Genetics of Congenital Heart Disease


Abstract
:1. Introduction
2. Congenital Heart Disease Classification and Prevalence
3. Developmental Processes in Formation of the Four-Chambered Heart
4. Role of Transcription Factors
4.1. NKX2-5
4.2. GATA Family
4.3. T-Box Family
4.4. Forkhead Box Family
4.5. Nuclear Receptor Family
4.6. HAND Family
5. Signaling Pathways Underlying CHD
5.1. Nodal Signaling
5.2. Notch Signaling
5.3. Wnt/β-Catenin Signaling
5.4. Bmp Signaling
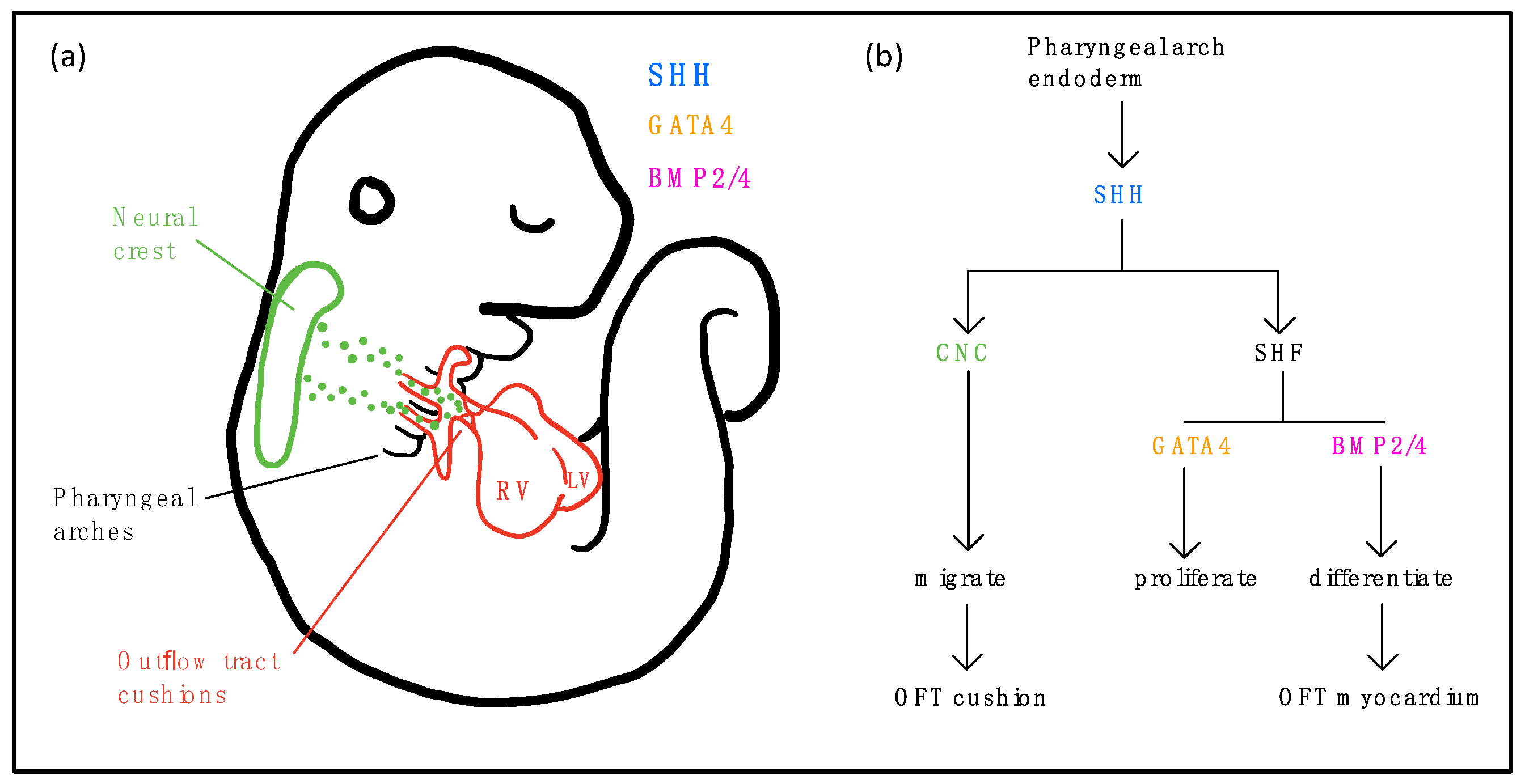
5.5. Sonic Hedgehog (SHH) Signaling
5.6. Ras/Mapk Signaling
5.7. Vegf Signaling
6. Myofilament and Extracellular Matrix Proteins
7. Chromatin Modifiers
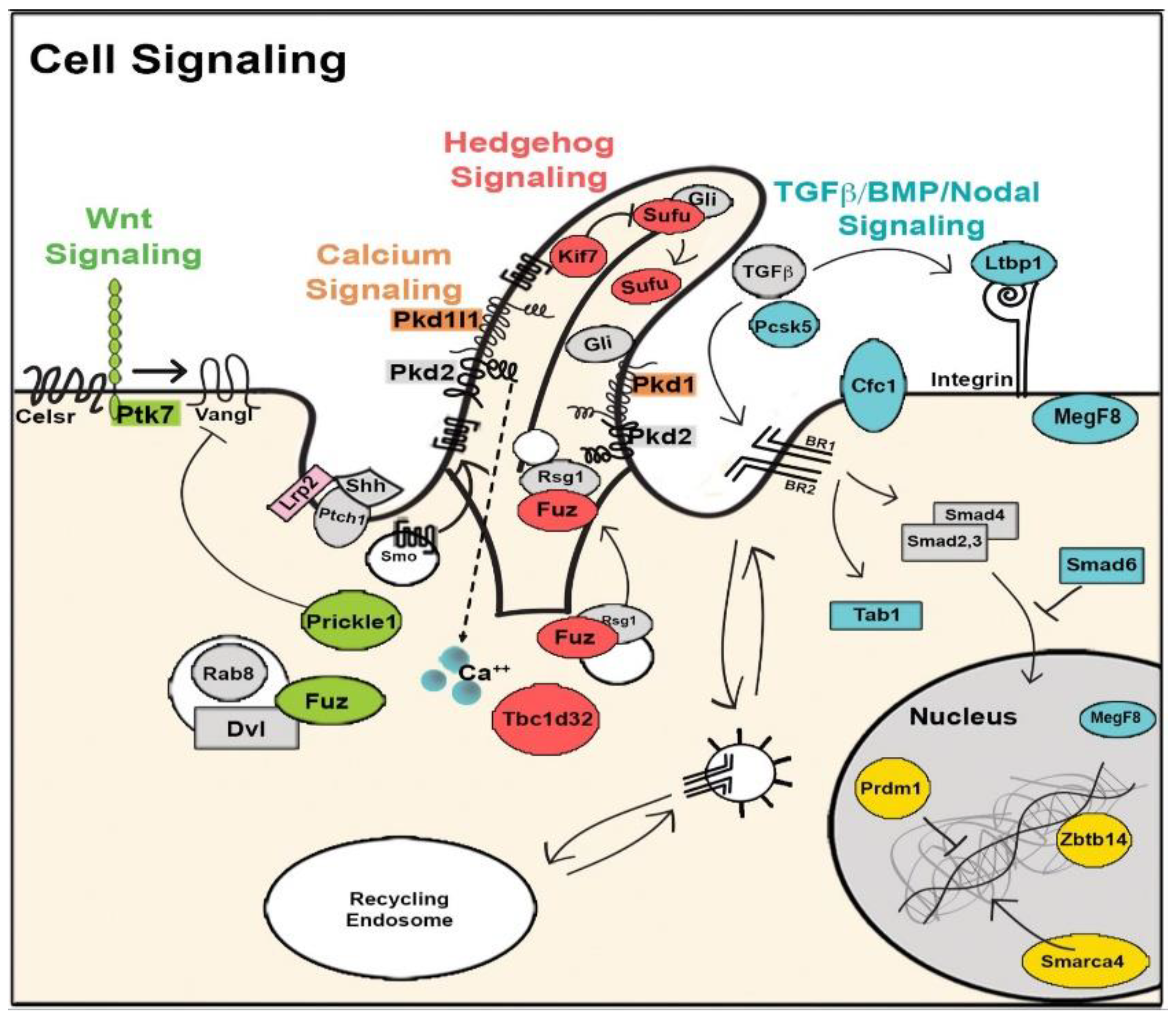
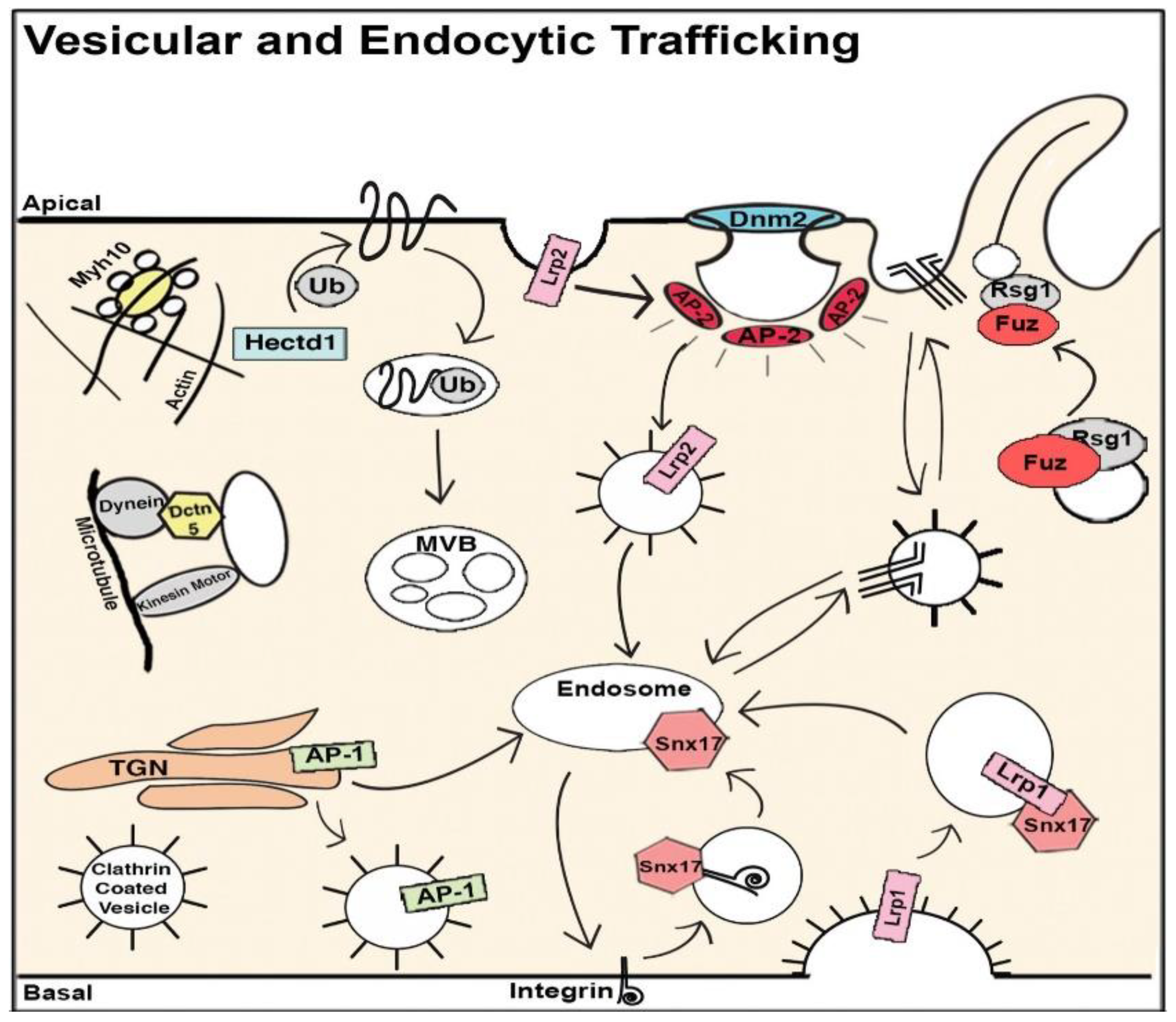
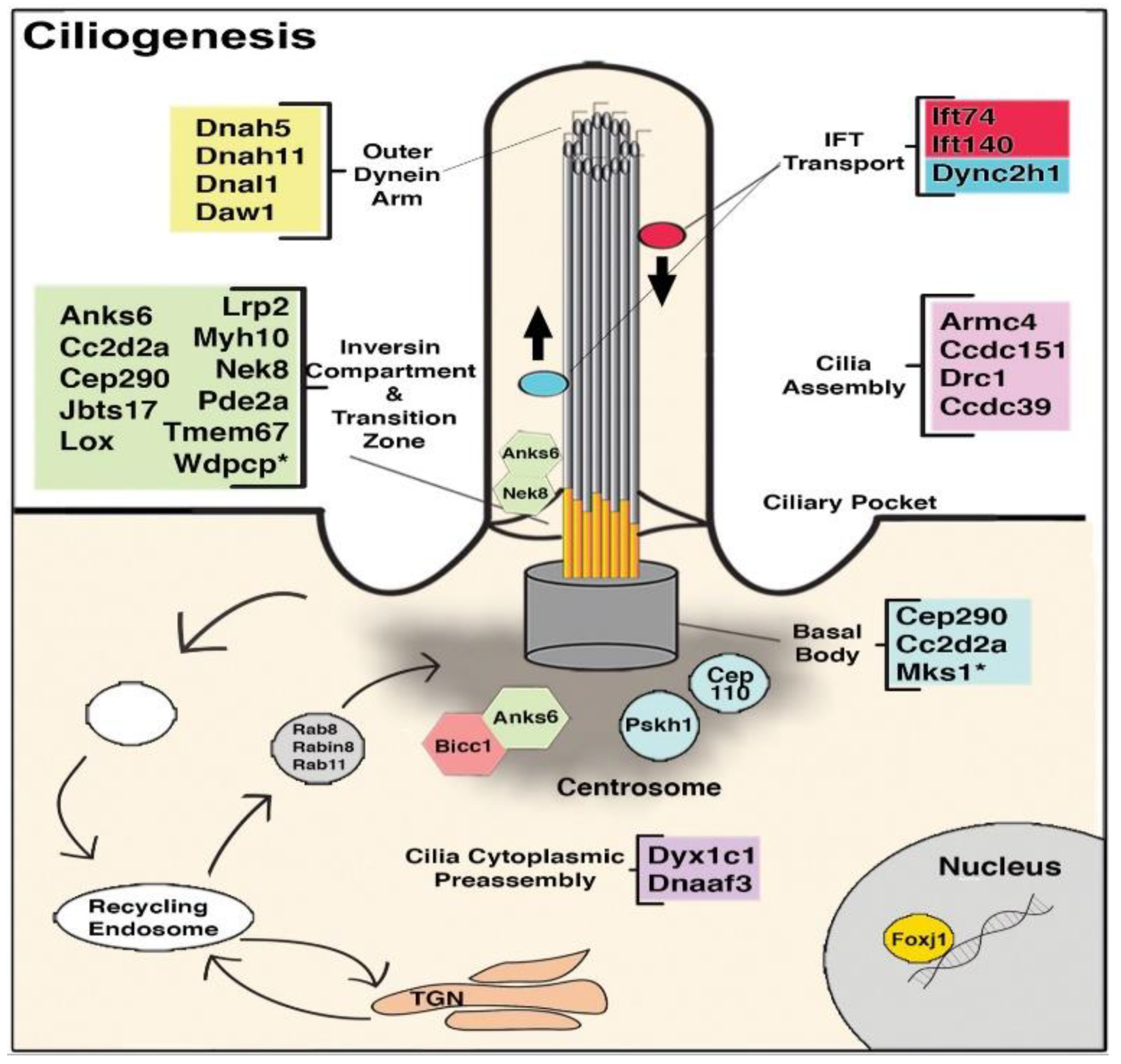
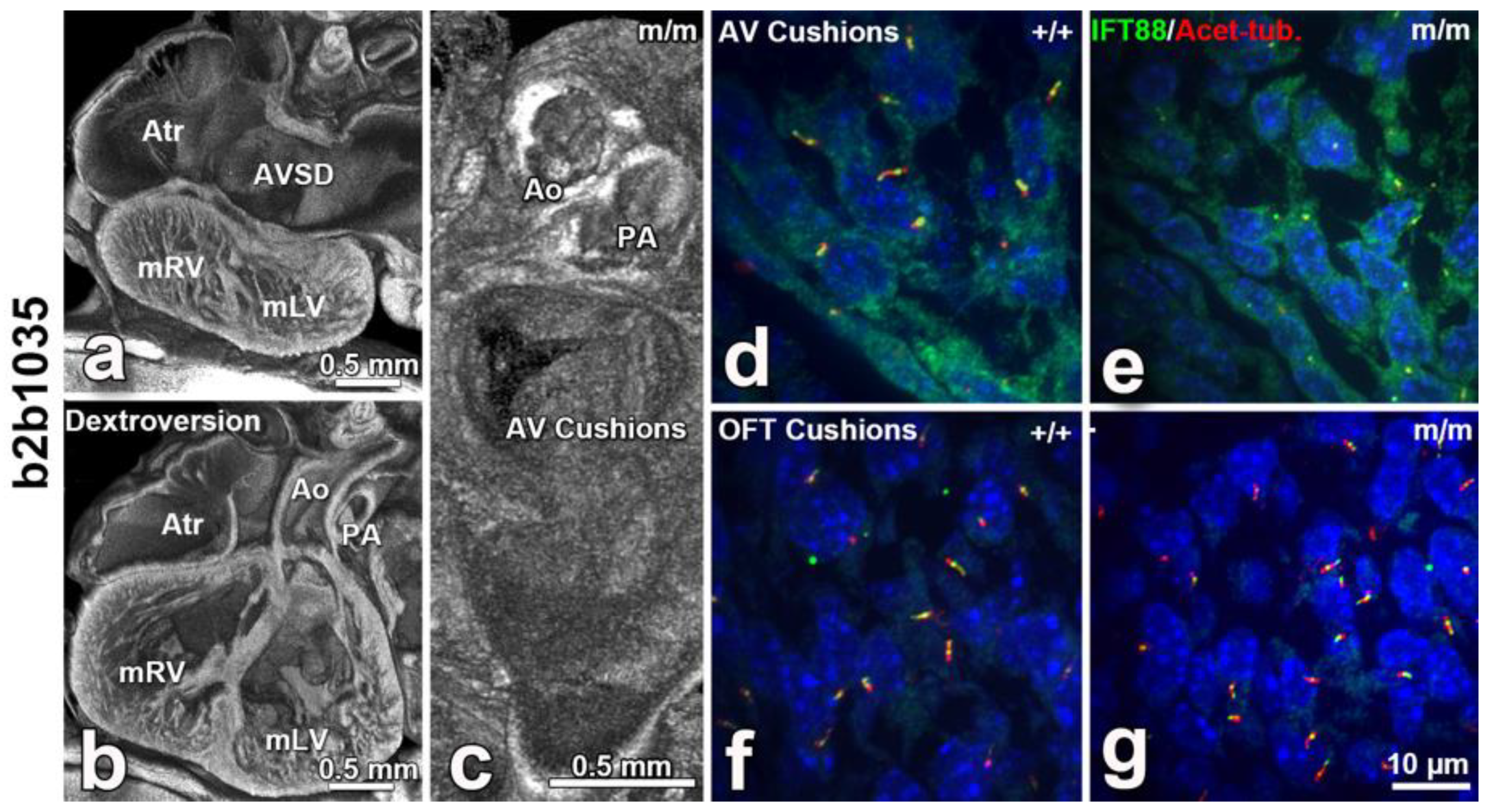
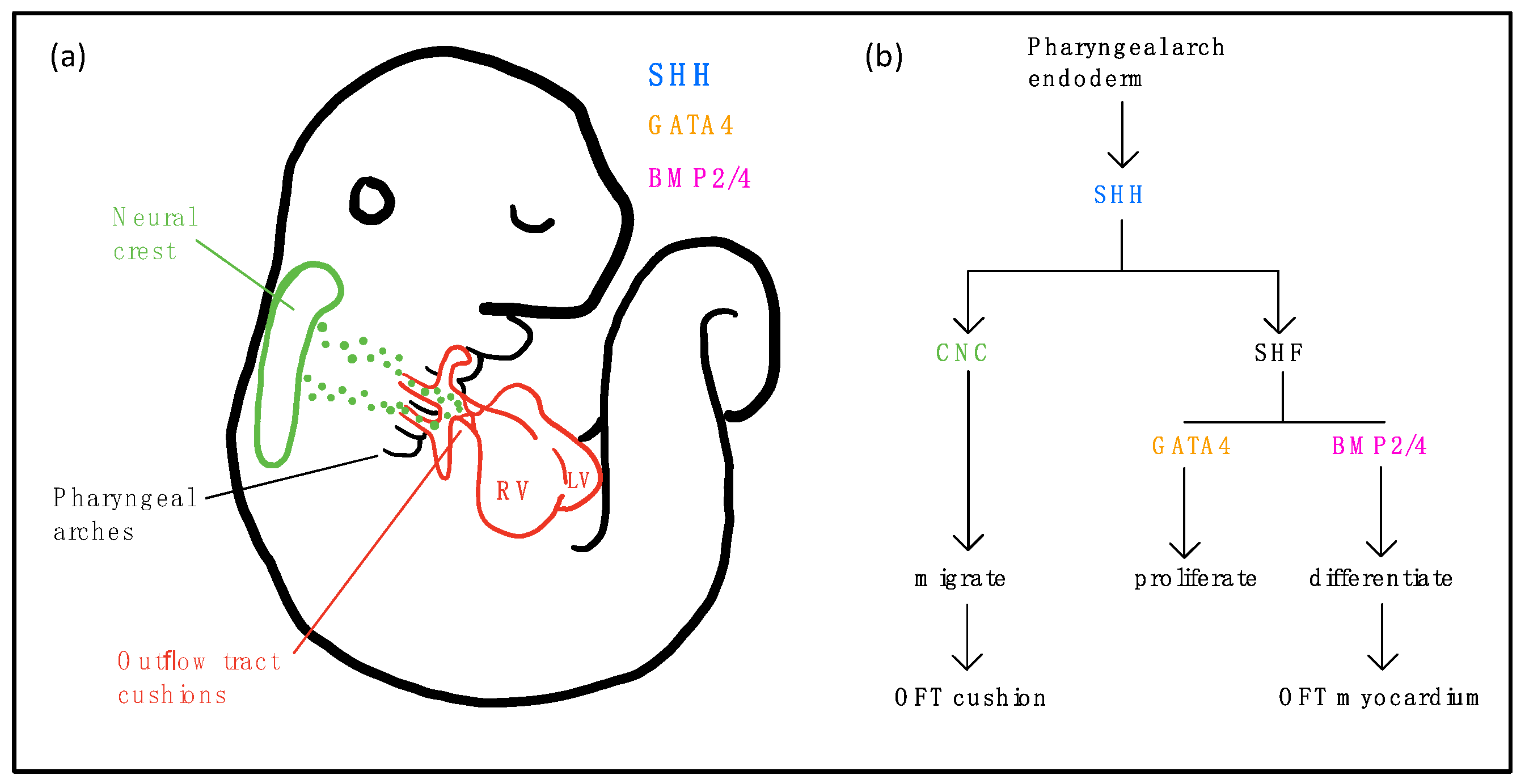
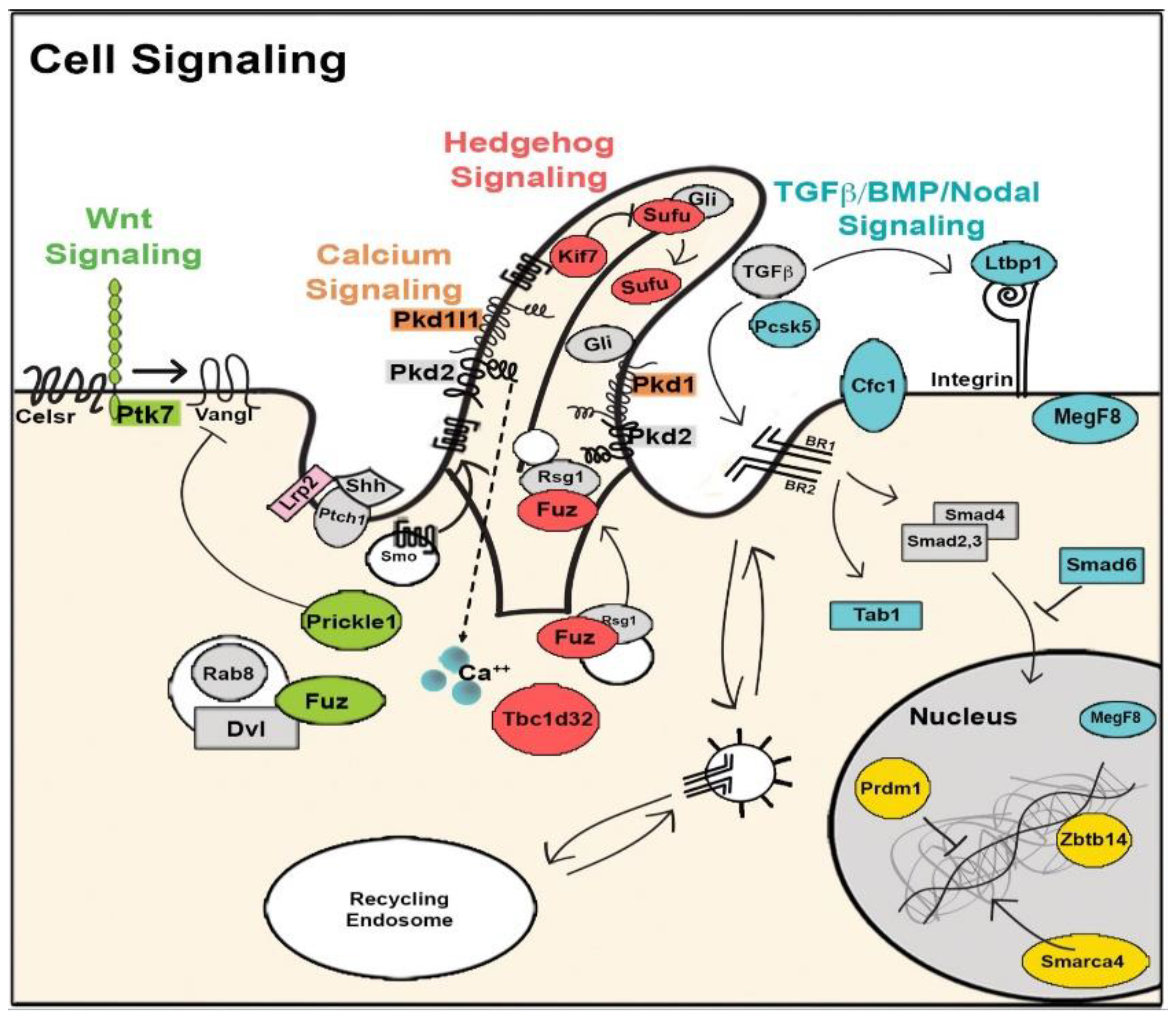
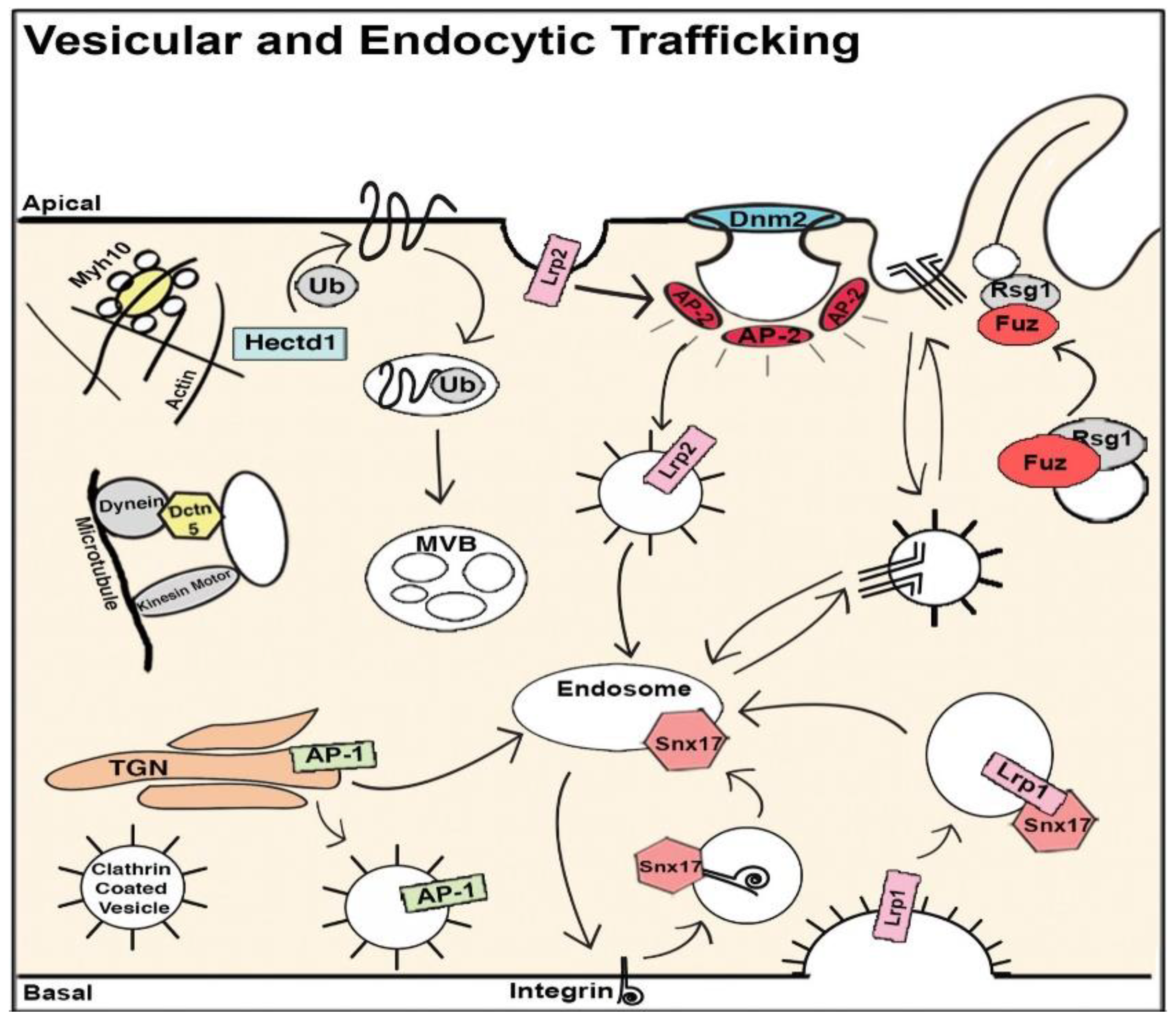
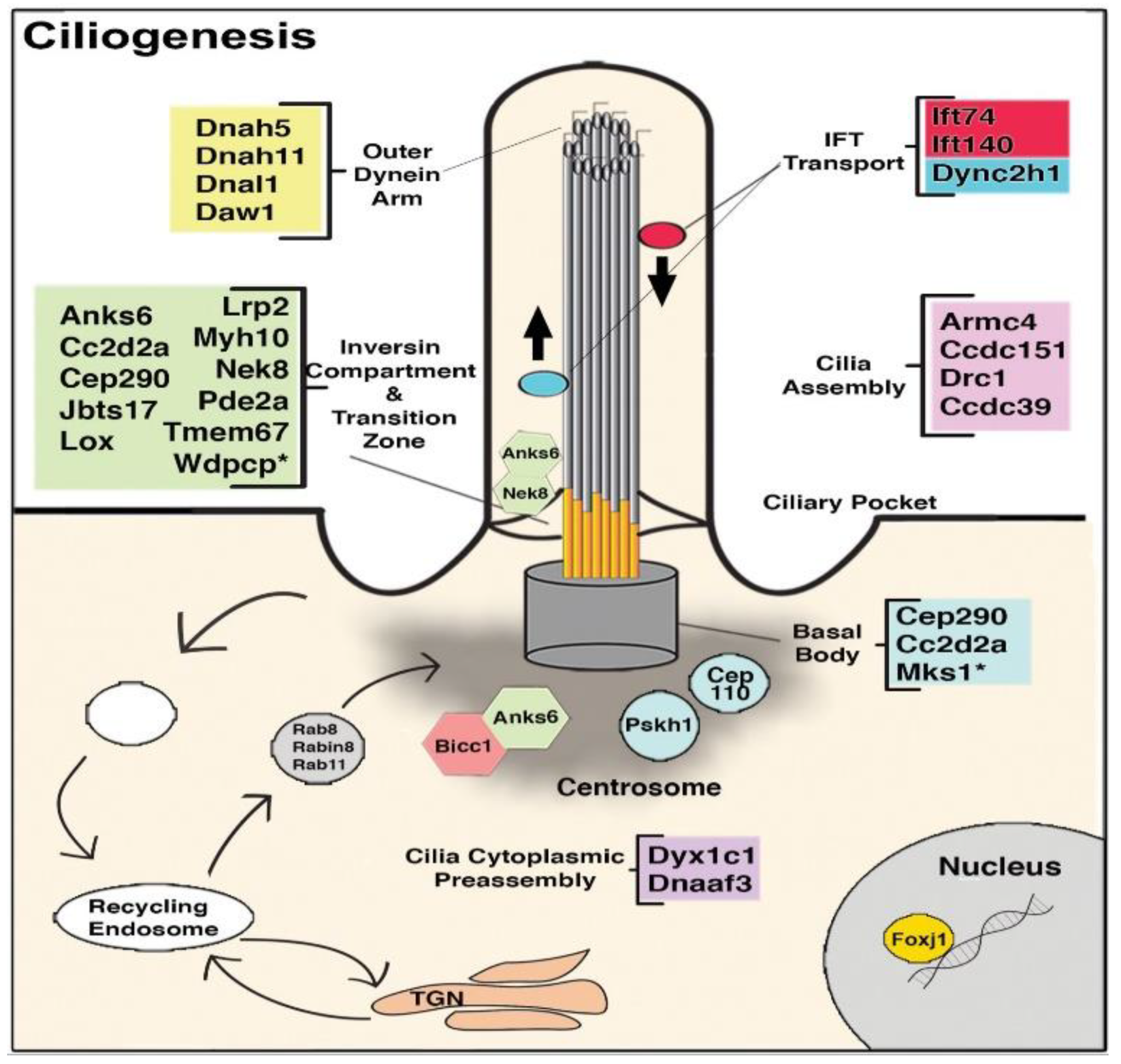
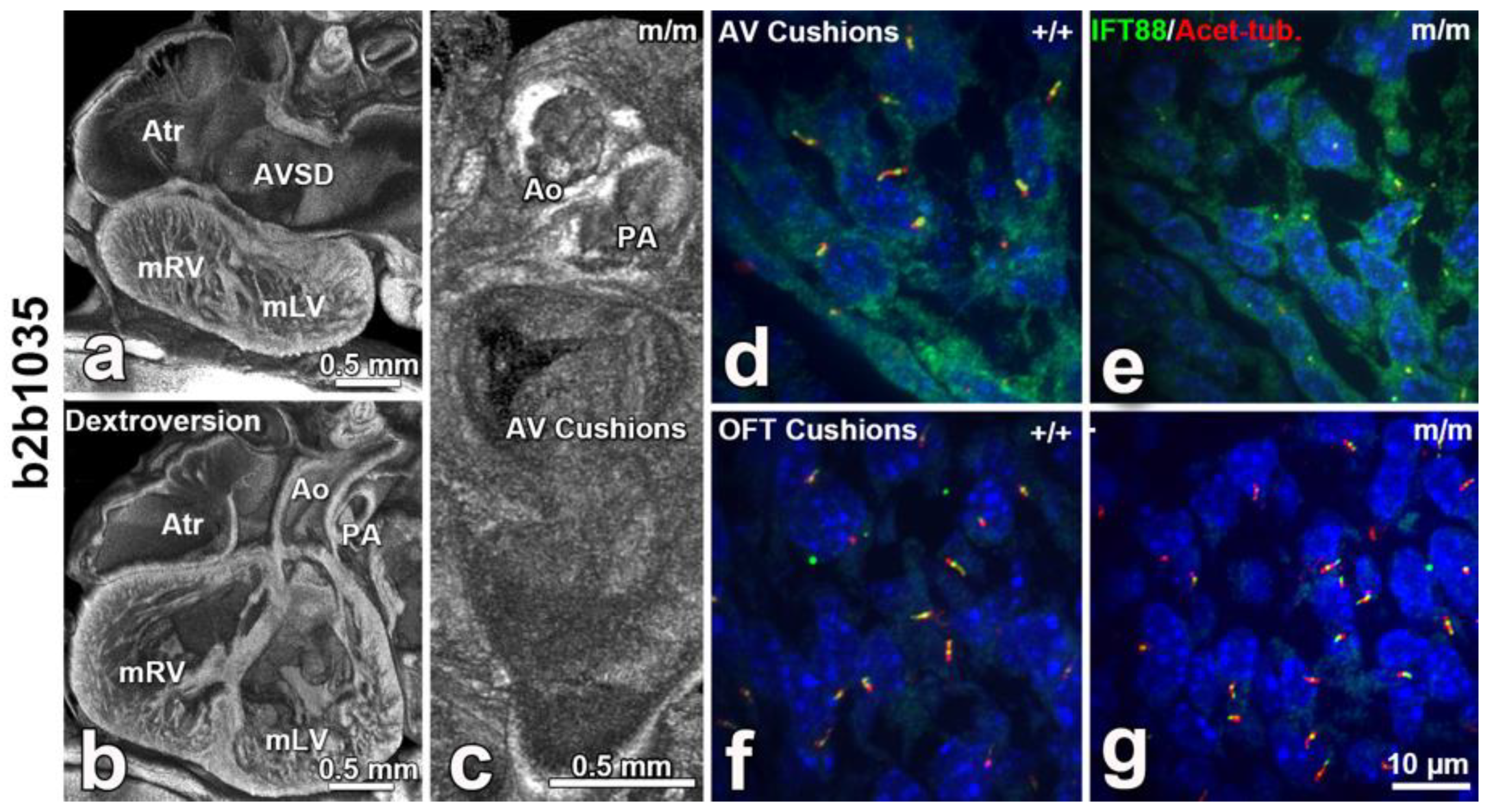
8. The Role of Cilia and Cilia-Transduced Cell Signaling During Cardiogenesis
9. Maternal Effects
10. Future Directions
Funding
Conflicts of Interest
References
- Mahler, G.J.; Butcher, J.T. Cardiac developmental toxicity. Birth Defects Res. Part C Embryo Today Rev. 2011, 93, 291–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenkins, K.J.; Correa, A.; Feinstein, J.A.; Botto, L.; Britt, A.E.; Daniels, S.R.; Elixson, M.; Warnes, C.A.; Webb, C.L. American Heart Association Council on Cardiovascular Disease in the Young Noninherited risk factors and congenital cardiovascular defects: Current knowledge: A scientific statement from the American Heart Association Council on Cardiovascular Disease in the Young: Endorsed by the American Academy of Pediatrics. Circulation 2007, 115, 2995–3014. [Google Scholar] [PubMed] [Green Version]
- Pierpont, M.E.; Brueckner, M.; Chung, W.K.; Garg, V.; Lacro, R.V.; McGuire, A.L.; Mital, S.; Priest, J.R.; Pu, W.T.; Roberts, A.; et al. Genetic Basis for Congenital Heart Disease: Revisited: A Scientific Statement from the American Heart Association. Circulation 2018, 138, 653–711. [Google Scholar] [CrossRef] [PubMed]
- Lage, K.; Greenway, S.C.; Rosenfeld, J.A.; Wakimoto, H.; Gorham, J.M.; Segre, A.V.; Roberts, A.E.; Smoot, L.B.; Pu, W.T.C.; Pereira, A.; et al. Genetic and environmental risk factors in congenital heart disease functionally converge in protein networks driving heart development. Proc. Natl. Acad. Sci. USA 2012, 109, 14035–14040. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Klena, N.T.; Gabriel, G.C.; Liu, X.; Kim, A.J.; Lemke, K.; Chen, Y.; Chatterjee, B.; Devine, W.; Damerla, R.R.; et al. Global genetic analysis in mice unveils central role for cilia in congenital heart disease. Nature 2015, 521, 520–524. [Google Scholar] [CrossRef] [Green Version]
- Zaidi, S.; Brueckner, M. Genetics and Genomics of Congenital Heart Disease. Circ. Res. 2017, 120, 923–940. [Google Scholar] [CrossRef]
- Costain, G.; Silversides, C.K.; Bassett, A.S. The importance of copy number variation in congenital heart disease. NPJ Genomic Med. 2016, 1, 16031. [Google Scholar] [CrossRef] [Green Version]
- Krishnan, A.; Samtani, R.; Dhanantwari, P.; Lee, E.; Yamada, S.; Shiota, K.; Donofrio, M.T.; Leatherbury, L.; Lo, C.W. A detailed comparison of mouse and human cardiac development. Pediatr. Res. 2014, 76, 500–507. [Google Scholar] [CrossRef] [Green Version]
- Botto, L.D.; Lin, A.E.; Riehle-Colarusso, T.; Malik, S.; Correa, A. Seeking causes: Classifying and evaluating congenital heart defects in etiologic studies. Birth Defects Res. Part Clin. Mol. Teratol. 2007, 79, 714–727. [Google Scholar] [CrossRef]
- Jin, S.C.; Homsy, J.; Zaidi, S.; Lu, Q.; Morton, S.; Depalma, S.R.; Zeng, X.; Qi, H.; Chang, W.; Sierant, M.C.; et al. Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat. Genet. 2017, 49, 1593–1601. [Google Scholar] [CrossRef] [Green Version]
- Houyel, L.; Khoshnood, B.; Anderson, R.H.; Lelong, N.; Thieulin, A.-C.; Goffinet, F.; Bonnet, D. Population-based evaluation of a suggested anatomic and clinical classification of congenital heart defects based on the International Paediatric and Congenital Cardiac Code. Orphanet. J. Rare Dis. 2011, 6, 64. [Google Scholar] [CrossRef] [PubMed]
- Ellesøe, S.G.; Workman, C.T.; Bouvagnet, P.; Loffredo, C.A.; McBride, K.L.; Hinton, R.B.; van Engelen, K.; Gertsen, E.C.; Mulder, B.J.M.; Postma, A.V.; et al. Familial co-occurrence of congenital heart defects follows distinct patterns. Eur. Heart, J. 2018, 39, 1015–1022. [Google Scholar] [CrossRef] [PubMed]
- Brodwall, K.; Greve, G.; Leirgul, E.; Tell, G.S.; Vollset, S.E.; Øyen, N. Recurrence of congenital heart defects among siblings—A nationwide study. Am. J. Med. Genet. A 2017, 173, 1575–1585. [Google Scholar] [CrossRef] [PubMed]
- Øyen, N.; Poulsen, G.; Boyd, H.A.; Wohlfahrt, J.; Jensen, P.K.A.; Melbye, M. Recurrence of congenital heart defects in families. Circulation 2009, 120, 295–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Chen, S.; Zühlke, L.; Black, G.C.; Choy, M.; Li, N.; Keavney, B.D. Global birth prevalence of congenital heart defects 1970–2017: Updated systematic review and meta-analysis of 260 studies. Int. J. Epidemiol. 2019, 48, 455–463. [Google Scholar] [CrossRef]
- Sifrim, A.; Hitz, M.-P.; Wilsdon, A.; Breckpot, J.; Al Turki, S.H.; Thienpont, B.; McRae, J.; Fitzgerald, T.W.; Singh, T.; Swaminathan, G.J.; et al. Distinct genetic architectures for syndromic and nonsyndromic congenital heart defects identified by exome sequencing. Nat. Genet. 2016, 48, 1–9. [Google Scholar] [CrossRef]
- Brade, T.; Pane, L.S.; Moretti, A.; Chien, K.R.; Laugwitz, K.-L. Embryonic Heart Progenitors and Cardiogenesis. Cold Spring Harb. Perspect. Med. 2013, 3, 13847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sylva, M.; Van den Hoff, M.J.B.; Moorman, A.F.M. Development of the human heart. Am. J. Med. Genet. A 2014, 164, 1347–1371. [Google Scholar] [CrossRef] [PubMed]
- Gittenberger-De Groot, A.C.; Bartelings, M.M.; Deruiter, M.C.; Poelmann, R.E. Basics of cardiac development for the understanding of congenital heart malformations. Pediatr. Res. 2005, 57, 169–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.J.; Lin, C.Y.; Chen, C.H.; Zhou, B.; Chang, C.P. Partitioning the heart: Mechanisms of cardiac septation and valve development. Dev. Camb. 2012, 139, 3277–3299. [Google Scholar] [CrossRef] [Green Version]
