The Plasma Factor XIII Heterotetrameric Complex Structure: Unexpected Unequal Pairing within a Symmetric Complex

 , ,
, , 

Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
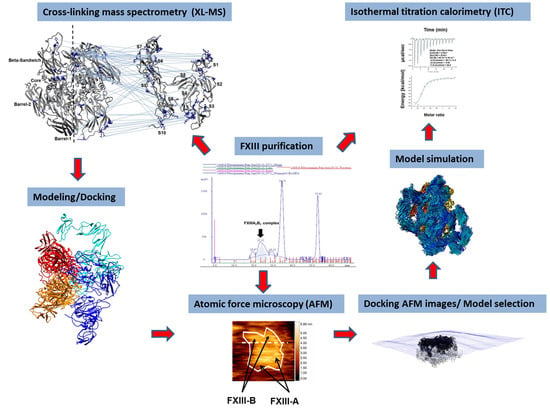
2. Materials and Methods
2.1. FXIII-A and FXIII-B Subunit, Cloning Expression, and Purification
2.2. Purification of FXIII-A2B2 Complex
2.3. Atomic Force Microscopy (AFM) of FXIII
2.4. XL-MS of FXIII-A2B2 Heterotetramer Complex
2.5. Generation of the FXIII-B Subunit Model
2.6. Generation of the FXIII-A2B2 All-Atom Model
2.7. Molecular Dynamics Simulations of the FXIII-A2B2 Heterotetramer Models
2.8. Modeling Transition States between the First FXIII-A2: FXIII-B2 Contacts and the Final FXIII-A2B2 Complex
2.9. Fitting and Docking Atomic Protein Structures on AFM Surface Topographs
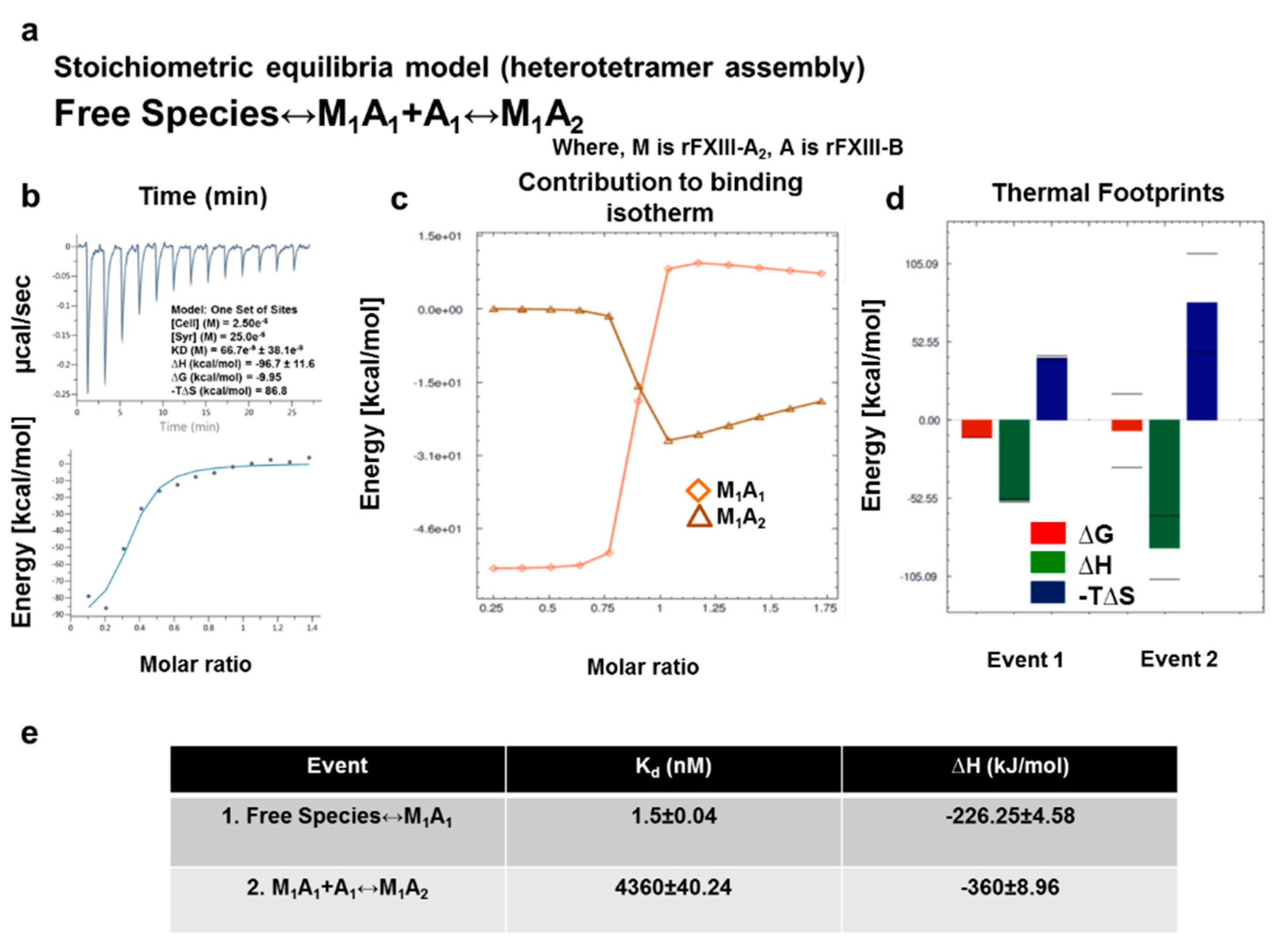
2.10. ITC-Based Thermodynamic Profiling of the Assembly and Dissociation of the FXIII-A2B2 Heterotetramer Complex
3. Results
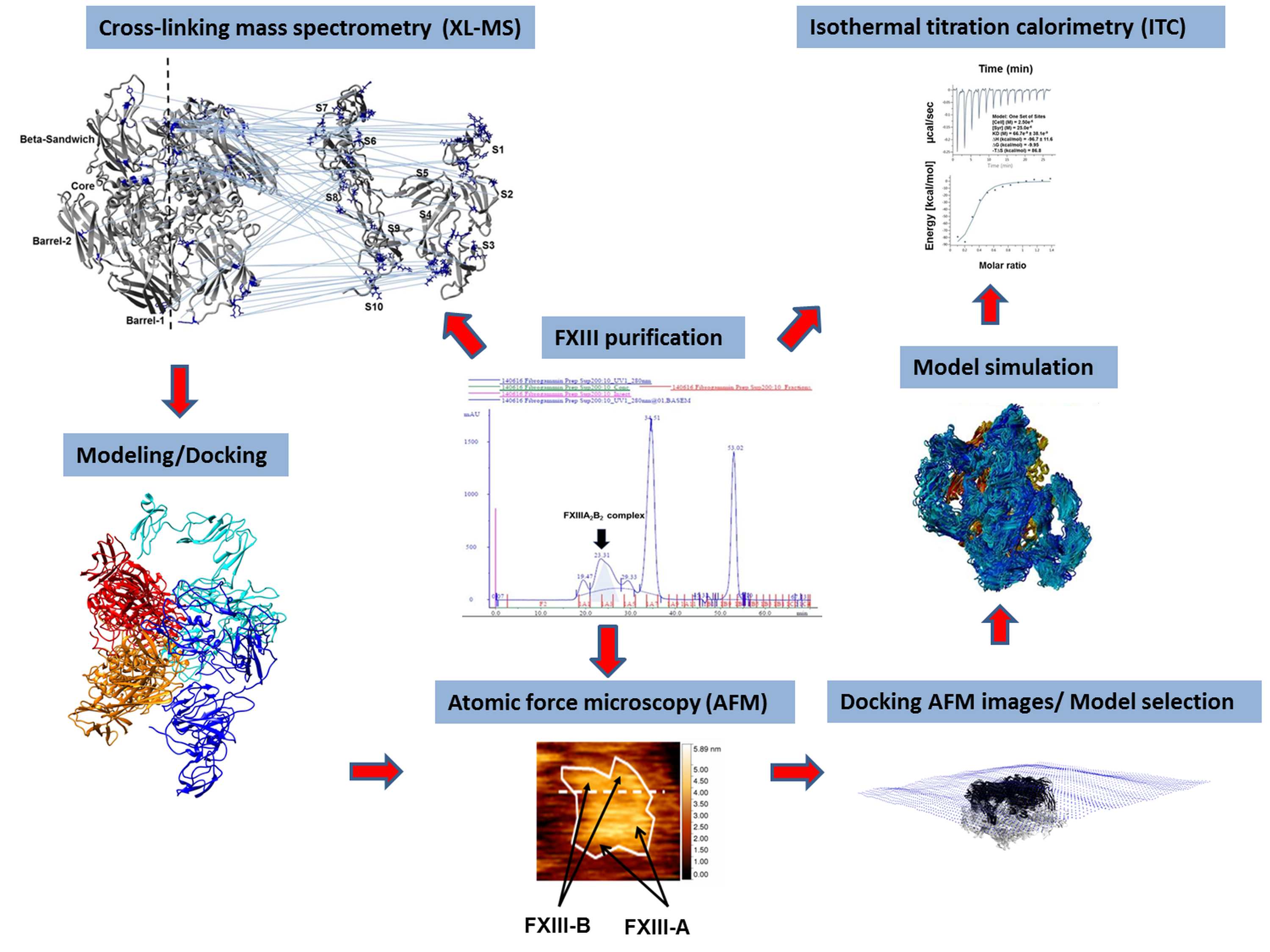
