. Funct. 1998, 23, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Juhász, G. Interpretation of bafilomycin, pH neutralizing or protease inhibitor treatments in autophagic flux experiments: Novel considerations. Autophagy 2012, 8, 1875–1876. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.K.; Vaidya, A.; Ravichandran, V.; Kashaw, S.K.; Agrawal, R.K. Recent developments and biological activities of thiazolidinone derivatives: A review. Bioorg. Med. Chem. 2012, 20, 3378–3395. [Google Scholar] [PubMed]
- Lehmann, J.M.; Moore, L.B.; Smith-Oliver, T.A.; Wilkison, W.O.; Willson, T.M.; Kliewer, S.A. An Antidiabetic Thiazolidinedione Is a High Affinity Ligand for Peroxisome Proliferator-activated Receptor γ (PPARγ). J. Biol. Chem. 1995, 270, 12953–12956. [Google Scholar] [CrossRef]
- Deeg, M.A.; Tan, M.H. Pioglitazone versus Rosiglitazone: Effects on Lipids, Lipoproteins, and Apolipoproteins in Head-to-Head Randomized Clinical Studies. PPAR Res. 2008, 2008, 520465. [Google Scholar] [CrossRef] [PubMed]
- Grygiel-Górniak, B. Peroxisome proliferator-activated receptors and their ligands: Nutritional and clinical implications—A review. Nutr. J. 2014, 13, 17. [Google Scholar] [CrossRef] [PubMed]
- Bleijerveld, O.B.; Brouwers, J.F.; Vaandrager, A.B.; Helms, J.B.; Houweling, M. The CDP-ethanolamine pathway and phosphatidylserine decarboxylation generate different phosphatidylethanolamine molecular species. J. Biol. Chem. 2007, 282, 28362–28372. [Google Scholar] [CrossRef]
- Ford, D.A. Separate myocardial ethanolamine phosphotransferase activities responsible for plasmenylethanolamine and phosphatidylethanolamine synthesis. J. Lipid Res. 2003, 44, 554–559. [Google Scholar] [CrossRef]
- Roberts, M.F. Phospholipases: Structural and functional motifs for working at an interface. FASEB J. 1996, 10, 1159–1172. [Google Scholar] [CrossRef]
- Jeucken, A.; Bernd Helms, J.; Brouwers, J.F. Cardiolipin synthases of Escherichia coli have phospholipid class specific phospholipase D activity dependent on endogenous and foreign phospholipids. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2018. [Google Scholar] [CrossRef] [PubMed]
- Brügger, B. Lipidomics: Analysis of the Lipid Composition of Cells and Subcellular Organelles by Electrospray Ionization Mass Spectrometry. Annu. Rev. Biochem. 2014, 83, 79–98. [Google Scholar] [CrossRef] [PubMed]
- Veldhuizen, E.J.A.; Haagsman, H.P. Role of pulmonary surfactant components in surface film formation and dynamics. Biochim. Biophys. Acta (BBA)-Biomembr. 2000, 1467, 255–270. [Google Scholar] [CrossRef]
- Macarron, R.; Banks, M.N.; Bojanic, D.; Burns, D.J.; Cirovic, D.A.; Garyantes, T.; Green, D.V.S.; Hertzberg, R.P.; Janzen, W.P.; Paslay, J.W.; et al. Impact of high-throughput screening in biomedical research. Nat. Rev. Drug Discov. 2011, 10, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Nyholm, T.K.M. Lipid-protein interplay and lateral organization in biomembranes. Chem. Phys. Lipids 2015, 189, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Dowhan, W. Understanding phospholipid function: Why are there so many lipids? J. Biol. Chem. 2017, 292, 10755–10766. [Google Scholar] [CrossRef] [PubMed]
- Kridel, S.J.; Axelrod, F.; Rozenkrantz, N.; Smith, J.W. Orlistat Is a Novel Inhibitor of Fatty Acid Synthase with Antitumor Activity. Cancer Res. 2004, 64, 2070–2075. [Google Scholar] [CrossRef]
- Santos, R.; Ursu, O.; Gaulton, A.; Bento, A.P.; Donadi, R.S.; Bologa, C.G.; Karlsson, A.; Al-Lazikani, B.; Hersey, A.; Oprea, T.I.; et al. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 2017, 16, 19–34. [Google Scholar] [CrossRef]
- Beloribi-Djefaflia, S.; Vasseur, S.; Guillaumond, F. Lipid metabolic reprogramming in cancer cells. Oncogenesis 2016, 5, e189. [Google Scholar] [CrossRef]
- Boroughs, L.K.; DeBerardinis, R.J. Metabolic pathways promoting cancer cell survival and growth. Nat. Cell Biol. 2015, 17, 351–359. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeucken, A.; Brouwers, J.F. High-Throughput Screening of Lipidomic Adaptations in Cultured Cells. Biomolecules 2019, 9, 42. https://doi.org/10.3390/biom9020042
Jeucken A, Brouwers JF. High-Throughput Screening of Lipidomic Adaptations in Cultured Cells. Biomolecules. 2019; 9(2):42. https://doi.org/10.3390/biom9020042
Chicago/Turabian StyleJeucken, Aike, and Jos F. Brouwers. 2019. "High-Throughput Screening of Lipidomic Adaptations in Cultured Cells" Biomolecules 9, no. 2: 42. https://doi.org/10.3390/biom9020042
APA StyleJeucken, A., & Brouwers, J. F. (2019). High-Throughput Screening of Lipidomic Adaptations in Cultured Cells. Biomolecules, 9(2), 42. https://doi.org/10.3390/biom9020042