- Kodo, K.; Takahashi, T.; Nakanishi, T.; Yamagishi, H.; Makino, S.; Fukuda, K.; Oda, M.; Nishizawa, T.; Furutani, M.; Arai, S.; et al. Genetic analysis of essential cardiac transcription factors in 256 patients with non-syndromic congenital heart defects. Circ. J. 2012, 76, 1703–1711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granados-Riveron, J.T.; Pope, M.; Bu’Lock, F.A.; Thornborough, C.; Eason, J.; Setchfield, K.; Ketley, A.; Kirk, E.P.; Fatkin, D.; Feneley, M.P.; et al. Combined mutation screening of NKX2-5, GATA4, and TBX5 in congenital heart disease: Multiple heterozygosity and novel mutations. Congenit. Heart Dis. 2012, 7, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Glessner, J.T.; Bick, A.G.; Ito, K.; Homsy, J.G.; Rodriguez-Murillo, L.; Fromer, M.; Mazaika, E.; Vardarajan, B.; Italia, M.; Leipzig, J.; et al. Increased Frequency of De Novo Copy Number Variants in Congenital Heart Disease by Integrative Analysis of Single Nucleotide Polymorphism Array and Exome Sequence Data. Circ. Res. 2014, 115, 884–896. [Google Scholar] [CrossRef] [PubMed]
- Prendiville, T.; Jay, P.Y.; Pu, W.T. Insights into the genetic structure of congenital heart disease from human and murine studies on monogenic disorders. Cold Spring Harb. Perspect. Med. 2014, 4, 13946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schott, J.-J.; Benson, D.W.; Basson, C.T.; Pease, W.; Silberbach, G.M.; Moak, J.P.; Maron, B.J.; Seidman, C.E.; Seidman, J.G. Congenital Heart Disease Caused by Mutations in the Transcription Factor NKX2-5. Science 1998, 281, 108–111. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Roelens, I.; Sluysmans, T.; Gewillig, M.; Devriendt, K.; Vikkula, M. Progressive AV-block and anomalous venous return among cardiac anomalies associated with two novel missense mutations in the CSX/NKX2-5 Gene. Hum. Mutat. 2002, 20, 75–76. [Google Scholar] [CrossRef]
- Winston, J.B.; Erlich, J.M.; Green, C.A.; Aluko, A.; Kaiser, K.A.; Takematsu, M.; Barlow, R.S.; Sureka, A.O.; Lapage, M.J.; Janss, L.L.; et al. Heterogeneity of genetic modifiers ensures normal cardiac development. Circulation 2010, 121, 1313–1321. [Google Scholar] [CrossRef] [Green Version]
- Gifford, C.A.; Ranade, S.S.; Samarakoon, R.; Salunga, H.T.; de Soysa, T.Y.; Huang, Y.; Zhou, P.; Elfenbein, A.; Wyman, S.K.; Bui, Y.K.; et al. Oligogenic inheritance of congenital heart disease involving a NKX2-5 modifier. Science 2019, 364, 865–870. [Google Scholar] [CrossRef]
- Zakariyah, A.F.; Rajgara, R.F.; Horner, E.; Cattin, M.E.; Blais, A.; Skerjanc, I.S.; Burgon, P.G. In Vitro Modeling of Congenital Heart Defects Associated with an NKX2-5 Mutation Revealed a Dysregulation in BMP/Notch-Mediated Signaling. Stem Cells 2018, 36, 514–526. [Google Scholar] [CrossRef] [Green Version]
- Pikkarainen, S.; Tokola, H.; Kerkelä, R.; Ruskoaho, H. GATA transcription factors in the developing and adult heart. Cardiovasc. Res. 2004, 63, 196–207. [Google Scholar] [CrossRef]
- Li, R.G.; Xu, Y.J.; Wang, J.; Liu, X.Y.; Yuan, F.; Huang, R.T.; Xue, S.; Li, L.; Liu, H.; Li, Y.J.; et al. GATA4 Loss-of-Function Mutation and the Congenitally Bicuspid Aortic Valve. Am. J. Cardiol. 2018, 121, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Zhou, L.; Wang, Q.; You, X.; Li, Y.; Zhao, Y.; Han, X.; Chang, Z.; He, X.; Cheng, C.; et al. NEXN inhibits GATA4 and leads to atrial septal defects in mice and humans. Cardiovasc. Res. 2014, 103, 228–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, R.; Watt, A.J.; Battle, M.A.; Li, J.; Bondow, B.J.; Duncan, S.A. Loss of both GATA4 and GATA6 blocks cardiac myocyte differentiation and results in acardia in mice. Dev. Biol. 2008, 317, 614–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, B.; Zhou, W.; Jiao, J.; Nielsen, J.B.; Mathis, M.R.; Heydarpour, M.; Lettre, G.; Folkersen, L.; Prakash, S.; Schurmann, C.; et al. Protein-altering and regulatory genetic variants near GATA4 implicated in bicuspid aortic valve. Nat. Commun. 2017, 8, 15481. [Google Scholar] [CrossRef] [Green Version]
- Kodo, K.; Nishizawa, T.; Furutani, M.; Arai, S.; Yamamura, E.; Joo, K.; Takahashi, T.; Matsuoka, R.; Yamagishi, H. GATA6 mutations cause human cardiac outflow tract defects by disrupting semaphorin-plexin signaling. Proc. Natl. Acad. Sci. USA 2009, 106, 13933–13938. [Google Scholar] [CrossRef] [Green Version]
- Maitra, M.; Koenig, S.N.; Srivastava, D.; Garg, V. Identification of GATA6 sequence variants in patients with congenital heart defects. Pediatr. Res. 2010, 68, 281–285. [Google Scholar] [CrossRef] [Green Version]
- Bonachea, E.M.; Zender, G.; White, P.; Corsmeier, D.; Newsom, D.; Fitzgerald-Butt, S.; Garg, V.; McBride, K.L. Use of a targeted, combinatorial next-generation sequencing approach for the study of bicuspid aortic valve. BMC Med. Genomics 2014, 7, 56. [Google Scholar] [CrossRef] [Green Version]
- Laforest, B.; Andelfinger, G.; Nemer, M. Loss of Gata5 in mice leads to bicuspid aortic valve. J. Clin. Invest. 2011, 121, 2876–2887. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Fulcoli, F.G.; Ferrentino, R.; Martucciello, S.; Illingworth, E.A.; Baldini, A. Transcriptional Control in Cardiac Progenitors: Tbx1 Interacts with the BAF Chromatin Remodeling Complex and Regulates Wnt5a. PLoS Genet. 2012, 8, e1002571. [Google Scholar] [CrossRef] [Green Version]
- Gao, W.; Higaki, T.; Eguchi-Ishimae, M.; Iwabuki, H.; Wu, Z.; Yamamoto, E.; Takata, H.; Ohta, M.; Imoto, I.; Ishii, E.; et al. DGCR6 at the proximal part of the DiGeorge critical region is involved in conotruncal heart defects. Hum. Genome Var. 2015, 2, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Greulich, F.; Rudat, C.; Kispert, A. Mechanisms of T-box gene function in the developing heart. Cardiovasc. Res. 2011, 91, 212–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reamon-Buettner, S.M.; Borlak, J. TBX5 mutations in non-Holt-Oram syndrome (HOS) malformed hearts. Hum. Mutat. 2004, 24, 104. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H. Forkhead box transcription factors in embryonic heart development and congenital heart disease. Life Sci. 2016, 144, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Stankiewicz, P.; Sen, P.; Bhatt, S.S.; Storer, M.; Xia, Z.; Bejjani, B.A.; Ou, Z.; Wiszniewska, J.; Driscoll, D.J.; Bolivar, J.; et al. Genomic and Genic Deletions of the FOX Gene Cluster on 16q24.1 and Inactivating Mutations of FOXF1 Cause Alveolar Capillary Dysplasia and Other Malformations. Am. J. Hum. Genet. 2009, 84, 780–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgenthau, A.; Frishman, W.H. Genetic Origins of Tetralogy of Fallot. Cardiol. Rev. 2018, 26, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Hilger, A.C.; Halbritter, J.; Pennimpede, T.; van der Ven, A.; Sarma, G.; Braun, D.A.; Porath, J.D.; Kohl, S.; Hwang, D.Y.; Dworschak, G.C.; et al. Targeted Resequencing of 29 Candidate Genes and Mouse Expression Studies Implicate ZIC3 and FOXF1 in Human VATER/VACTERL Association. Hum. Mutat. 2015, 36, 1150–1154. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Liu, S.; Chen, W.; Yuan, Y.; Gu, R.; Song, Y.; Li, J.; Cao, Y.; Lin, Y.; Xu, J.; et al. A novel ZIC3 gene mutation identified in patients with heterotaxy and congenital heart disease. Sci. Rep. 2018, 8, 12386. [Google Scholar] [CrossRef]