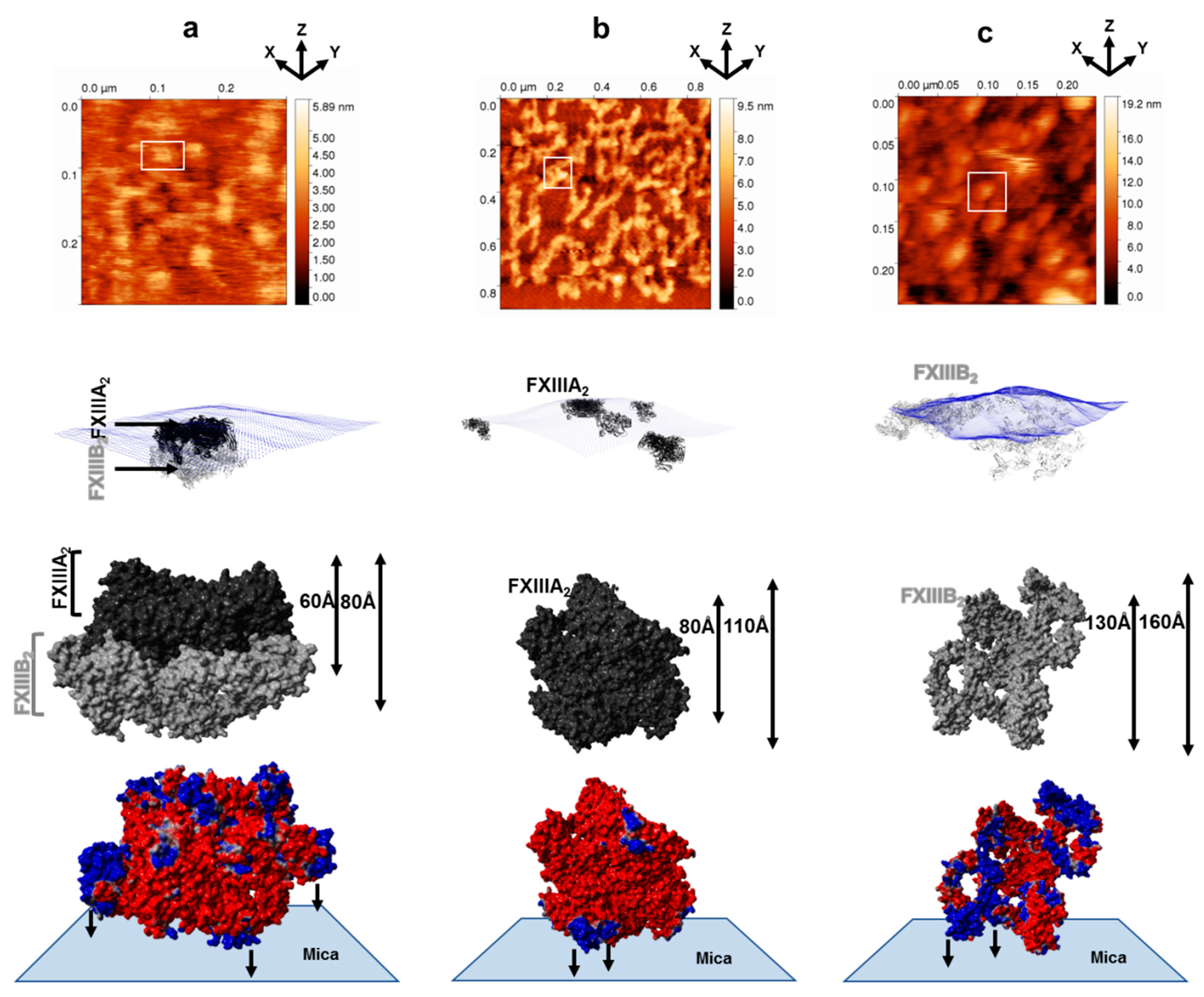
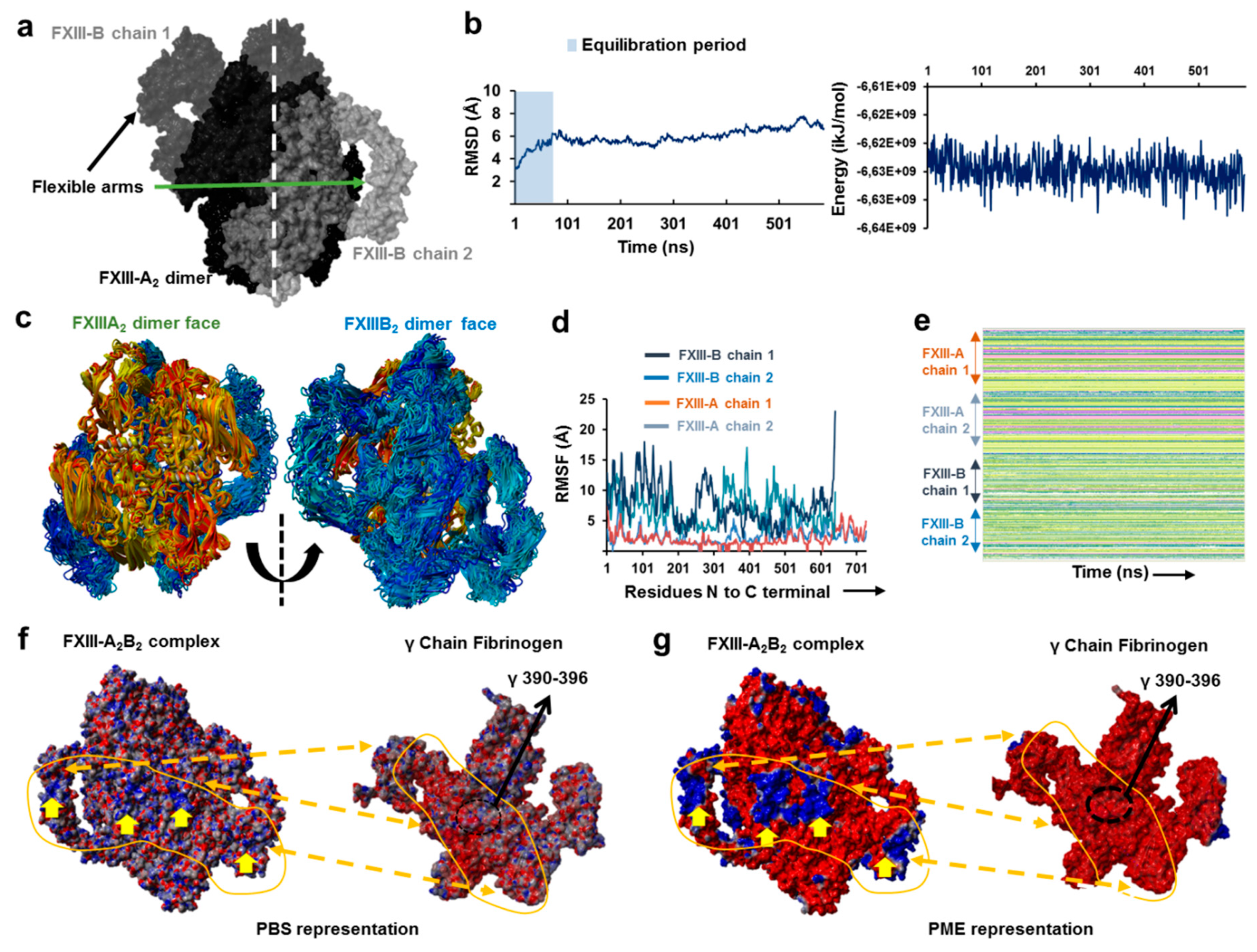
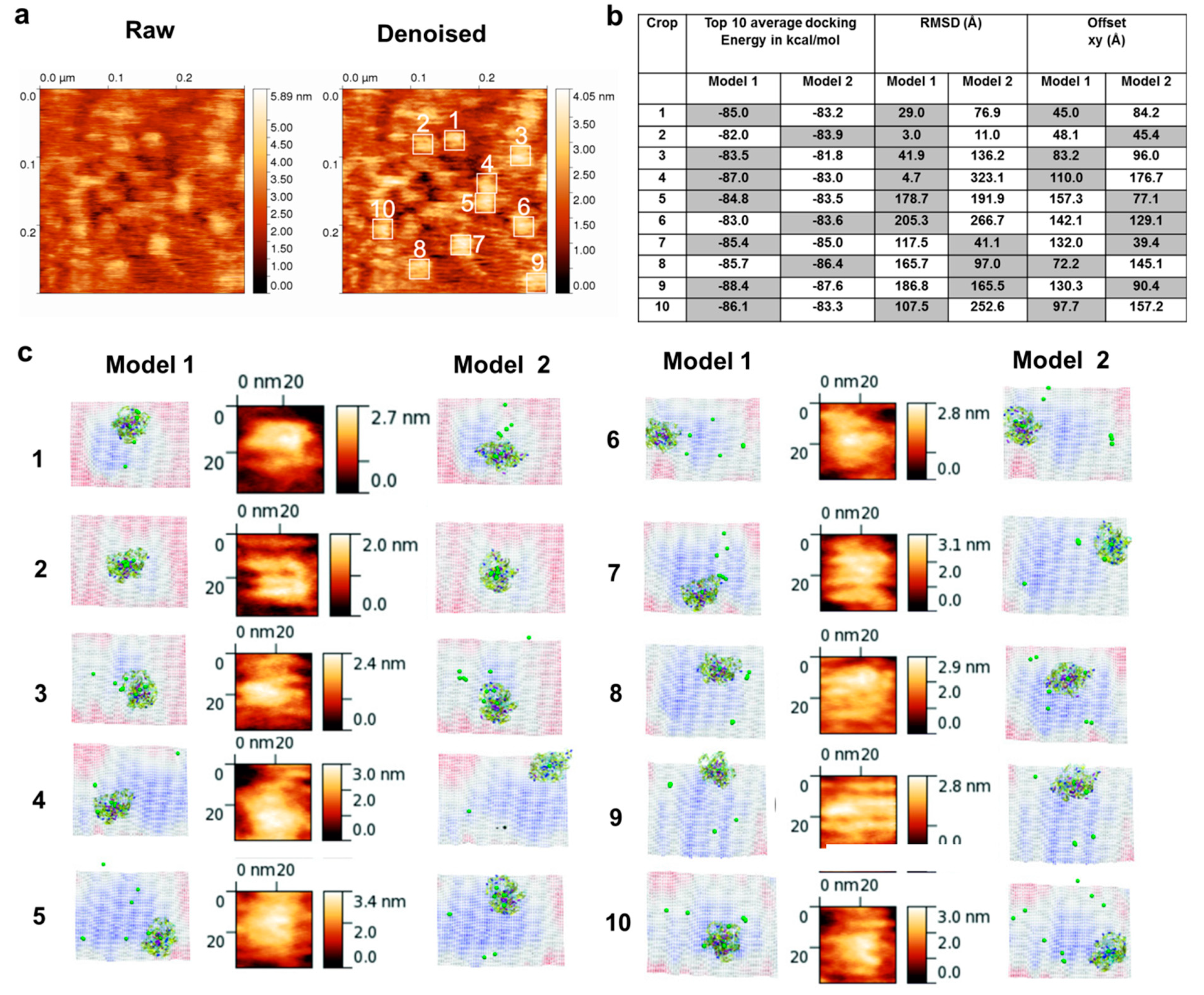
3.1. AFM Topographs Indicates Complex Formation Restricts the Conformational Flexibility of FXIII-B
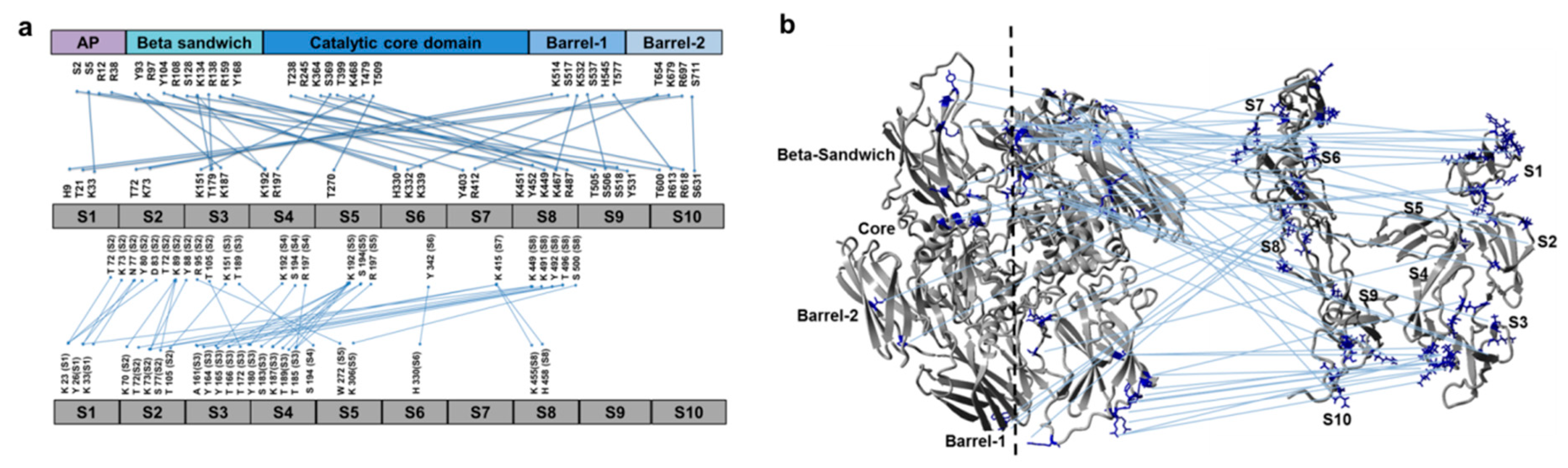
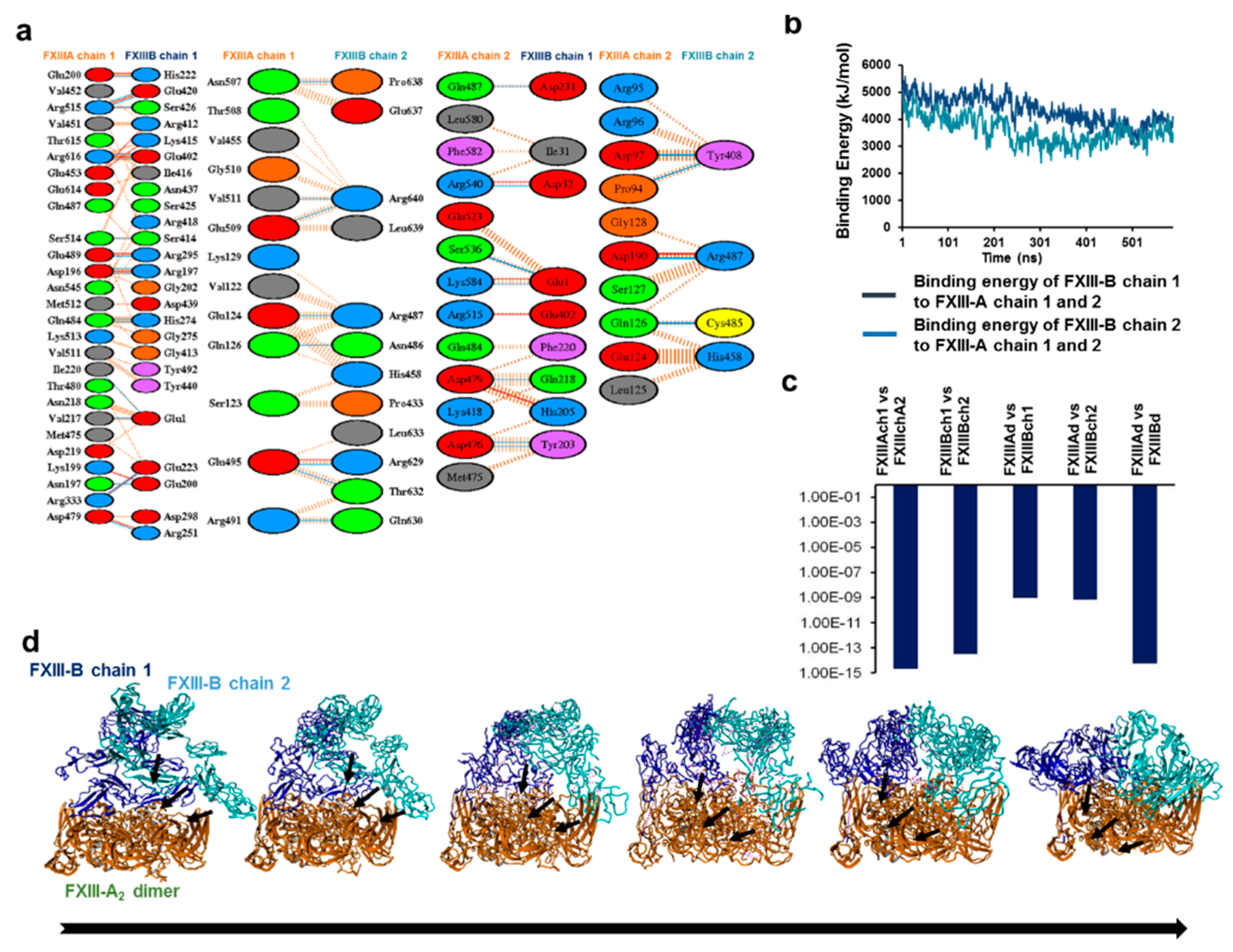
3.2. Cross-Links in the FXIII-A2B2 Complex Interface Expose Reverse, N-to C-Terminal Symmetry between FXIII-A and FXIII-B Subunits
3.3. Molecular Docking Reveals a Stoichiometrically-Symmetrical, Bi-Partite, FXIII-A2B2 Complex
3.4. Molecular Docking into the AFM Topographs Identifies the Best Model Representative of the Native FXIII Complex
3.5. Unequal Pairing within the Bi-Partite FXIII-A2B2 Complex Influences Dissociation of Subunits during FXIII Activation
3.6. Transition State Analysis Suggests Stepwise Binding Events during the Association of FXIII-A: FXIII-B Subunits
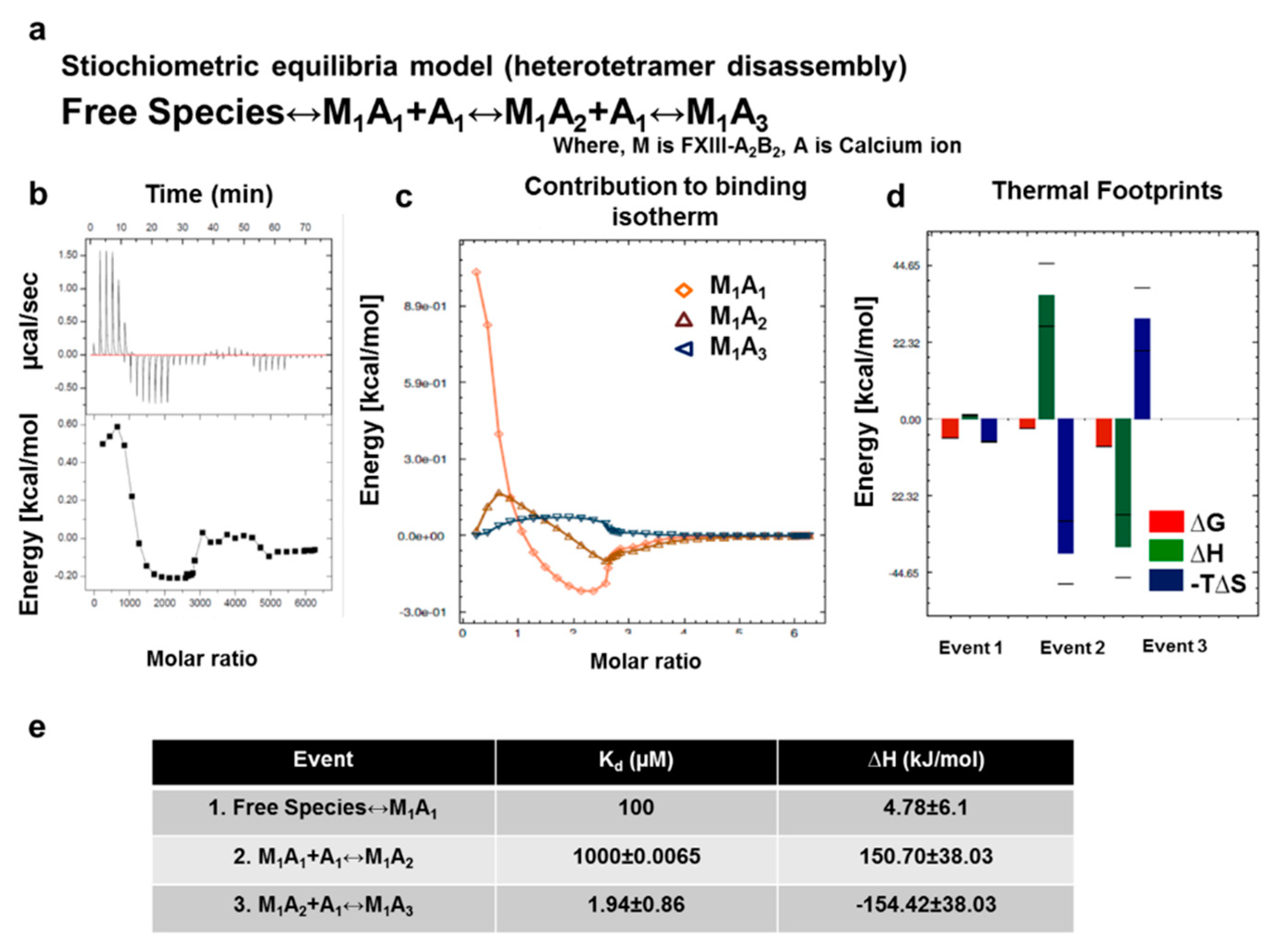
3.7. Thermodynamic Patterns Underlying FXIII-A2B2 Complex Assembly and Dissociation Suggest Stepwise Models for Both Events
4. Discussion
4.1. IH Approaches Reveal a Unique FXIII Complex Structure
4.2. Assembly of FXIII-A2B2 is a Two-Step Process Aided by the Conformational Flexibility of FXIII-B Subunit
4.3. Unequal Pairing within the FXIII Complex May Generate a Transient FXIII-AB Species during the Activation Induced Complex Disassembly
4.4. Is Complex Interface a Potential Underlying Driver of Unexplained Heterozygous FXIII Mutations Observed in Mild FXIII Deficiency?