- Wang, B.; Yan, J.; Mi, R.; Zhou, S.; Xie, X.; Wang, J.; Ma, X. Forkhead box H1 (FOXH1) sequence variants in ventricular septal defect. Int. J. Cardiol. 2010, 145, 83–85. [Google Scholar] [CrossRef]
- De Luca, A.; Sarkozy, A.; Consoli, F.; Ferese, R.; Guida, V.; Dentici, M.L.; Mingarelli, R.; Bellacchio, E.; Tuo, G.; Limongelli, G.; et al. Familial transposition of the great arteries caused by multiple mutations in laterality genes. Heart 2010, 96, 673–677. [Google Scholar] [CrossRef]
- Priest, J.R.; Osoegawa, K.; Mohammed, N.; Nanda, V.; Kundu, R.; Schultz, K.; Lammer, E.J.; Girirajan, S.; Scheetz, T.; Waggott, D.; et al. De Novo and Rare Variants at Multiple Loci Support the Oligogenic Origins of Atrioventricular Septal Heart Defects. PLoS Genet. 2016, 12, e1005963. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Cheng, C.-M.; Lanz, R.B.; Wang, T.; Respress, J.L.; Ather, S.; Chen, W.; Tsai, S.-J.; Wehrens, X.H.T.; Tsai, M.-J.; et al. Atrial Identity Is Determined by a COUP-TFII Regulatory Network. Dev. Cell 2013, 25, 417–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiao, X.-H.; Wang, Q.; Wang, J.; Liu, X.-Y.; Xu, Y.-J.; Huang, R.-T.; Xue, S.; Li, Y.-J.; Zhang, M.; Qu, X.-K.; et al. A novel NR2F2 loss-of-function mutation predisposes to congenital heart defect. Eur. J. Med. Gene. 2017, 61, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Al Turki, S.; Manickaraj, A.K.; Mercer, C.L.; Gerety, S.S.; Hitz, M.-P.; Lindsay, S.; D’Alessandro, L.C.A.; Swaminathan, G.J.; Bentham, J.; Arndt, A.-K.; et al. Rare Variants in NR2F2 Cause Congenital Heart Defects in Humans. Am. J. Hum. Genet. 2014, 94, 574–585. [Google Scholar] [CrossRef] [Green Version]
- McFadden, D.G.; Barbosa, A.C.; Richardson, J.A.; Schneider, M.D.; Srivastava, D.; Olson, E.N. The Hand1 and Hand2 transcription factors regulate expansion of the embryonic cardiac ventricles in a gene dosage-dependent manner. Dev. Camb. Engl. 2005, 132, 189–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Firulli, B.A.; Toolan, K.P.; Harkin, J.; Millar, H.; Pineda, S.; Firulli, A.B. The HAND1 frameshift A126FS mutation does not cause hypoplastic left heart syndrome in mice. Cardiovasc. Res. 2017, 113, 1732–1742. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.M.; Wang, J.; Qiu, X.B.; Yuan, F.; Li, R.G.; Xu, Y.J.; Qu, X.K.; Shi, H.Y.; Hou, X.M.; Huang, R.T.; et al. A HAND2 loss-of-function mutation causes familial ventricular septal defect and pulmonary stenosis. G3 Genes Genomes Genet. 2016, 6, 987–992. [Google Scholar] [CrossRef] [Green Version]
- McCulley, D.J.; Black, B.L. Transcription factor pathways and congenital heart disease. Curr. Top. Dev. Biol. 2012, 100, 253–277. [Google Scholar]
- Zhu, X.; Deng, X.; Huang, G.; Wang, J.; Yang, J. A Novel Mutation of Hyaluronan Synthase 2 Gene in Chinese Children with Ventricular Septal Defect. PLoS ONE 2014, 9, 87437. [Google Scholar] [CrossRef] [Green Version]
- Neeb, Z.; Lajiness, J.D.; Bolanis, E.; Conway, S.J. Cardiac outflow tract anomalies. Wiley Interdiscip. Rev. Dev. Biol. 2013, 2, 499–530. [Google Scholar] [CrossRef] [Green Version]
- Andersen, T.A.; Troelsen, K.; Troelsen, K.d.L.; Larsen, L.A. Of mice and men: Molecular genetics of congenital heart disease. Cell. Mol. Life Sci. 2014, 71, 1327–1352. [Google Scholar] [CrossRef] [Green Version]
- Lalani, S.R.; Belmont, J.W. Genetic basis of congenital cardiovascular malformations. Eur. J. Med. Genet. 2014, 57, 402–413. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.-P.; Bruneau, B.G. Epigenetics and Cardiovascular Development. Annu. Rev. Physiol. 2012, 74, 41–68. [Google Scholar] [CrossRef]
- Azhar, M.; Ware, S.M. Genetic and Developmental Basis of Cardiovascular Malformations. Clin. Perinatol. 2016, 43, 39–53. [Google Scholar] [CrossRef] [Green Version]
- Fahed, A.C.; Gelb, B.D.; Seidman, J.G.; Seidman, C.E. Genetics of Congenital Heart Disease: The Glass Half Empty. Circ. Res. 2013, 112, 707–720. [Google Scholar] [CrossRef] [Green Version]
- Sutherland, M.J.; Ware, S.M. Disorders of left–right asymmetry: Heterotaxy and situs inversus. Am. J. Med. Genet. C Semin. Med. Genet. 2009, 151, 307–317. [Google Scholar] [CrossRef]
- Lebo, M.S.; Baxter, S.M. New molecular genetic tests in the diagnosis of heart disease. Clin. Lab. Med. 2014, 34, 137–156. [Google Scholar] [CrossRef]
- Reuter, M.S.; Jobling, R.; Chaturvedi, R.R.; Manshaei, R.; Costain, G.; Heung, T.; Curtis, M.; Hosseini, S.M.; Liston, E.; Lowther, C.; et al. Haploinsufficiency of vascular endothelial growth factor related signaling genes is associated with tetralogy of Fallot. Genet. Med. 2019, 21, 1001–1007. [Google Scholar] [CrossRef] [Green Version]
- Muntean, I.; Togănel, R.; Benedek, T. Genetics of Congenital Heart Disease: Past and Present. Biochem. Genet. 2017, 55, 105–123. [Google Scholar] [CrossRef]
- Girdauskas, E.; Geist, L.; Disha, K.; Kazakbaev, I.; Groß, T.; Schulz, S.; Ungelenk, M.; Kuntze, T.; Reichenspurner, H.; Kurth, I. Genetic abnormalities in bicuspid aortic valve root phenotype: Preliminary results. Eur. J. Cardiothorac. Surg. 2017, 52, 156–162. [Google Scholar] [CrossRef] [Green Version]
- Finsterer, J.; Stöllberger, C.; Towbin, J.A. Left ventricular noncompaction cardiomyopathy: Cardiac, neuromuscular, and genetic factors. Nat. Rev. Cardiol. 2017, 14, 224–237. [Google Scholar] [CrossRef]
- Wessels, M.; Willems, P. Genetic factors in non-syndromic congenital heart malformations. Clin. Genet. 2010, 78, 103–123. [Google Scholar] [CrossRef]
- Togi, K.; Kawamoto, T.; Yamauchi, R.; Yoshida, Y.; Kita, T.; Tanaka, M. Role of Hand1/eHAND in the dorso-ventral patterning and interventricular septum formation in the embryonic heart. Mol. Cell. Biol. 2004, 24, 4627–4635. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Wang, J.; Liu, X.Y.; Liu, H.; Shi, H.Y.; Yang, X.X.; Li, N.; Li, Y.J.; Huang, R.T.; Xue, S.; et al. HAND1 loss-of-function mutation contributes to congenital double outlet right ventricle. Int. J. Mol. Med. 2017, 39, 711–718. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, D.; Olson, E.N. A genetic blueprint for cardiac development. Nature 2000, 407, 221. [Google Scholar] [CrossRef]
- Li, A.H.; Hanchard, N.A.; Furthner, D.; Fernbach, S.; Azamian, M.; Nicosia, A.; Rosenfeld, J.; Muzny, D.; D’Alessandro, L.C.A.; Morris, S.; et al. Whole exome sequencing in 342 congenital cardiac left sided lesion cases reveals extensive genetic heterogeneity and complex inheritance patterns. Genome Med. 2017, 9, 95. [Google Scholar] [CrossRef]
- Silversides, C.K.; Lionel, A.C.; Costain, G.; Merico, D.; Migita, O.; Liu, B.; Yuen, T.; Rickaby, J.; Thiruvahindrapuram, B.; Marshall, C.R.; et al. Rare Copy Number Variations in Adults with Tetralogy of Fallot Implicate Novel Risk Gene Pathways. PLoS Genet. 2012, 8, e1002843. [Google Scholar] [CrossRef]
- Lincoln, J.; Garg, V. Etiology of valvular heart disease-genetic and developmental origins. Circ. J. Off. J. Jpn. Circ. Soc. 2014, 78, 1801–1807. [Google Scholar]
- MacGrogan, D.; Luxán, G.; de la Pompa, J.L. Genetic and functional genomics approaches targeting the Notch pathway in cardiac development and congenital heart disease. Brief Funct. Genomics 2014, 13, 15–27. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Yagi, H.; Saeed, S.; Bais, A.S.; Gabriel, G.C.; Chen, Z.; Peterson, K.A.; Li, Y.; Schwartz, M.C.; Reynolds, W.T.; et al. The complex genetics of hypoplastic left heart syndrome. Nat. Genet. 2017, 49, 1152–1159. [Google Scholar] [CrossRef]
- Reiter, J.F.; Leroux, M.R. Genes and molecular pathways underpinning ciliopathies. Nat. Rev. Mol. Cell Biol. 2017, 18, 533–547. [Google Scholar] [CrossRef]
- Xie, H.M.; Werner, P.; Stambolian, D.; Bailey-Wilson, J.E.; Hakonarson, H.; White, P.S.; Taylor, D.M.; Goldmuntz, E. Rare copy number variants in patients with congenital conotruncal heart defects. Birth Defects Res. 2017, 109, 271–295. [Google Scholar] [CrossRef] [Green Version]
- Warburton, D.; Ronemus, M.; Kline, J.; Jobanputra, V.; Williams, I.; Anyane-Yeboa, K.; Chung, W.; Yu, L.; Wong, N.; Awad, D.; et al. The contribution of de novo and rare inherited copy number changes to congenital heart disease in an unselected sample of children with conotruncal defects or hypoplastic left heart disease. Hum. Genet. 2014, 133, 11–27. [Google Scholar] [CrossRef] [Green Version]
- Cast, A.E.; Gao, C.; Amack, J.D.; Ware, S.M. An essential and highly conserved role for Zic3 in left–right patterning, gastrulation and convergent extension morphogenesis. Dev. Biol. 2012, 364, 22–31. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Liu, L.; Zhou, J.; Wang, C. Heterogeneity Analysis and Diagnosis of Complex Diseases Based on Deep Learning Method. Sci. Rep. 2018, 8, 6155. [Google Scholar] [CrossRef]
- Zhao, C.M.; Peng, L.Y.; Li, L.; Liu, X.Y.; Wang, J.; Zhang, X.L.; Yuan, F.; Li, R.G.; Qiu, X.B.; Yang, Y.Q. PITX2 loss-of-function mutation contributes to congenital endocardial cushion defect and Axenfeld–Rieger syndrome. PLoS ONE 2015, 10, e0124409. [Google Scholar] [CrossRef] [Green Version]
- MacGrogan, D.; Münch, J.; de la Pompa, J.L. Notch and interacting signalling pathways in cardiac development, disease, and regeneration. Nat. Rev. Cardiol. 2018, 15, 685–704. [Google Scholar] [CrossRef]
- Timmerman, L.A.; Grego-Bessa, J.; Raya, A.; Bertrán, E.; Pérez-Pomares, J.M.; Díez, J.; Aranda, S.; Palomo, S.; McCormick, F.; Izpisúa-Belmonte, J.C.; et al. Notch promotes epithelial-mesenchymal transition during cardiac development and oncogenic transformation. Genes Dev. 2004, 18, 99–115. [Google Scholar] [CrossRef] [Green Version]
- Blue, G.M.; Kirk, E.P.; Giannoulatou, E.; Dunwoodie, S.L.; Ho, J.W.K.; Hilton, D.C.K.; White, S.M.; Sholler, G.F.; Harvey, R.P.; Winlaw, D.S. Targeted Next-Generation Sequencing Identifies Pathogenic Variants in Familial Congenital Heart Disease. J. Am. Coll. Cardiol. 2014, 64, 2498–2506. [Google Scholar] [CrossRef] [Green Version]
- Kerstjens-Frederikse, W.S.; Van De Laar, I.M.B.H.; Vos, Y.J.; Verhagen, J.M.A.; Berger, R.M.F.; Lichtenbelt, K.D.; Klein Wassink-Ruiter, J.S.; Van Der Zwaag, P.A.; Du Marchie Sarvaas, G.J.; Bergman, K.A.; et al. Cardiovascular malformations caused by NOTCH1 mutations do not keep left: Data on 428 probands with left-sided CHD and their families. Genet. Med. 2016, 18, 914–923. [Google Scholar] [CrossRef]
- Helle, E.; Córdova-Palomera, A.; Ojala, T.; Saha, P.; Potiny, P.; Gustafsson, S.; Ingelsson, E.; Bamshad, M.; Nickerson, D.; Chong, J.X.; et al. Loss of function, missense, and intronic variants in NOTCH1 confer different risks for left ventricular outflow tract obstructive heart defects in two European cohorts. Genet. Epidemiol. 2019, 43, 215–226. [Google Scholar] [CrossRef]
- Iascone, M.; Ciccone, R.; Galletti, L.; Marchetti, D.; Seddio, F.; Lincesso, A.R.; Pezzoli, L.; Vetro, A.; Barachetti, D.; Boni, L.; et al. Identification of de novo mutations and rare variants in hypoplastic left heart syndrome. Clin. Genet. 2012, 81, 542–554. [Google Scholar] [CrossRef]
- Zahavich, L.; Bowdin, S.; Mital, S. Use of Clinical Exome Sequencing in Isolated Congenital Heart Disease. Circ. Cardiovasc. Genet. 2017, 10, e001581. [Google Scholar] [CrossRef] [Green Version]
- Preuss, C.; Capredon, M.; Wunnemann, F.; Chetaille, P.; Prince, A.; Godard, B.; Leclerc, S.; Sobreira, N.; Ling, H.; Awadalla, P.; et al. Family Based Whole Exome Sequencing Reveals the Multifaceted Role of Notch Signaling in Congenital Heart Disease. PLoS Genet. 2016, 12, e1006335. [Google Scholar] [CrossRef]
- Li, B.; Yu, L.; Liu, D.; Yang, X.; Zheng, Y.; Gui, Y.; Wang, H. MIB1 mutations reduce Notch signaling activation and contribute to congenital heart disease. Clin. Sci. Lond. Engl. 1979 2018, 132, 2483–2491. [Google Scholar] [CrossRef] [Green Version]
- Mommersteeg, M.T.M.; Yeh, M.L.; Parnavelas, J.G.; Andrews, W.D. Disrupted Slit-Robo signalling results in membranous ventricular septum defects and bicuspid aortic valves. Cardiovasc. Res. 2015, 106, 55–66. [Google Scholar] [CrossRef] [Green Version]
- Rochais, F.; Mesbah, K.; Kelly, R.G. Signaling pathways controlling second heart field development. Circ. Res. 2009, 104, 933–942. [Google Scholar] [CrossRef] [Green Version]
- Homsy, J.; Zaidi, S.; Shen, Y.; Ware, J.S.; Samocha, K.E.; Karczewski, K.J.; DePalma, S.R.; McKean, D.; Wakimoto, H.; Gorham, J.; et al. De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science 2015, 350, 1262–1266. [Google Scholar] [CrossRef] [Green Version]