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Katona, E.; Pénzes, K.; Csapó, A.; Fazakas, F.; Udvardy, M.L.; Bagoly, Z.; Orosz, Z.Z.; Muszbek, L. Interaction of factor XIII subunits. Blood 2014, 123, 1757–1763. [Google Scholar] [CrossRef] [PubMed]
- Radek, J.T.; Jeong, J.M.; Wilson, J.; Lorand, L. Association of the A subunits of recombinant placental factor XIII with the native carrier B subunits from human plasma. Biochemistry 1993, 32, 3527–3534. [Google Scholar] [CrossRef] [PubMed]
- Biswas, A.; Ivaskevicius, V.; Thomas, A.; Oldenburg, J. Coagulation factor XIII deficiency. Diagnosis, prevalence and management of inherited and acquired forms. Hamostaseologie 2014, 34, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Stieler, M.; Weber, J.; Hils, M.; Kolb, P.; Heine, A.; Büchold, C.; Pasternack, R.; Klebe, G. Structure of active coagulation factor XIII triggered by calcium binding: Basis for the design of next-generation anticoagulants. Angew. Chem. Int. Ed. Engl. 2013, 52, 11930–11934. [Google Scholar] [CrossRef]
- Fox, B.A.; Yee, V.C.; Pedersen, L.C.; Le Trong, I.; Bishop, P.D.; Stenkamp, R.E.; Teller, D.C. Identification of the calcium binding site and a novel ytterbium site in blood coagulation factor XIII by x-ray crystallography. J. Biol. Chem. 1999, 274, 4917–4923. [Google Scholar] [CrossRef]
- Souri, M.; Kaetsu, H.; Ichinose, A. Sushi domains in the B subunit of factor XIII responsible for oligomer assembly. Biochemistry 2008, 47, 8656–8664. [Google Scholar] [CrossRef]
- Seelig, G.F.; Folk, J.E. Noncatalytic subunits of human blood plasma coagulation factor XIII. Preparation and partial characterization of modified forms. J. Biol. Chem. 1980, 255, 8881–8886. [Google Scholar]
- Gupta, S.; Biswas, A.; Akhter, M.S.; Krettler, C.; Reinhart, C.; Dodt, J.; Reuter, A.; Philippou, H.; Ivaskevicius, V.; Oldenburg, J. Revisiting the mechanism of coagulation factor XIII activation and regulation from a structure/functional perspective. Sci. Rep. 2016, 6, 30105. [Google Scholar] [CrossRef]
- Bullock, J.M.A.; Sen, N.; Thalassinos, K.; Topf, M. Modeling Protein Complexes Using Restraints from Crosslinking Mass Spectrometry. Structure 2018, 26, 1015–1024. [Google Scholar] [CrossRef]
- Zheng, J.; Corzo, C.; Chang, M.R.; Shang, J.; Lam, V.Q.; Brust, R.; Blayo, A.-L.; Bruning, J.B.; Kamenecka, T.M.; Kojetin, D.J.; et al. Chemical Crosslinking Mass Spectrometry Reveals the Conformational Landscape of the Activation Helix of PPARgamma; a Model for Ligand-Dependent Antagonism. Structure 2018, 26, 1431–1439.e6. [Google Scholar] [CrossRef]
- Yang, L.; Yang, D.; de Graaf, C.; Moeller, A.; West, G.M.; Dharmarajan, V.; Wang, C.; Siu, F.Y.; Song, G.; Reedtz-Runge, S.; et al. Conformational states of the full-length glucagon receptor. Nat. Commun. 2015, 6, 7859. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, X.; Zhang, Z.; Cai, G. Structure and Functional Interactions of INO80 Actin/Arp Module. J. Mol. Cell Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Chaves, R.C.; Dahmane, S.; Odorico, M.; Nicolaes, G.A.F.; Pellequer, J.-L. Factor Va alternative conformation reconstruction using atomic force microscopy. Thromb. Haemost. 2014, 112, 1167–1173. [Google Scholar] [CrossRef] [PubMed]
- Doerr, A. A home for integrative structural models. Nat. Methods 2018, 15, 409. [Google Scholar] [CrossRef]
- Burley, S.K.; Kurisu, G.; Markley, J.L.; Nakamura, H.; Velankar, S.; Berman, H.M.; Sali, A.; Schwede, T.; Trewhella, J. PDB-Dev: A Prototype System for Depositing Integrative/Hybrid Structural Models. Structure 2017, 25, 1317–1318. [Google Scholar] [CrossRef]
- Thomas, A.; Biswas, A.; Ivaskevicius, V.; Oldenburg, J. Structural and functional influences of coagulation factor XIII subunit B heterozygous missense mutants. Mol. Genet. Genom. Med. 2015, 3, 258–271. [Google Scholar] [CrossRef]
- Kaniyappan, S.; Chandupatla, R.R.; Mandelkow, E. Purification and Characterization of Low-n Tau Oligomers. Methods Mol. Biol. 2018, 1779, 99–111. [Google Scholar] [CrossRef]
- Xu, D.; Jaroszewski, L.; Li, Z.; Godzik, A. AIDA: Ab initio domain assembly server. Nucleic Acids Res. 2014, 42, W308–W313. [Google Scholar] [CrossRef]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef]
- Pierce, B.; Tong, W.; Weng, Z. M-ZDOCK: A grid-based approach for Cn symmetric multimer docking. Bioinformatics 2005, 21, 1472–1478. [Google Scholar] [CrossRef]
- van Zundert, G.C.P.; Rodrigues, J.P.G.L.M.; Trellet, M.; Schmitz, C.; Kastritis, P.L.; Karaca, E.; Melquiond, A.S.J.; van Dijk, M.; de Vries, S.J.; Bonvin, A.M.J.J. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef]
- Mattson, G.; Conklin, E.; Desai, S.; Nielander, G.; Savage, M.D.; Morgensen, S. A practical approach to crosslinking. Mol. Biol. Rep. 1993, 17, 167–183. [Google Scholar] [CrossRef]
- Bonvin, A.M.J.J.; Karaca, E.; Kastritis, P.L.; Rodrigues, J.P.G.L.M. Defining distance restraints in HADDOCK. Nat. Protoc. 2018, 13, 1503. [Google Scholar] [CrossRef]
- de Vries, S.J.; van Dijk, M.; Bonvin, A.M.J.J. The HADDOCK web server for data-driven biomolecular docking. Nat. Protoc. 2010, 5, 883–897. [Google Scholar] [CrossRef]