- Ye, B.; Hou, N.; Xiao, L.; Xu, Y.; Boyer, J.; Xu, H.; Li, F. APC controls asymmetric Wnt/β-catenin signaling and cardiomyocyte proliferation gradient in the heart. J. Mol. Cell. Cardiol. 2015, 89, 287–296. [Google Scholar] [CrossRef] [Green Version]
- Cantù, C.; Felker, A.; Zimmerli, D.; Prummel, K.D.; Cabello, E.M.; Chiavacci, E.; Méndez-Acevedo, K.M.; Kirchgeorg, L.; Burger, S.; Ripoll, J.; et al. Mutations in Bcl9 and Pygo genes cause congenital heart defects by tissue-specific perturbation of Wnt/β-catenin signaling. Genes Dev. 2018, 32, 1443–1458. [Google Scholar] [CrossRef] [Green Version]
- Phillips, M.D.; Mukhopadhyay, M.; Poscablo, C.; Westphal, H. Dkk1 and Dkk2 regulate epicardial specification during mouse heart development. Int. J. Cardiol. 2011, 150, 186–192. [Google Scholar] [CrossRef] [Green Version]
- May-Simera, H.L.; Kelley, M.W. Cilia, Wnt signaling, and the cytoskeleton. Cilia 2012, 1, 7. [Google Scholar] [CrossRef] [Green Version]
- Caron, A.; Xu, X.; Lin, X. Wnt/β-catenin signaling directly regulates Foxj1 expression and ciliogenesis in zebrafish Kupffer’s vesicle. Development 2012, 139, 514–524. [Google Scholar] [CrossRef] [Green Version]
- Merks, A.M.; Swinarski, M.; Meyer, A.M.; Müller, N.V.; Özcan, I.; Donat, S.; Burger, A.; Gilbert, S.; Mosimann, C.; Abdelilah-Seyfried, S.; et al. Planar cell polarity signalling coordinates heart tube remodelling through tissue-scale polarisation of actomyosin activity. Nat. Commun. 2018, 9, 2161. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Shi, S.; Acosta, L.; Li, W.; Lu, J.; Bao, S.; Chen, Z.; Yang, Z.; Schneider, M.D.; Chien, K.R.; et al. BMP10 is essential for maintaining cardiac growth during murine cardiogenesis. Development 2004, 131, 2219–2231. [Google Scholar] [CrossRef] [Green Version]
- Qian, B.; Mo, R.; Da, M.; Peng, W.; Hu, Y.; Mo, X. Common variations in BMP4 confer genetic susceptibility to sporadic congenital heart disease in a Han Chinese population. Pediatr. Cardiol. 2014, 35, 1442–1447. [Google Scholar] [CrossRef] [Green Version]
- Tan, H.L.; Glen, E.; Töpf, A.; Hall, D.; O’Sullivan, J.J.; Sneddon, L.; Wren, C.; Avery, P.; Lewis, R.J.; ten Dijke, P.; et al. Nonsynonymous variants in the SMAD6 gene predispose to congenital cardiovascular malformation. Hum. Mutat. 2012, 33, 720–727. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.; Essalmani, R.; Szumska, D.; Creemers, J.W.M.; Roebroek, A.J.M.; D’Orleans-Juste, P.; Bhattacharya, S.; Seidah, N.G.; Prat, A. Loss of Endothelial Furin Leads to Cardiac Malformation and Early Postnatal Death. Mol. Cell. Biol. 2012, 32, 3382–3391. [Google Scholar] [CrossRef] [Green Version]
- Zaidi, S.; Choi, M.; Wakimoto, H.; Ma, L.; Jiang, J.; Overton, J.D.; Romano-Adesman, A.; Bjornson, R.D.; Breitbart, R.E.; Brown, K.K.; et al. De novo mutations in histone-modifying genes in congenital heart disease. Nature 2013, 498, 220–223. [Google Scholar] [CrossRef] [Green Version]
- Rahman, M.S.; Akhtar, N.; Jamil, H.M.; Banik, R.S.; Asaduzzaman, S.M. TGF-β/BMP signaling and other molecular events: Regulation of osteoblastogenesis and bone formation. Bone Res. 2015, 3, 15005. [Google Scholar] [CrossRef] [Green Version]
- Granadillo, J.L.; Chung, W.K.; Hecht, L.; Corsten-Janssen, N.; Wegner, D.; Nij Bijvank, S.W.A.; Toler, T.L.; Pineda-Alvarez, D.E.; Douglas, G.; Murphy, J.J.; et al. Variable cardiovascular phenotypes associated with SMAD2 pathogenic variants. Hum. Mutat. 2018, 39, 1875–1884. [Google Scholar] [CrossRef]
- Tian, E.; Stevens, S.R.; Guan, Y.; Springer, D.A.; Anderson, S.A.; Starost, M.F.; Patel, V.; Hagen, K.G.T.; Tabak, L.A. Galnt1 is required for normal heart valve development and cardiac function. PLoS ONE 2015, 10, e0115861. [Google Scholar] [CrossRef]
- Dykes, I.M.; Van Bueren, K.L.; Ashmore, R.J.; Floss, T.; Wurst, W.; Szumska, D.; Bhattacharya, S.; Scambler, P.J. HIC2 is a novel dosage-dependent regulator of cardiac development located within the distal 22q11 deletion syndrome region. Circ. Res. 2014, 115, 23–31. [Google Scholar] [CrossRef] [Green Version]
- Dyer, L.A.; Kirby, M.L. Sonic hedgehog maintains proliferation in secondary heart field progenitors and is required for normal arterial pole formation. Dev. Biol. 2009, 330, 305–317. [Google Scholar] [CrossRef] [Green Version]
- Washington Smoak, I.; Byrd, N.A.; Abu-Issa, R.; Goddeeris, M.M.; Anderson, R.; Morris, J.; Yamamura, K.; Klingensmith, J.; Meyers, E.N. Sonic hedgehog is required for cardiac outflow tract and neural crest cell development. Dev. Biol. 2005, 283, 357–372. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Cheng, H.; Xiang, M.; Zhou, L.; Wu, B.; Moskowitz, I.P.; Zhang, K.; Xie, L. Gata4 regulates hedgehog signaling and Gata6 expression for outflow tract development. PLoS Genet. 2019, 15, e1007711. [Google Scholar] [CrossRef] [Green Version]
- Dyer, L.A.; Makadia, F.A.; Scott, A.; Pegram, K.; Hutson, M.R.; Kirby, M.L. BMP signaling modulates hedgehog-induced secondary heart field proliferation. Dev. Biol. 2010, 348, 167–176. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Wang, J.; Guo, C.; Chang, W.; Zhuang, J.; Zhu, P.; Li, X. Temporally Distinct Six2-Positive Second Heart Field Progenitors Regulate Mammalian Heart Development and Disease. Cell Rep. 2017, 18, 1019–1032. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Alpert, D.; Francis, R.; Chatterjee, B.; Yu, Q.; Tansey, T.; Sabol, S.L.; Cui, C.; Bai, Y.; Koriabine, M.; et al. Massively parallel sequencing identifies the gene Megf8 with ENU-induced mutation causing heterotaxy. Proc. Natl. Acad. Sci. USA 2009, 106, 3219–3224. [Google Scholar] [CrossRef] [Green Version]
- Pusapati, G.V.; Kong, J.H.; Patel, B.B.; Krishnan, A.; Sagner, A.; Kinnebrew, M.; Briscoe, J.; Aravind, L.; Rohatgi, R. CRISPR Screens Uncover Genes that Regulate Target Cell Sensitivity to the Morphogen Sonic Hedgehog. Dev. Cell 2018, 44, 113–129.e8. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Cao, R.; Xu, Y.; Li, T.; Li, F.; Chen, S.; Xu, R.; Sun, K. Rare copy number variants analysis identifies novel candidate genes in heterotaxy syndrome patients with congenital heart defects. Genome Med. 2018, 10, 40. [Google Scholar] [CrossRef] [Green Version]
- Higgins, E.M.; Bos, J.M.; Mason-Suares, H.; Tester, D.J.; Ackerman, J.P.; MacRae, C.A.; Sol-Church, K.; Gripp, K.W.; Urrutia, R.; Ackerman, M.J. Elucidation of MRAS-mediated Noonan syndrome with cardiac hypertrophy. JCI Insight 2017, 2, 91225. [Google Scholar] [CrossRef]