- Krieger, E.; Vriend, G. YASARA View—Molecular graphics for all devices—From smartphones to workstations. Bioinformatics 2014, 30, 2981–2982. [Google Scholar] [CrossRef]
- Krieger, E.; Koraimann, G.; Vriend, G. Increasing the precision of comparative models with YASARA NOVA--a self-parameterizing force field. Proteins 2002, 47, 393–402. [Google Scholar] [CrossRef]
- Pierce, B.G.; Wiehe, K.; Hwang, H.; Kim, B.-H.; Vreven, T.; Weng, Z. ZDOCK server: Interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinformatics 2014, 30, 1771–1773. [Google Scholar] [CrossRef]
- Franklin, J.; Koehl, P.; Doniach, S.; Delarue, M. MinActionPath: Maximum likelihood trajectory for large-scale structural transitions in a coarse-grained locally harmonic energy landscape. Nucleic Acids Res. 2007, 35, W477–W482. [Google Scholar] [CrossRef]
- Chaves, R.C.; Teulon, J.-M.; Odorico, M.; Parot, P.; Chen, S.-W.W.; Pellequer, J.-L. Conformational dynamics of individual antibodies using computational docking and AFM. J. Mol. Recognit. 2013, 26, 596–604. [Google Scholar] [CrossRef]
- Chaves, R.C.; Pellequer, J.-L. DockAFM: Benchmarking protein structures by docking under AFM topographs. Bioinformatics 2013, 29, 3230–3231. [Google Scholar] [CrossRef]
- Anokhin, B.A.; Stribinskis, V.; Dean, W.L.; Maurer, M.C. Activation of factor XIII is accompanied by a change in oligomerization state. FEBS J. 2017, 284, 3849–3861. [Google Scholar] [CrossRef] [PubMed]
- Godon, C.; Teulon, J.-M.; Odorico, M.; Basset, C.; Meillan, M.; Vellutini, L.; Chen, S.-W.W.; Pellequer, J.-L. Conditions to minimize soft single biomolecule deformation when imaging with atomic force microscopy. J. Struct. Biol. 2017, 197, 322–329. [Google Scholar] [CrossRef] [PubMed]
- Souri, M.; Osaki, T.; Ichinose, A. The Non-catalytic B Subunit of Coagulation Factor XIII Accelerates Fibrin Cross-linking. J. Biol. Chem. 2015, 290, 12027–12039. [Google Scholar] [CrossRef] [PubMed]
- Byrnes, J.R.; Wilson, C.; Boutelle, A.M.; Brandner, C.B.; Flick, M.J.; Philippou, H.; Wolberg, A.S. The interaction between fibrinogen and zymogen FXIII-A2B2 is mediated by fibrinogen residues gamma390-396 and the FXIII-B subunits. Blood 2016, 128, 1969–1978. [Google Scholar] [CrossRef]
- Kohler, H.P. Interaction between FXIII and fibrinogen. Blood 2013, 121, 1931–1932. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.A.; Pease, R.J.; Avery, C.A.; Brown, J.M.; Adamson, P.J.; Cooke, E.J.; Neergaard-Petersen, S.; Cordell, P.A.; Ariens, R.A.S.; Fishwick, C.W.G.; et al. The activation peptide cleft exposed by thrombin cleavage of FXIII-A(2) contains a recognition site for the fibrinogen alpha chain. Blood 2013, 121, 2117–2126. [Google Scholar] [CrossRef][Green Version]
- Wolberg, A.S. Fibrinogen and factor XIII: Newly recognized roles in venous thrombus formation and composition. Curr. Opin. Hematol. 2018, 25, 358–364. [Google Scholar] [CrossRef]
- Xue, L.C.; Rodrigues, J.P.; Kastritis, P.L.; Bonvin, A.M.; Vangone, A. PRODIGY: A web server for predicting the binding affinity of protein-protein complexes. Bioinformatics 2016, 32, 3676–3678. [Google Scholar] [CrossRef]
- Hitomi, K.; Kojima, S.; Fesus, L.; Máté, À. Demény, Ilma Korponay-Szabo, and Laszlo Fésüs. In Chapter 1; Structure of Transglutaminases: Unique Features Serve Diverse Functions; Hitomi, K., Kojima, S., Fesus, L., Eds.; Springer: Tokyo, Japan, 2015. [Google Scholar] [CrossRef]
- Kristiansen, G.K.; Andersen, M.D. Reversible activation of cellular factor XIII by calcium. J. Biol. Chem. 2011, 286, 9833–9839. [Google Scholar] [CrossRef]
- Ambrus, A.; Bányai, I.; Weiss, M.S.; Hilgenfeld, R.; Keresztessy, Z.; Muszbek, L.; Fésüs, L. Calcium binding of transglutaminases: A 43Ca NMR study combined with surface polarity analysis. J. Biomol. Struct. Dyn. 2001, 19, 59–74. [Google Scholar] [CrossRef]
- Dragan, A.I.; Read, C.M.; Crane-Robinson, C. Enthalpy-entropy compensation: The role of solvation. Eur. Biophys. J. 2017, 46, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Olsson, T.S.G.; Ladbury, J.E.; Pitt, W.R.; Williams, M.A. Extent of enthalpy-entropy compensation in protein-ligand interactions. Protein Sci. 2011, 20, 1607–1618. [Google Scholar] [CrossRef] [PubMed]
- Benfield, A.P.; Teresk, M.G.; Plake, H.R.; DeLorbe, J.E.; Millspaugh, L.E.; Martin, S.F. Ligand Preorganization May Be Accompanied by Entropic Penalties in Protein–Ligand Interactions. Angew. Chem. Int. Ed. 2006, 45, 6830–6835. [Google Scholar] [CrossRef] [PubMed]
- Al-Horani, R.A.; Karuturi, R.; Lee, M.; Afosah, D.K.; Desai, U.R. Allosteric Inhibition of Factor XIIIa. Non-Saccharide Glycosaminoglycan Mimetics, but Not Glycosaminoglycans, Exhibit Promising Inhibition Profile. PLoS ONE 2016, 11, e0160189. [Google Scholar] [CrossRef]
- Avery, C.A.; Pease, R.J.; Smith, K.; Boothby, M.; Buckley, H.M.; Grant, P.J.; Fishwick, C.W.G. (±) cis-Bisamido epoxides: A novel series of potent FXIII-A inhibitors. Eur. J. Med. Chem. 2015, 98, 49–53. [Google Scholar] [CrossRef]
- Badarau, E.; Collighan, R.J.; Griffin, M. Recent advances in the development of tissue transglutaminase (TG2) inhibitors. Amino Acids 2013, 44, 119–127. [Google Scholar] [CrossRef]