- ELM Vissers, L.; Bonetti, M.; Paardekooper Overman, J.; Nillesen, W.M.; Frints, S.G.; de Ligt, J.; Zampino, G.; Justino, A.; Machado, J.C.; Schepens, M.; et al. Heterozygous germline mutations in A2ML1 are associated with a disorder clinically related to Noonan syndrome. Eur. J. Hum. gene. 2014, 23, 317. [Google Scholar] [CrossRef]
- Ackerman, C.; Locke, A.E.; Feingold, E.; Reshey, B.; Espana, K.; Thusberg, J.; Mooney, S.; Bean, L.J.H.; Dooley, K.J.; Cua, C.L.; et al. An excess of deleterious variants in VEGF-A pathway genes in down-syndrome-associated atrioventricular septal defects. Am. J. Hum. Genet. 2012, 91, 646–659. [Google Scholar] [CrossRef] [Green Version]
- Bjornsson, T.; Thorolfsdottir, R.B.; Sveinbjornsson, G.; Sulem, P.; Norddahl, G.L.; Helgadottir, A.; Gretarsdottir, S.; Magnusdottir, A.; Danielsen, R.; Sigurdsson, E.L.; et al. A rare missense mutation in MYH6 associates with non-syndromic coarctation of the aorta. Eur. Heart J. 2018, 39, 3243–3249. [Google Scholar] [CrossRef]
- England, J.; Granados-Riveron, J.; Polo-Parada, L.; Kuriakose, D.; Moore, C.; Brook, J.D.; Rutland, C.S.; Setchfield, K.; Gell, C.; Ghosh, T.K.; et al. Tropomyosin 1: Multiple roles in the developing heart and in the formation of congenital heart defects. J. Mol. Cell. Cardiol. 2017, 106, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.K.; Qiu, G.R.; Li-Ling, J.; Xin, N.; Sun, K.L. Reduced ACTC1 expression might play a role in the onset of congenital heart disease by inducing cardiomyocyte apoptosis. Circ. J. 2010, 74, 2410–2418. [Google Scholar] [CrossRef] [Green Version]
- Matsson, H.; Eason, J.; Bookwalter, C.S.; Klar, J.; Gustavsson, P.; Sunnegårdh, J.; Enell, H.; Jonzon, A.; Vikkula, M.; Gutierrez, I.; et al. Alpha-cardiac actin mutations produce atrial septal defects. Hum. Mol. Genet. 2008, 17, 256–265. [Google Scholar] [CrossRef]
- Rexiati, M.; Sun, M.; Guo, W. Muscle-Specific Mis-Splicing and Heart Disease Exemplified by RBM20. Genes 2018, 9, 18. [Google Scholar] [CrossRef] [Green Version]
- van den Hoogenhof, M.M.G.; Beqqali, A.; Amin, A.S.; van der Made, I.; Aufiero, S.; Khan, M.A.F.; Schumacher, C.A.; Jansweijer, J.A.; van Spaendonck-Zwarts, K.Y.; Remme, C.A.; et al. RBM20 Mutations Induce an Arrhythmogenic Dilated Cardiomyopathy Related to Disturbed Calcium Handling. Circulation 2018, 138, 1330–1342. [Google Scholar] [CrossRef]
- Mo, F.-E.; Lau, L.F. The Matricellular Protein CCN1 Is Essential for Cardiac Development. Circ. Res. 2006, 99, 961–969. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Li, Y.; He, X.; Shao, X.; Yang, F.; Zhao, M.; Wu, C.; Zhang, C.; Zhou, L. Mutational and functional analysis of the BVES gene coding region in Chinese patients with non-syndromic tetralogy of Fallot. Int. J. Mol. Med. 2013, 31, 899–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reamon-Buettner, S.M.; Borlak, J. HEY2 mutations in malformed hearts. Hum. Mutat. 2006, 27, 118. [Google Scholar] [CrossRef] [PubMed]
- Aburawi, E.H.; Aburawi, H.E.; Bagnall, K.M.; Bhuiyan, Z.A. Molecular insight into heart development and congenital heart disease: An update review from the Arab countries. Trends Cardiovasc. Med. 2015, 25, 291–301. [Google Scholar] [CrossRef]
- Chaix, M.A. Genetic testing in congenital heart disease: A clinical approach. World J. Cardiol. 2016, 8, 180. [Google Scholar] [CrossRef]
- Hassed, S.; Li, S.; Mulvihill, J.; Aston, C.; Palmer, S. Adams-Oliver syndrome review of the literature: Refining the diagnostic phenotype. Am. J. Med. Genet. A 2017, 173, 790–800. [Google Scholar] [CrossRef]
- Durst, R.; Sauls, K.; Peal, D.S.; deVlaming, A.; Toomer, K.; Leyne, M.; Salani, M.; Talkowski, M.E.; Brand, H.; Perrocheau, M.; et al. Mutations in DCHS1 cause mitral valve prolapse. Nature 2015, 525, 109–113. [Google Scholar] [CrossRef]
- Grunert, M.; Dorn, C.; Schueler, M.; Dunkel, I.; Schlesinger, J.; Mebus, S.; Alexi-Meskishvili, V.; Perrot, A.; Wassilew, K.; Timmermann, B.; et al. Rare and private variations in neural crest, apoptosis and sarcomere genes define the polygenic background of isolated Tetralogy of Fallot. Hum. Mol. Genet. 2014, 23, 3115–3128. [Google Scholar] [CrossRef] [Green Version]
- Richards, A.A.; Garg, V. Genetics of Congenital Heart Disease. Curr. Cardiol. Rev. 2010, 6, 91–97. [Google Scholar] [CrossRef] [Green Version]
- Li, A.H.; Hanchard, N.A.; Azamian, M.; D’Alessandro, L.C.A.; Coban-Akdemir, Z.; Lopez, K.N.; Hall, N.J.; Dickerson, H.; Nicosia, A.; Fernbach, S.; et al. Genetic architecture of laterality defects revealed by whole exome sequencing. Eur. J. Hum. Genet. 2019, 27, 563. [Google Scholar] [CrossRef]
- Molck, M.C.; Simioni, M.; Paiva Vieira, T.; Sgardioli, I.C.; Paoli Monteiro, F.; Souza, J.; Fett-Conte, A.C.; Félix, T.M.; Lopes Monlléo, I.; Gil-da-Silva-Lopes, V.L. Genomic imbalances in syndromic congenital heart disease. J. Pediatr. 2017, 93, 497–507. [Google Scholar] [CrossRef]
- D’Alessandro, L.C.A.; Al Turki, S.; Manickaraj, A.K.; Manase, D.; Mulder, B.J.M.; Bergin, L.; Rosenberg, H.C.; Mondal, T.; Gordon, E.; Lougheed, J.; et al. Exome sequencing identifies rare variants in multiple genes in atrioventricular septal defect. Genet. Med. 2016, 18, 189–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, H.; Xia, H.; Deng, S. Genetic basis of human left–right asymmetry disorders. Expert Rev. Mol. Med. 2014, 16, 19. [Google Scholar] [CrossRef] [PubMed]
- Obler, D.; Juraszek, A.L.; Smoot, L.B.; Natowicz, M.R. Double outlet right ventricle: Aetiologies and associations. J. Med. Genet. 2008, 45, 481–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaHaye, S.; Lincoln, J.; Garg, V. Genetics of valvular heart disease. Curr. Cardiol. Rep. 2014, 16, 487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattassi, R.; Manara, E.; Colombo, P.G.; Manara, S.; Porcella, A.; Bruno, G.; Bruson, A.; Bertelli, M. Variant discovery in patients with Mendelian vascular anomalies by next-generation sequencing and their use in patient clinical management. J. Vasc. Surg. 2018, 67, 922–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, J.; Guo, J.; Huang, L.; Wu, Q.; Yin, K.; Wang, L.; Zhang, T.; Quan, L.; Zhao, Q.; Cheng, J. Genetic diagnosis of acute aortic dissection in South China Han population using next-generation sequencing. Int. J. Legal Med. 2018, 132, 1273–1280. [Google Scholar] [CrossRef]
- Pierpont, M.E.; Basson, C.T.; Benson, D.W.; Gelb, B.D.; Giglia, T.M.; Goldmuntz, E.; McGee, G.; Sable, C.A.; Srivastava, D.; Webb, C.L.; et al. Genetic basis for congenital heart defects: Current knowledge: A scientific statement from the American Heart Association Congenital Cardiac Defects Committee, Council on Cardiovascular Disease in the Young: Endorsed by the American Academy of Pediatrics. Circulation 2007, 115, 3015–3038. [Google Scholar] [CrossRef] [Green Version]
- Jindal, G.A.; Goyal, Y.; Burdine, R.D.; Rauen, K.A.; Shvartsman, S.Y. RASopathies: Unraveling mechanisms with animal models. DMM Dis. Models Mech. 2015, 8, 769–782. [Google Scholar] [CrossRef] [Green Version]
- Aoki, Y.; Niihori, T.; Inoue, S.; Matsubara, Y. Recent advances in RASopathies. J. Hum. Genet. 2016, 61, 33–39. [Google Scholar] [CrossRef]
- Araki, T.; Chan, G.; Newbigging, S.; Morikawa, L.; Bronson, R.; Neel, B.G. Noonan syndrome cardiac defects are caused by PTPN11 acting in endocardium to enhance endocardial-mesenchymal transformation. Proc. Natl. Acad. Sci. USA 2009, 106, 4736–4741. [Google Scholar] [CrossRef] [Green Version]
- Bruneau, B.G. The developmental genetics of congenital heart disease. Nature 2008, 451, 943–948. [Google Scholar] [CrossRef] [PubMed]
- Ang, S.Y.; Uebersohn, A.; Ian Spencer, C.; Huang, Y.; Lee, J.E.; Ge, K.; Bruneau, B.G. KMT2D regulates specific programs in heart development via histone H3 lysine 4 di-methylation. Dev. Camb. 2016, 143, 810–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, R.T.; Xue, S.; Wang, J.; Gu, J.Y.; Xu, J.H.; Li, Y.J.; Li, N.; Yang, X.X.; Liu, H.; Zhang, X.D.; et al. CASZ1 loss-of-function mutation associated with congenital heart disease. Gene 2016, 595, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, R.L.; Davis, C.A.; Potthoff, M.J.; Haberland, M.; Fielitz, J.; Qi, X.; Hill, J.A.; Richardson, J.A.; Olson, E.N. Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes Dev. 2007, 21, 1790–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, D.; Wang, H.; Hao, L.; Guo, X.; Ma, X.; Qian, Y.; Chen, H.; Ma, J.; Zhang, J.; Sheng, W.; et al. The roles of SMYD4 in epigenetic regulation of cardiac development in zebrafish. PLOS Genet. 2018, 14, e1007578. [Google Scholar] [CrossRef] [Green Version]
- Yagi, H.; Liu, X.Q.; Gabriel, G.C.; Wu, Y.; Peterson, K.; Murray, S.A.; Bruce, A.; Martin, L.J.; Benson, D.W. The Genetic Landscape of Hypoplastic Left Heart Syndrome. Pediatr. Cardiol. 2018, 39, 1069–1081. [Google Scholar] [CrossRef]
- von Gise, A.; Pu, W.T. Endocardial and epicardial epithelial to mesenchymal transitions in heart development and disease. Circ. Res. 2012, 110, 1628–1645. [Google Scholar] [CrossRef]
- Koefoed, K.; Veland, I.R.; Bang, L.; Lars, P.; Larsen, A.; Christensen, S.T. Cilia and coordination of signaling networks during heart development. Organogenesis 2014, 10, 108–125. [Google Scholar] [CrossRef] [Green Version]
- Burnicka-Turek, O.; Steimle, J.D.; Huang, W.; Felker, L.; Kamp, A.; Kweon, J.; Peterson, M.; Reeves, R.H.; Maslen, C.L.; Gruber, P.J.; et al. Cilia gene mutations cause atrioventricular septal defects by multiple mechanisms. Hum. Mol. Genet. 2016, 25, 3011–3028. [Google Scholar] [CrossRef]
- Willaredt, M.A.; Gorgas, K.; Gardner, H.A.R.; Tucker, K.L. Multiple essential roles for primary cilia in heart development. Cilia 2012, 1, 23. [Google Scholar] [CrossRef] [Green Version]
- Clement, C.A.; Kristensen, S.G.; Møllgård, K.; Pazour, G.J.; Yoder, B.K.; Larsen, L.A.; Christensen, S.T. The primary cilium coordinates early cardiogenesis and hedgehog signaling in cardiomyocyte differentiation. J. Cell Sci. 2009, 122, 3070–3082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toomer, K.A.; Fulmer, D.; Guo, L.; Drohan, A.; Peterson, N.; Swanson, P.; Brooks, B.; Mukherjee, R.; Body, S.; Lipschutz, J.H.; et al. A role for primary cilia in aortic valve development and disease. Dev. Dyn. 2017, 246, 625–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slough, J.; Cooney, L.; Brueckner, M. Monocilia in the embryonic mouse heart suggest a direct role for cilia in cardiac morphogenesis. Dev. Dyn. 2008, 237, 2304–2314. [Google Scholar] [CrossRef] [PubMed]
- Fung, A.; Manlhiot, C.; Naik, S.; Rosenberg, H.; Smythe, J.; Lougheed, J.; Mondal, T.; Chitayat, D.; McCrindle, B.W.; Mital, S. Impact of Prenatal Risk Factors on Congenital Heart Disease in the Current Era. J. Am. Heart Assoc. 2013, 2, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Zhu, C.; Sun, M.; Maimaiti, R.; Ford, S.P.; Nathanielsz, P.W.; Ren, J.; Guo, W. Maternal obesity impairs fetal cardiomyocyte contractile function in sheep. FASEB J. 2018, 33, 2587–2598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Chen, X.; Chen, H.; Ma, Z.; Zhou, J. Maternal Alcohol Consumption before and during Pregnancy and the Risks of Congenital Heart Defects in Offspring: A Systematic Review and Meta-analysis. Congenit. Heart Dis. 2015, 10, 216–224. [Google Scholar] [CrossRef]
- Ye, Z.; Wang, L.; Yang, T.; Chen, L.; Wang, T.; Chen, L.; Zhao, L.; Zhang, S.; Zheng, Z.; Luo, L.; et al. Maternal Viral Infection and Risk of Fetal Congenital Heart Diseases: A Meta-Analysis of Observational Studies. J. Am. Heart Assoc. 2019, 8, 11264. [Google Scholar]
- Courtney, J.A.; Cnota, J.F.; Jones, H.N. The Role of Abnormal Placentation in Congenital Heart Disease; Cause, Correlate, or Consequence? Front. Physiol. 2018, 9, 1045. [Google Scholar] [CrossRef]
- Cole, C.R.; Yutzey, K.E.; Brar, A.K.; Goessling, L.S.; VanVickle-Chavez, S.J.; Cunningham, M.W.; Eghtesady, P. Congenital Heart Disease Linked to Maternal Autoimmunity against Cardiac Myosin. J. Immunol. 2014, 192, 4074–4082. [Google Scholar] [CrossRef] [Green Version]
- Mao, B.; Qiu, J.; Zhao, N.; Shao, Y.; Dai, W.; He, X.; Cui, H.; Lin, X.; Lv, L.; Tang, Z.; et al. Maternal folic acid supplementation and dietary folate intake and congenital heart defects. PLoS ONE 2017, 12, e0187996. [Google Scholar] [CrossRef]
- Coppedè, F. The genetics of folate metabolism and maternal risk of birth of a child with Down syndrome and associated congenital heart defects. Front. Genet. 2015, 6, 223. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Human Phenotype | Mouse Phenotype | References |
|---|---|---|---|
| CITED2 | AS, PS, SIT, Dextrocardia, TGA, TOF, RVOTO, TAPVR, ASD, VSD | DORV, PTA, OA, AA, PAA anomaly, ASD, VSD | [20,57,58,59] |
| CREBBP | Rubinstein-Taybi syndrome | CHD | [60,61] |
| EP300 | Rubinstein-Taybi syndrome | Hypotrabeculation, Thin myocardium, ASD, VSD | [60,62] |
| ETS1 | DORV, HLHS, ASD, VSD | ASD, VSD | [20,63] |
| FOXC1 | HLHS, OA, PA, PAH, PDA, Bilateral SVC, VSD, Axenfeld–Rieger syndrome, ASD | Aortic arch defects, IAA, Inflow tract defects, OFT defects, RV defects, Semilunar valve defects, VSD | [20,43,57,61] |
| FOXC2 | HLHS, TOF, OA, PA, PDA, PAH, TAPVR, Bilateral SVC, ASD, VSD | Aortic arch defects, IAA, Inflow tract defects, OFT defects, PTA, RV defects, Semilunar valve defects, VSD | [20,43,57,61] |