- Novakovic, J.; Wodzinska, J.; Tesoro, A.; Thiessen, J.J.; Spino, M. Pharmacokinetic studies of a novel 1,2,4-thiadiazole derivative, inhibitor of Factor XIIIa, in the rabbit by a validated HPLC method. J. Pharm. Biomed. Anal. 2005, 38, 293–297. [Google Scholar] [CrossRef]
- Protopopova, A.D.; Ramirez, A.; Klinov, D.V.; Litvinov, R.I.; Weisel, J.W. Factor XIII topology: Organization of B subunits and changes with activation studied with single-molecule atomic force microscopy. J. Thromb. Haemost. 2019, 17, 737–748. [Google Scholar] [CrossRef]
- Chang, C.-E.A.; McLaughlin, W.A.; Baron, R.; Wang, W.; McCammon, J.A. Entropic contributions and the influence of the hydrophobic environment in promiscuous protein-protein association. Proc. Natl. Acad. Sci. USA 2008, 105, 7456–7461. [Google Scholar] [CrossRef]
- Kurochkin, I.V.; Procyk, R.; Bishop, P.D.; Yee, V.C.; Teller, D.C.; Ingham, K.C.; Medved, L.V. Domain structure, stability and domain-domaininteractions in recombinant factor XIII. J. Mol. Biol. 1995, 248, 414–430. [Google Scholar] [CrossRef]
- Kanchan, K.; Ergülen, E.; Király, R.; Simon-Vecsei, Z.; Fuxreiter, M.; Fésüs, L. Identification of a specific one amino acid change in recombinant human transglutaminase 2 that regulates its activity and calcium sensitivity. Biochem. J. 2013, 455, 261–272. [Google Scholar] [CrossRef]
- Gopal, S.M.; Klumpers, F.; Herrmann, C.; Schäfer, L.V. Solvent effects on ligand binding to a serine protease. Phys. Chem. Chem. Phys. 2017, 19, 10753–10766. [Google Scholar] [CrossRef]
- Ben-Amotz, D. Water-Mediated Hydrophobic Interactions. Annu. Rev. Phys. Chem. 2016, 67, 617–638. [Google Scholar] [CrossRef]
- Song, M.; Hwang, H.; Im, C.Y.; Kim, S.-Y. Recent Progress in the Development of Transglutaminase 2 (TGase2) Inhibitors. J. Med. Chem. 2017, 60, 554–567. [Google Scholar] [CrossRef]
- Ivaškevičius, V.; Biswas, A.; Garly, M.-L.; Oldenburg, J. Comparison of F13A1 gene mutations in 73 patients treated with recombinant FXIII-A2. Haemophilia 2017, 23, e194–e203. [Google Scholar] [CrossRef]
- Thomas, A.; Biswas, A.; Dodt, J.; Philippou, H.; Hethershaw, E.; Ensikat, H.J.; Ivaskevicius, V.; Oldenburg, J. Coagulation Factor XIIIA Subunit Missense Mutations Affect Structure and Function at the Various Steps of Factor XIII Action. Hum. Mutat. 2016, 37, 1030–1041. [Google Scholar] [CrossRef]
- Biswas, A.; Ivaskevicius, V.; Thomas, A.; Varvenne, M.; Brand, B.; Rott, H.; Haussels, I.; Ruehl, H.; Scholz, U.; Klamroth, R.; et al. Eight novel F13A1 gene missense mutations in patients with mild FXIII deficiency: In silico analysis suggests changes in FXIII-A subunit structure/function. Ann. Hematol. 2014, 93, 1665–1676. [Google Scholar] [CrossRef]
- Ivaskevicius, V.; Biswas, A.; Bevans, C.; Schroeder, V.; Kohler, H.P.; Rott, H.; Halimeh, S.; Petrides, P.E.; Lenk, H.; Krause, M.; et al. Identification of eight novel coagulation factor XIII subunit A mutations: Implied consequences for structure and function. Haematologica 2010, 95, 956–962. [Google Scholar] [CrossRef]
- David, A.; Sternberg, M.J.E. The Contribution of Missense Mutations in Core and Rim Residues of Protein-Protein Interfaces to Human Disease. J. Mol. Biol. 2015, 427, 2886–2898. [Google Scholar] [CrossRef]
- Ivaskevicius, V.; Biswas, A.; Loreth, R.; Schroeder, V.; Ohlenforst, S.; Rott, H.; Krause, M.; Kohler, H.-P.; Scharrer, I.; Oldenburg, J. Mutations affecting disulphide bonds contribute to a fairly common prevalence of F13B gene defects: Results of a genetic study in 14 families with factor XIII B deficiency. Haemophilia 2010, 16, 675–682. [Google Scholar] [CrossRef]
- Hashiguchi, T.; Ichinose, A. Molecular and cellular basis of deficiency of the b subunit for factor XIII secondary to a Cys430-Phe mutation in the seventh Sushi domain. J. Clin. Investig. 1995, 95, 1002–1008. [Google Scholar] [CrossRef][Green Version]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, S.; Nazabal, A.; Kaniyappan, S.; Pellequer, J.-L.; Wolberg, A.S.; Imhof, D.; Oldenburg, J.; Biswas, A. The Plasma Factor XIII Heterotetrameric Complex Structure: Unexpected Unequal Pairing within a Symmetric Complex. Biomolecules 2019, 9, 765. https://doi.org/10.3390/biom9120765
Singh S, Nazabal A, Kaniyappan S, Pellequer J-L, Wolberg AS, Imhof D, Oldenburg J, Biswas A. The Plasma Factor XIII Heterotetrameric Complex Structure: Unexpected Unequal Pairing within a Symmetric Complex. Biomolecules. 2019; 9(12):765. https://doi.org/10.3390/biom9120765
Chicago/Turabian StyleSingh, Sneha, Alexis Nazabal, Senthilvelrajan Kaniyappan, Jean-Luc Pellequer, Alisa S. Wolberg, Diana Imhof, Johannes Oldenburg, and Arijit Biswas. 2019. "The Plasma Factor XIII Heterotetrameric Complex Structure: Unexpected Unequal Pairing within a Symmetric Complex" Biomolecules 9, no. 12: 765. https://doi.org/10.3390/biom9120765
APA StyleSingh, S., Nazabal, A., Kaniyappan, S., Pellequer, J.-L., Wolberg, A. S., Imhof, D., Oldenburg, J., & Biswas, A. (2019). The Plasma Factor XIII Heterotetrameric Complex Structure: Unexpected Unequal Pairing within a Symmetric Complex. Biomolecules, 9(12), 765. https://doi.org/10.3390/biom9120765