| FOXH1 | TOF, TGA, HTX, VSD | Disorganized myocardium, OFT defects, RV defects | [43,60,64,65,66] |
| FOXJ1 | CHD | Complex CHD with HTX | [5,44] |
| FOXO1 | TOF | Endocardial cushion defects, Reduced trabeculations | [43,67] |
| FOXP1 | CHD | Defects in ventricular/OFT septation, valve formation, myocardial proliferation | [43,60] |
| GATA4 | Dextrocardia, AVSD, DORV, TOF, BAV, CoA, AR, PAPVR, PDA, PS, ASD, VSD | Acardia, Cardia bifida, AVSD, DORV, PTA, ASD, VSD | [20,57,63,64] |
| GATA5 | AVSD, DORV, LVNC, BAV, CoA | BAV | [20,68,69,70] |
| GATA6 | AVSD, TOF, PDA, PTA, PS, ASD, VSD | Acardia, AVSD, DORV, PTA, IAA, PAA anomaly, ASD, VSD | [33,57,63,64] |
| HAND1 | AVSD, DORV, HLHS, HLV, HRV, ASD, VSD | Arrest at looping stage, VSD and hypoplastic AV valves, Absent ventricular septum and thin compact myocardium | [54,55,57,71,72,73] |
| HAND2 | TOF, LVNC, VSD | DORV, HRV, PAA anomaly, PS, VSD | [20,59,64,70,74] |
| JARID2 | Left-sided lesions | DORV, Hypertrabeculation, Myocardial defects, Noncompaction, VSD | [20,62,75] |
| MSX1 | BAV, CoA | DORV, TOF, PTA, Hypoplastic valves, VSD | [20,37,59] |
| NFATC1 | TOF, LVNC, BAV, CoA, TA, VSD | Absent valves, Blunting of AV/OFT valves, VSD | [20,37,57,70,76] |
| NKX2-5 | ASD, AVSD, BAV, CoA, Dextrocardia, DORV, Ebstein’s anomaly, HTX, HLHS, IAA, LVNC, Mitral valve anomalies, PA, PAPVR, PDA, PS, SVAS, TA, TAPVR, TGA, TOF, PTA, VSD | AVSD, Looping defect, ASD, VSD | [25,57,61,63,64,74] |
| NR1D2 | AVSD | AVSD | [50] |
| NR2F2 | AVSD, DORV with VSD | Hypoplastic atria, Ventricularized atria | [51,52,53,74] |
| RBPJ | HLHS | Defective EMT, Hypoplastic endocardial cushions, Impaired trabeculation, VSD | [20,77,78,79] |
| RFX3 | PTA | HTX | [80,81] |
| SMAD6 | HLHS, AS, BAV, CoA | DORV, TGA, PTA, IAA, RAA, Hypoplastic pulmonary artery, Aortic valve dysplasia, Hyperplastic valves, VSD | [5,24,64,79] |
| TBX1 | DORV, TOF, IAA, PTA, VSD, DiGeorge syndrome, Velocardiofacial syndrome | AVSD, DORV, TGA, TOF, PTA, PAA anomaly, VSD | [20,57,60,63,64] |
| TBX2 | CHD | DORV, Hypoplastic endocardial cushions, PAA anomaly | [20,60] |
| TBX20 | DORV, HLV, LVNC, DCM, CoA, MS, PDA, ASD, VSD | AVSD, DORV, PTA, Hypoplastic right heart, ASD, VSD | [20,59,63,70,71] |
| TBX3 | Ulnar-Mammary syndrome | DORV, TGA, PAA anomaly, VSD | [61,80] |
| TBX5 | AVSD, TOF, BAV, CoA, ASD, VSD, Holt-Oram syndrome | ASD, VSD | [37,45,57,61,63] |
| ZFPM2 | AVSD, DORV, TOF, VSD | Alignment defects, Coronary artery defects, OA, PS, TA, ASD, VSD | [20,50,64,74,81] |
| Gene | Human Phenotype | Mouse Phenotype | References |
|---|---|---|---|
| Notch Signaling | |||
| ADAM17 | AVSD | CHD | [50] |
| HES1 | TGA | OA, PAA anomalies, VSD | [59,78,81] |
| HEY2 | AVSD | TOF, HRV, OA, TA, PS, Thickened mitral valve, ASD, VSD | [20,78,132] |
| JAG1 | Aortic dextroposition, TOF, BAV, CoA, PS, VSD, Alagille syndrome | DORV, PTA, TOF, IAA, OA, AAAD, PS, Thickened or calcified valves, ASD, VSD | [20,37,57,61,74,78,133] |
| NOTCH1 | HTX, AVSD, TOF, HLHS, LVNC, BAV, CoA, AS, MS, VSD | Aberrant trabeculation, DORV, HRV, Hypoplastic endocardial cushions, Impaired EMT, IAA, PAA anomalies, PS, PTA, TA, Valve defects, ASD, VSD | [10,20,24,37,50,61,63,64,66,71,77,78,134,135] |
| NOTCH2 | AVSD, TOF, BAV, CoA, PS, Alagille syndrome | PS, Reduced compact myocardium, ASD, VSD | [20,37,50,61,68,78,133] |
| WNT/β-Catenin Signaling | |||
| APC | BAV, CoA | Ventricular hyperplasia | [37,98] |
| BCL9 | CHD | Septal defects, Valve defects | [99,108] |
| DCHS1 | LVNC, Mitral valve prolapse | Prolapsed, thickened mitral leaflets | [70,136] |
| DVL1 | LVNC, PDA | CHD | [60,64] |
| EDN1 | TOF | DORV, PTA, PAA anomaly, VSD | [20,74,137] |
| PCDHA9 | HLHS | HLHS, BAV, Aortic hypoplasia/stenosis | [79] |
| TGF-β/BMP/Nodal Signaling | |||
| ACVR1 | HTX, AVSD, DORV, TGA, Left-sided lesions, ASD | PTA, PAA anomaly, ASD, VSD | [20,71,75,138] |
| ACVR2B | HTX, Dextrocardia, AVSD, DORV, TGA, HLHS, LSVC, PS, Venous anomaly | HTX, TGA, DORV, AA | [59,61,64,66,139] |
| BMPR1A | AVSD | Hypoplastic endocardial cushion, Impaired EMT, PTA, ASD, VSD | [20,77,140,141] |
| BMPR2 | AVSD, PDA, PAPVR, ASD, VSD | Absent OFT valves, AV cushion defect, DORV, PTA, IAA, OA, Thickened valve leaflets, ASD, VSD | [59,61,91,138] |
| GDF1 | HTX, AVSD, DORV, TGA, TOF | HTX, DORV, TGA, TOF | [10,59,63,71,142] |
| SMAD6 | HLHS, AS, BAV, CoA | DORV, TGA, PTA, IAA, RAA, Hypoplastic pulmonary artery, Aortic valve dysplasia, Hyperplastic valves, VSD | [5,20,24,64,79,134] |
| TGFB2 | VSD, Loeys-Dietz syndrome | DORV, DILV, PTA, Hypoplastic endocardial cushions, Hypoplastic aortic arch, OA, PAA anomaly, TAAD, BAV, Abnormal AV valves, Hyperplastic valves, VSD | [20,59,63,77,81,143] |
| TGFB3 | Loeys-Dietz syndrome | VSD | [20,63] |
| TGFBR1 | BAV, Myxomatous mitral valve, TAAD, Loeys-Dietz syndrome, Marfan syndrome | Hypoplastic endocardial cushions, PTA, PAA anomaly, VSD | [20,61,63,66,69,138,144,145] |
| TGFBR2 | HTX, Mitral valve prolapse, Myxomatous mitral valve, TAAD, Loeys-Dietz syndrome, Marfan syndrome | DORV, PTA, OA, PAA anomaly, Tricuspid valve defect, ASD, VSD | [20,61,63,66,138,142,144,145,146,147] |
| RAS/MAPK Signaling | |||
| BRAF | Cardiofaciocutaneous syndrome, Costello syndrome, LEOPARD syndrome, Noonan syndrome | Cardiac defects modeling cardiofaciocutaneous syndrome | [61,63,68,133,148,149] |
| PTPN11 | AVSD, CoA, AS, PS, Cardiofaciocutaneous syndrome, Costello syndrome, LEOPARD syndrome, Noonan syndrome | AVSD, DORV, PTA, Valve defects, ASD, VSD | [20,60,61,63,68,134,149,150,151] |
| SOS1 | AVSD, PS, Cardiofaciocutaneous syndrome, Costello syndrome, LEOPARD syndrome, Noonan syndrome | Valve defects | [60,61,63,64,68,108,134,144,148] |
| VEGF Signaling | |||
| ETS1 | DORV, HLHS, ASD, VSD | ASD, VSD | [20,57,63,81] |
| VEGFA | TOF, PDA, PTA, AS, BAV, CoA, IAA, VSD | EMT defects, DORV, TOF, Blunted AV valves, VSD | [20,24,64,71] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Williams, K.; Carson, J.; Lo, C. Genetics of Congenital Heart Disease. Biomolecules 2019, 9, 879. https://doi.org/10.3390/biom9120879
Williams K, Carson J, Lo C. Genetics of Congenital Heart Disease. Biomolecules. 2019; 9(12):879. https://doi.org/10.3390/biom9120879
Chicago/Turabian StyleWilliams, Kylia, Jason Carson, and Cecilia Lo. 2019. "Genetics of Congenital Heart Disease" Biomolecules 9, no. 12: 879. https://doi.org/10.3390/biom9120879
APA StyleWilliams, K., Carson, J., & Lo, C. (2019). Genetics of Congenital Heart Disease. Biomolecules, 9(12), 879. https://doi.org/10.3390/biom9120879